

Abstract
Irreversible HER/erbB inhibitors selectively inhibit HER-family kinases by targeting a unique cysteine residue located within the ATP-binding pocket. Sequence alignment reveals that this rare cysteine is also present in ten other protein kinases including all five Tec-family members. We demonstrate that the Tec-family kinase Bmx is potently inhibited by irreversible modification at Cys496 by clinical stage EGFR-inhibitors such as CI-1033. This cross-reactivity may have significant clinical implications.
The activities of HER/erbB family tyrosine kinases (HER-TKs) are abnormally activated in various human epidermal cancers, and several anticancer drugs have been designed to specifically target HER-TKs.1 Two clinically approved quinazoline scaffold-based drugs, gefitinib (Iressa™) and erlotinib (Tarceva™) (Fig. 1), are selective, ATP-competitive EGFR (Epidermal Growth Factor Receptor, HER1) inhibitors that reversibly bind to the adenosine triphosphate (ATP) binding site of the enzyme with a high affinity.
Figure 1.

Chemical structures of quinazoline scaffold-EGFR inhibitors.
In the late 1990s, rational drug design efforts resulted in the identification of the prototypical potent irreversible HER1/2 kinase inhibitor PD168393.2 A reactive acrylamide moiety was incorporated at the 6-position of a 4-anilinoquinazoline in order to affect a Michael-addition reaction with a relatively unique cysteine residue (Cys773) within the ATP binding pocket of EGFR. Notably, the irreversible nature of the inhibitor turned out to be advantageous in achieving superior antitumor activity in vivo, due to its rapid reaction and long-lasting inactivation of the enzyme. Moreover, a recent study demonstrated that other irreversible EGFR inhibitors such as EKB-569 and CI-1033 can strongly inhibit a gefitinib- and erlotinib-resistant gatekeeper mutant of EGFR (T790M), demonstrating further therapeutic utility for irreversible inhibitors.3, 4 Currently, six irreversible HER-TK inhibitors are in clinical development for the treatment of a variety of cancers. However, a recent clinical phase II study reported that CI-1033 is associated with severe toxicity, suggesting that further development of the drug seems unlikely.5, 6 Here, we report that such irreversible HER-TK inhibitors are also capable of potently inhibiting the Tec-family kinase Bmx/Etk. To our knowledge, this is also the first report of a sub-micromolar Bmx/Etk inhibitor.
As both the reversible and irreversible quinazoline inhibitors have been reported to be highly selective to HER-TKs, we hypothesized that other kinases bearing this unique cysteine residue might also be targeted by these inhibitors. We first performed a sequence alignment to determine which other protein kinases possess an equivalently positioned cysteine to Cys773 of EGFR. In addition to the other HER-TKs HER2 and HER4, eight more kinases were identified, including Jak3, Blk, Lkb1, and the Tec-family kinases (Bmx, Btk, Itk, Tec, and Txk)6 (Fig. 2a). In order to determine which of these kinases might also be targeted by irreversible quinazoline inhibitors, we tested PD168393 and CI-1033 (Fig. 1) against a panel of 37 different Tel-tyrosine kinase transformed Ba/F3 cell lines.7 PD168393 was also tested against a panel of sixty biochemical kinase assays. Of the reactive cysteine-containing kinases, the biochemical panel included EGFR, HER2/4, Jak-3, Blk, Lkb-1, Bmx, BTK, and Itk, while the cellular panel contained Jak3, Bmx, and Blk. While moderate to no inhibition was seen for the majority of the kinases tested, CI-1033 demonstrated potent inhibitory activity against Bmx and Blk in both biochemical and cellular assays, whereas PD168393 exhibited only moderate potency against Bmx (Fig. 2b and Supplementary Table 1 and 2). The observation that only a subset of the kinases possessing an equivalently positioned reactive cysteine are significantly inhibited by these compounds demonstrates that potent inhibition requires a combination of non-covalent recognition of the scaffold by the kinase and proper positioning of the reactive cysteine nucleophile. Indeed, similar principles were recently used to design cysteine alkylating inhibitors for Rsk,8 VEGFR,9 and Tec-family kinase BTK.6
Figure 2.

Irreversible quinazoline EGFR inhibitors cross-react with other tyrosine kinases that possess a conserved reactive cysteine in their kinase domain. (a) The sequence of EGFR active site and its alignment with ten other kinases. The common putative nucleophilic cysteine residue is highlighted in bold. (b) Inhibition (IC50, μM) of kinases by PD168393 and CI-1033 examined by biochemical enzymatic assays and cytotoxicity assays using Tel-tyrosine kinase transformed Ba/F3 cells (ND = Not Determined).
We next used mass spectrometry to confirm the covalent nature of the inhibition, the stoichiometry of binding, and the site(s) of modification of the inhibitors on recombinantly expressed Bmx kinase domain. After incubation with PD168393, an average mass of 31,732 Da was measured for the Bmx kinase domain. This represents an increase of 369 Da, which is consistent with the covalent addition of a single inhibitor molecule (Fig. 3a and 3b). A second observed species corresponds to the covalent addition of the singly phosphorylated form of the domain by a single inhibitor molecule. Similar results were obtained upon incubation with CI-1033, as the expected mass increase of 485 Da was observed for both protein signals (Fig. 3c).
Figure 3.
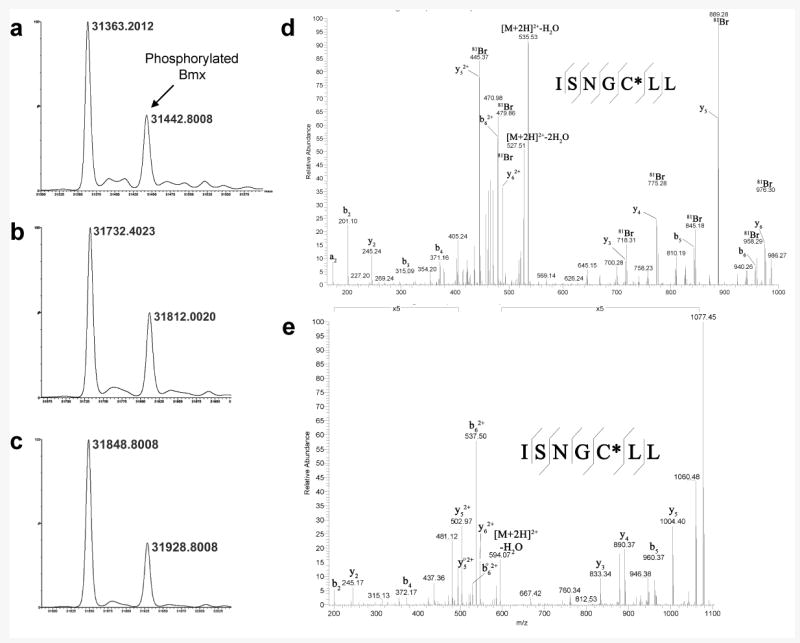
MS analysis of irreversible EGFR inhibitors binding to Bmx kinase domain. MS spectra of (a) Bmx kinase domain, and the protein after addition of (b) PD168393 or (c) CI-1033 showing mass increases of 369 Da and 485 Da respectively. Tandem mass spectra of peptide ISNGCLL resulting from chymotryptic digestion of Bmx kinase domain after addition of (d) PD168393 or (e) CI-1033, confirming modification of cysteine 496.
To determine the site(s) of modification, the inhibitor-modified samples were reduced with dithiothreitol, alkylated with iodoacetamide, and subjected to chymotryptic digestion. Subsequent analysis by LC/MS/MS identified several overlapping species that suggest Cys496 as the dominant nucleophilic site, which is analogous to Cys773 of EGFR in the sequence alignment. Fig. 3d and 3e show the tandem MS spectra of the chymotryptic peptide ISNGCLL modified with PD168393 or CI-1033 respectively. It should be noted that partial modification of Cys516 by PD168393 (Supplementary Fig. 1a) was also observed. However, the signal for this species was more than 400 times lower than the signal for species containing modified Cys496 (Supplementary Fig. 1b). Furthermore, iodoacetamide-modified peptides of high signal intensity were identified for every cysteine residue in the Bmx kinase domain except Cys496 (data not shown).
To confirm that Cys496 of Bmx is important for the cellular activity of the inhibitors, we created a Ba/F3 cell line stably transformed with a Tel-C496S Bmx mutant kinase domain. This substitution resulted in almost 30-fold less sensitivity to PD168393 (IC50 = 8.58 and 0.324 μM for mutant and WT respectively, Fig. 4). We next examined the capability of the compounds to inhibit full-length Bmx in a cellular context. Flag-tagged Bmx was transfected to human prostate cancer LNCaP cells and an in vitro kinase assay was performed using immunoprecipitated Flag-Bmx in the presence or absence of reversible and irreversible clinical EGFR inhibitors (Fig. 5a).10 The in vitro autophosphorylation of Bmx, monitored by measuring incorporation of labeled phosphate from 32P-ATP to the immunoprecipitated Flag-Bmx, was almost completely inhibited upon treatment with either PD168393 (10 μM) or CI-1033 (10 μM), whereas it was not affected by treatment of the potent reversible EGFR inhibitors gefitinib (10 μM) and erlotinib (10 μM). The same result was obtained when Flag-Bmx was immunopurified from the cells that were pre-treated with the drugs for 4 hrs. Immunoblot analysis using anti-phosphotyrosine antibody demonstrated only the irreversible but not the reversible inhibitors were able to inhibit phosphorylation (Fig. 5b). These results demonstrate that both PD168393 and CI-1033 inhibit the activity of Bmx in cells in a Cys496-dependent fashion.
Figure 4.

Cellular IC50 curves of PD168393 against Tel-wild-type and Tel-mutant Bmx (C496S) transformed Ba/F3 cells.
Figure 5.

Irreversible EGFR inhibitors inhibit autophosphorylation of overexpressed Bmx in LNCaP cells, whereas reversible inhibitors show no effect. (a) Immunoprecipitated Flag-Bmx was pre-incubated with different drugs at 4°C for 2 hrs, and in vitro kinase assay using 32P-ATP was performed. (b) In vitro kinase assay using Flag-Bmx immunopurified from the cells that were pre-treated with drugs for 4 hrs. Autophosphorylation was examined by using both radiography and phosphotyrosine immunoblot analysis.
Unlike other Tec-family kinases that are preferentially expressed in hematopoietic cells, Bmx/Etk is expressed in other cell types as well, including endothelial, epithelial, and importantly metastatic carcinoma cells.11 As Bmx is involved in the migration of both epithelial and endothelial cells,12 Bmx inhibitors could potentially be used for antimetastasis and antiangiogenesis therapies. Blk is a key player in signaling B-cell growth inhibition in response to surface IgM cross-linking. Thus, Blk inhibitors could also prevent B-cells from apoptosis13 and be used to boost immune functions. Here we have demonstrated that kinases such as Bmx and Blk can potently be inhibited by covalent inhibitors from the 4-anilinoquinazoline scaffold class. Our results suggest that it will be important to test the numerous other irreversible clinical EGFR inhibitors for their propensity to cross-react with the cysteine-containing kinases highlighted in this study.
Supplementary Material
Supplementary materials, figures, and tables associated with this article can be found, in the online version, at
Supplementary Figure 1. Alkylation of cysteine 516 within Bmx kinase domain by PD168393 (a) Tandem mass spectra of peptide DVCEGMAF resulting from chymotryptic digestion of Bmx kinase domain after addition of PD168393, confirming partial modification of cysteine 516. (b) Single ion chromatograms of covalently-modified peptides resulting from chymotryptic digestion of Bmx kinase domain after addition of PD168393, demonstrating more than 450 times higher signal of C496-modified species compared to C516-modified species. No enzyme specificity was set for the custom database search that allowed for variable modification of cysteine residues.
Acknowledgments
We thank Dr. Dario Alessi for his help on Lkb1 biology, and Mr. Scott Brittain for his help with the intact protein mass measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sharma SV, Bell DW, Settleman J, Haber DA. Nat Rev Cancer. 2007;7:169. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 2.Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, McNamara DJ, Nelson JM, Sherwood V, Smaill JB, Trumpp-Kallmeyer S, Dobrusin EM. Proc Natl Acad Sci USA. 1998;95:12022. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, Sharma SV, Isselbacher KJ, Settleman J, Haber DA. Proc Natl Acad Sci USA. 2005;102:7665. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, Floyd M, Ford JM, Grotzfeld RM, Herrgard S, Insko DE, Mehta SA, Patel HK, Pao W, Sawyers CL, Varmus H, Zarrinkar PP, Lockhart DJ. Proc Natl Acad Sci USA. 2005;102:11011. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campos S, Hamid O, Seiden MV, Oza A, Plante M, Potkul RK, Lenehan PF, Kaldjian EP, Varterasian ML, Jordan C, Charbonneau C, Hirte H. J Clin Oncol. 2005;23:5597. doi: 10.1200/JCO.2005.08.091. [DOI] [PubMed] [Google Scholar]
- 6.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, Young PR, Dalrymple SA, Palmer JT. Chem Med Chem. 2007;2:58. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 7.Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, Beigi R, Xia G, Harig RA, Asatryan H, Yan SF, Zhou Y, Gu XJ, Saadat A, Zhou V, King FJ, Shaw CM, Su AI, Downs R, Gray NS, Schultz PG, Warmuth M, Caldwell JS. Proc Natl Acad Sci USA. 2006;103:3153. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen MS, Zhang C, Shokat KM, Taunton J. Science. 2005;308:1318. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wissner A, Floyd MB, Johnson BD, Fraser H, Ingalls C, Nittoli T, Dushin RG, Discafani C, Nilakantan R, Marini J, Ravi M, Cheung K, Tan X, Musto S, Annable T, Siegel MM, Loganzo F. J Med Chem. 2005;48:7560. doi: 10.1021/jm050559f. [DOI] [PubMed] [Google Scholar]
- 10.Jiang X, Borgesi RA, McKnight NC, Kaur R, Carpenter CL, Balk SP. J Biol Chem. 2007;282:32689. doi: 10.1074/jbc.M703412200. [DOI] [PubMed] [Google Scholar]
- 11.Qiu Y, Robinson D, Pretlow TG, Kung HJ. Proc Natl Acad Sci USA. 1998;95:3644. doi: 10.1073/pnas.95.7.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan S, An P, Zhang R, He X, Yin G, Min W. Mol Cell Biol. 2002;22:7512. doi: 10.1128/MCB.22.21.7512-7523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao XR, Scott DW. Proc Natl Acad Sci USA. 1993;90:7946. doi: 10.1073/pnas.90.17.7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials, figures, and tables associated with this article can be found, in the online version, at
Supplementary Figure 1. Alkylation of cysteine 516 within Bmx kinase domain by PD168393 (a) Tandem mass spectra of peptide DVCEGMAF resulting from chymotryptic digestion of Bmx kinase domain after addition of PD168393, confirming partial modification of cysteine 516. (b) Single ion chromatograms of covalently-modified peptides resulting from chymotryptic digestion of Bmx kinase domain after addition of PD168393, demonstrating more than 450 times higher signal of C496-modified species compared to C516-modified species. No enzyme specificity was set for the custom database search that allowed for variable modification of cysteine residues.