

Abstract
Chromosome 17 is unusual among the human chromosomes in many respects. It is the largest human autosome with orthology to only a single mouse chromosome1, mapping entirely to the distal half of mouse chromosome 11. Chromosome 17 is rich in protein-coding genes, having the second highest gene density in the genome2,3. It is also enriched in segmental duplications, ranking third in density among the autosomes4. Here we report a finished sequence for human chromosome 17, as well as a structural comparison with the finished sequence for mouse chromosome 11, the first finished mouse chromosome. Comparison of the orthologous regions reveals striking differences. In contrast to the typical pattern seen in mammalian evolution5,6, the human sequence has undergone extensive intrachromosomal rearrangement, whereas the mouse sequence has been remarkably stable. Moreover, although the human sequence has a high density of segmental duplication, the mouse sequence has a very low density. Notably, these segmental duplications correspond closely to the sites of structural rearrangement, demonstrating a link between duplication and rearrangement. Examination of the main classes of duplicated segments provides insight into the dynamics underlying expansion of chromosome-specific, low-copy repeats in the human genome.
Human chromosome 17 is implicated in a wide range of human genetic diseases. It is home to genes involved in early-onset breast cancer (BRCA1), neurofibromatosis (NF1) and the DNA damage response (TP53 encoding the p53 protein). The complex rearrangement and duplication structure of chromsome 17 predisposes it to non-allelic homologous recombination (NAHR), resulting in DNA rearrangements that cause several well-studied microdeletion disorders. These include herditary neuropathy with pressure palsies (HNPP)7 at 17p12, and Smith–Magenis syndrome (SMS) deletions at 17p11.2 (refs 8, 9). Microduplication counterparts for both of these conditions are also known, famously in the case of Charcot–Marie–Tooth disease type 1A (CMT1A) at 17p12 (refs 10, 11), the most common inherited peripheral neuropathy (see ref. 12 for a review), and more recently for SMS13. Further, the complex architecture in this region, consisting of ∼50-kb subunits of direct and inverted repeats able to form cruciform structures, is also responsible for susceptibility to one of the most common somatic rearrangement events characterized, isodicentric 17q, which is associated with several cancers and signifies poor prognosis14. Finally, all cases of Miller–Dieker syndrome12 and many cases of isolated lissencephaly sequence12 are associated with haploinsufficiency effects of deletions at 17p13.3.
As part of the Human Genome Project2, we generated a finished sequence of human chromosome 17, which comprises ∼2.8% of the euchromatic genome. The finished sequence contains 78,839,971 bases and is interrupted by nine euchromatic gaps and one gap containing the centromere region (Fig. 1). The total size of the euchromatic gaps is estimated at 854 kb (see Methods and Supplementary Table S1). The frequency and size of gaps is slightly higher than the human genome average2. (It is also significantly higher than for mouse chromosome 11; see below.) This is largely due to segmentally duplicated or unclonable regions on human chromosome 17 (see Supplementary Information). A clone list for the most recent version of the sequence is provided in Supplementary Table S2.
Figure 1. Landscapes of human chromosome 17 and mouse chromosome 11.
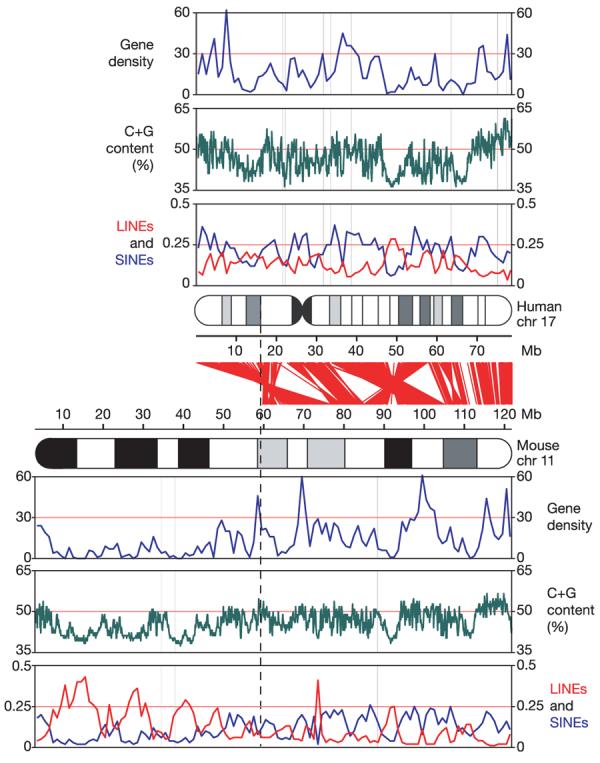
Approximate alignments of ideograms of the two chromosomes are shown in the centre of the figure, with red lines showing the relationships between orthologous genes. From top to bottom for each organism, tracks show gene density (in genes per Mb), G+C content on a scale from 35–65%, and densities of LINEs (red) and SINEs (blue) (fraction of bases). The vertical dashed line represents the approximate boundary between the distal region of mouse chromosome 11, which has shared synteny with human chromosome 17, and the proximal region, which does not. Chr, chromosome.
An overview of chromosome 17 is shown in Fig. 1. The chromosome has features characteristic of gene-rich regions2, including a high average G+C content (45.5%) and a collection of transposable element fossils (45.3% overall) with a relative excess of short interspersed elements (SINEs, 22.3%) and a deficit of long interspersed elements (LINEs, 14.4%). It also has a large excess of segmental duplications (defined as having >90% identity and being >1 kb in length15). Such segmental duplications constitute 8.6% (7 Mb) of the finished sequence, in contrast to a median of 3.9% for human chromosomes and an average of 4.3% across autosomal sequences. As with other duplication-rich chromosomes, chromosome 17 has a large proportion of intrachromosomal duplication: 62.3% of the duplicated sequence is strictly intrachromosomal, 17.4% is both intra- and inter-chromosomal, and 20.3% is solely interchromosomal. We elaborate below on the segmental duplication content of the chromosome.
We have produced a manually curated catalogue of protein-coding genes on chromosome 17 (see Methods), annotating 1,266 gene loci and 274 pseudogene loci (Supplementary Table S3). This catalogue includes all of the 1,079 genes on the chromosome in the RefSeq database (see Methods and Supplementary Information). The gene density (16.2 genes per Mb) is substantially higher than the genome average2 and ranks only behind chromosome 19 (26.2 genes per Mb; ref. 3). There is evidence of extensive alternative splicing, with gene loci having an average of five distinct transcripts, and 76.6% having at least two transcripts, a proportion comparable to recent reports3,16-19. Of the 274 pseudogenes on the chromosome, 73.0% are processed (intronless) pseudogenes arising from retroposition. In addition to protein-coding genes, we identified 42 transfer RNA genes on the chromosome (Supplementary Table S4).
As part of the Mouse Genome Sequencing Consortium (MGSC), a finished sequence of mouse chromosome 11 has been produced (Supplementary Table S5). The sequence was derived from the C57BL/6J strain and has a finished euchromatic length of 118,816,080 bases, representing ∼4.6% of the mouse genome. The finished sequence is interrupted by only two euchromatic gaps, with an estimated total size of 97 kb, and one additional gap containing the acrocentric heterochromatin (Fig. 1, Supplementary Table S1 and Methods). See Supplementary Table S5 for a clone list of the most up-to-date version of mouse chromosome 11.
Mouse chromosome 11 can be divided into two parts on the basis of conserved synteny with human chromosomes (Fig. 1). Although the proximal 59 Mb shows conserved synteny with five different human chromosomes (1, 2, 5, 7 and 22), the distal ∼60 Mb has conserved synteny exclusively with human chromosome 17 (see Fig. 1 and Supplementary Fig. S1). The distal portion of mouse chromosome 11 is similar to human chromosome 17 in various respects, including having a high G+C content (45.5%), high gene density (18.7 genes per Mb), an excess of SINEs (14.4%), and a deficit of LINEs (8.2%). The ratio of SINEs to LINEs is similar to that in human, although the absolute density is lower, consistent with the fraction of detectable transposon sequence in mouse. In contrast, the proximal portion of mouse chromosome 11 has a lower G+C content (41.9%), low gene density (8.2 genes per Mb), and an opposite ratio of SINEs and LINEs (7.8% versus 19.1%). Although it is similar to human chromosome 17 in most other respects, the distal region of mouse chromosome 11 differs sharply with respect to segmental duplications: such sequences constitute only 1.1% of the distal region. Indeed, mouse chromosome 11 has the lowest rate of segmental duplication (1.4%) among all mouse chromosomes.
The block of undirected synteny (corresponding regions of orthology between the species, in which order may be disrupted by internal rearrangements) between human chromosome 17 and mouse chromosome 11 is the second largest such autosomal block between human and mouse (see Supplementary Information). Within this block, there are 23 segments of directed (collinear) synteny of >100 kb, indicating frequent inversion. To better understand this relationship, we reconstructed the chromosomal structure of the primate–rodent ancestor of human chromosome 17 (Supplementary Fig. S2) on the basis of genome sequences from human, mouse, dog and opossum (see Supplementary Information). The primate–rodent ancestor could be resolved except for one small segment, and also represents the state of the boreoeutherian ancestor except in two regions of conserved directed synteny between human and mouse that differ from dog and opossum (see Supplementary Information and Supplementary Fig. S2).
Comparison of the modern chromosomes with our ancestral reconstruction (Fig. 2a) shows that the mouse structure is virtually unchanged, whereas significant rearrangements have occurred in human. There are 20 rearrangement breakpoints in human but only three in mouse. (The terminal break and one internal break are used for different events in both species, leading to only 22 observed differences in the map of direct human–mouse conserved synteny; see Supplementary Fig S1.) This is contrary to the general pattern for the human and mouse genomes, which typically shows many more rearrangements along the rodent lineage5,6 (see Supplementary Information). The mouse-specific rearrangements are confined to a single region orthologous to the Smith–Magenis region in human, which shows an even more complex pattern of rearrangements at finer scale (Fig. 2b).
Figure 2. Syntenic relationship between mouse, human and the ancestral chromosome.

a, Conserved syntenic blocks of 100 kb or more between human chromosome 17 (Hs17), mouse chromosome 11 (Mm11), and a most parsimonious primate–rodent ancestor (Anc) reconstructed using dog and opossum as outgroups. b, An enlargement of the CMT1A/SMS region and the mouse orthologous sequence (human 14–22 Mb, mouse 59–64 Mb), including conserved syntenic blocks of 10 kb or more between human and mouse, and human segmental duplications of 20 kb or more. Segmental duplications are shown above the human CMT1A/SMS line. Duplications with copies outside the enlarged region connect to 17q in a. Coloured blocks marked ‘C’ represent CMT1A duplicons, and those marked ‘S’ represent SMS duplicons. In both sections, direct (reference strand to reference strand) blocks of conserved synteny or segmental duplication are shown in blue; inverted blocks are shown in green. Chromosome blocks are labelled in megabases. The black circle indicates the location of the human chromosome 17 centromere.
What accounts for the extreme rearrangement of human chromosome 17? The answer seems to be related to the extensive segmental duplication on the human chromosome. The human-specific breaks in conserved synteny are highly correlated with regions of intrachromosomal duplication (Fig. 3). Roughly 74% of the duplicated bases reside in the breakpoint regions between stretches of conserved synteny, which comprise only 7.3% of the bases in the chromosome sequence.
Figure 3. Duplication landscape of chromosome 17 and its association with breaks in conserved synteny.
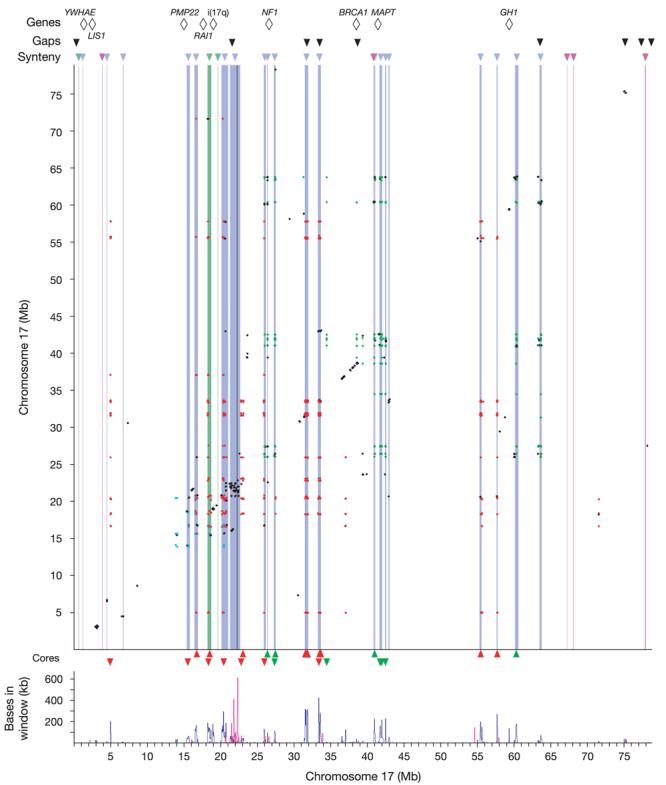
The top line shows genes/breakpoints of interest for rearrangements mentioned in the text. Black triangles show the locations of the nine remaining gaps in the build. Below that, coloured triangles show breaks in synteny (blue for human, green for mouse, pink for dog). Intrachromosomal duplications are shown as an all-versus-all dot plot, coloured by class (class 1 in red, class 2 in green, class 3 in blue, all other classes in black; see Methods). Breaks in conserved synteny are represented as vertical bands coloured as above. The centromere is shown as a vertical dark grey band. Triangles at the bottom show core element locations (class 1 core elements in red, class 2 core elements in green), with direction showing orientation (up, forward strand; down, reverse strand). The panel at the bottom shows a density plot of intrachromosomal (blue) and interchromosomal (red) duplication, using 50-kb non-overlapping sliding windows.
To further explore the nature of these duplicated sequences, we clustered the duplicons into classes on the basis of shared sequence (see Methods). After clustering, most of the duplicated sequences fall into one of three classes (1–3), which account for 51.6%, 19.9% and 3.1%, respectively, of the intrachromosomal duplications (Fig. 3 and Table 1). For each duplication class, we then sought to define a ‘core element', corresponding to the maximum length of sequence occurring in the largest number of duplicons (see Methods). These core elements can serve as useful markers for identifying class members and may provide insight into the origins of the duplications20.
Table 1. Duplication classes on human chromosome 17.
| Class | No. of duplications |
No. of duplications containing core element |
No. of duplications in syntenic break regions |
Core element size (bp) |
Total bases in duplicons |
Duplicon bases in syntenic break regions |
Total duplicon bases in syntenic break regions (%) |
|---|---|---|---|---|---|---|---|
| Class 1 | 529 | 225 | 509 | 10,734 | 1,837,890 | 1,581,557 | 86.05 |
| Class 2 | 204 | 65 | 202 | 4,185 | 979,083 | 735,413 | 75.11 |
| Class 3 | 32 | 0 | 30 | NA | 125,758 | 105,741 | 84.08 |
| Other (132)* | 260 | 0 | 196 | NA | 2,449,945 | 1,574,952 | 64.29 |
NA, not applicable.
Each of these 132 duplicon classes has fewer than 15 events.
The class 1 core element (∼11 kb in length) contains a TBC1 domain and is found in 12 of the 19 human-specific breakpoint regions. Several of the class 1 core elements are actively transcribed, including six very recently duplicated genes from the TBC1D3 family (at 31–33 Mb) and the USP6 oncogene (also known as TRE-2), which is a fusion of a TBC1D3 gene and the USP32 gene21. The TBC1D3 family genes encode proteins with TBC1 domains, and are believed to be GTPase-activating proteins specific to RAB5, a RAS-related gene involved in vesicle trafficking. The TBC1D3 gene family has no known homologues outside the primates, and we could find no similar sequence in dog or mouse, making it impossible to identify an ancestral copy. However, the element certainly pre-dates the primate clade: human copies show substantial divergence (∼34%) (Supplementary Fig. S3) and copies can be identified in ring-tailed lemur by polymerase chain reaction (PCR) (see Methods). The element is also duplicated in macaque, and a phylogeny of the elements in human and macaque (Supplementary Fig. S3) shows that at least some of these duplications pre-date the divergence of Old World monkeys, as some macaque copies have a human copy as their nearest neighbour.
The class 2 core element (∼4 kb in length) is found in 9 of the 19 human-specific breakpoint regions (Fig. 3). The ancestral copy is probably the copy immediately upstream of the NSF gene at 41.95 Mb, as this is the only copy with an orthologous sequence in mouse. The class 2 duplicons occur primarily in three regions of chromosome 17q, at 26–27 Mb, 40–42 Mb and 60–63 Mb. These regions are all sites of extensive, small local inversion events that have occurred since the primate–rodent ancestor. A phylogenetic tree of the core elements (Supplementary Fig. S4) shows two main clusters, one for the copies at 26–27 Mb and another for those at 40–42 and 60–63 Mb. Notably, the reconstruction of the ancestral chromosome order (Supplementary Fig. S2) places these latter regions nearly adjacent to one another, suggesting that the core elements dispersed in a local region that was then disrupted by an inversion with breakpoints at 33 and 57 Mb. This ancestral chromosomal order seems to correspond to the modern karyotype of orangutan22, which has a paracentric inversion with respect to the human karyotype on 17q, with nearly coincident breakpoints. This would imply that the inversion in the human lineage is a fairly recent event. Notably, two class 2 core elements (the ancestral copy and a copy at 40.97 Mb) flank the boundaries of a 900-kb polymorphic inversion in the human population23 and also the boundaries of a human rearrangement with respect to the ancestral chromosome (Fig. 2 and Supplementary Fig. S2), indicating a reuse of these breakpoints within the human lineage (see Supplementary Information).
The class 3 duplicons all reside in the CMT1A/SMS region (13.8–20.4 Mb), which seems to be a region of remarkable genomic fragility. The class is too small to define a meaningful core element, but examination of the longest copy shows that it is a previously identified low-copy repeat, LCRA110. This element is the result of a fusion of two distinct 3′-UTR elements from elsewhere in the human genome. Although at least one of the individual sub-elements comprising the fusion is found in mouse and dog, the fusion product is not seen in these species, but only in primates. One of the two sub-elements is actively transcribed (see Supplementary Information).
Previous studies8,10 of the CMT1A/SMS region have identified large, highly similar local repeat blocks. The blocks are themselves fusions of multiple duplicons (including class 1 core elements) that do not occur together elsewhere in the genome. None of these duplicated sequences has a mouse orthologue within the region (Fig. 2b), having all come from the q arm of 17, indicating that this region may be a site of multiple inversion. Indeed, cytogentic mapping of primate rearrangements22 shows the CMT1A/SMS region to be the site of an inversion breakpoint in orangutan and of a translocation between chromosomes 17 and 5 in gorilla24.
Certain common characteristics emerge from the study of segmental duplication on chromosome 17, as well as from our recent study of segmental duplications on chromosome 15 (ref. 20). First, the vast majority of duplicons in these classes fall in breaks of conserved synteny. Second, segmentally duplicated sequence seems to spread readily within local regions. Local clustering is evident for all duplicon classes studied on chromosomes 15 and 17. Third, segmentally duplicated sequence then seems to spread over larger distances by chromosomal inversions. In two cases (class 2 on chromosome 17 and the main class on chromosome 15; ref. 20), such dispersal is confirmed on the basis of both sequence phylogeny and reconstruction of ancestral chromosome structure. With additional comparative data on lower primates, it should be possible to reconstruct the dispersal of most of the segmentally duplicated sequences in the human genome. Finally, the core elements underlying the main classes of segmentally duplicated sequence on chromosomes 15 and 17 all show active transcription from a large number of the duplicons10,25,26. This observation suggests a link between active transcription, duplication and rearrangement. It is possible that the nature of transcription, the chromatin structure or the transcripts at these loci may render the regions more susceptible to breakage, duplication or non-allelic recombination.
Our comparison of human chromosome 17 and distal mouse chromosome 11 shows that a genomic region can undergo strikingly different rearrangements in different lineages. There has been remarkably little interchromosomal rearrangement in the region orthologous to human chromosome 17 across all mammals, which may perhaps reflect an inherent stability of the region or a selective constraint on contiguity. In contrast, the region shows extensive internal rearrangement in the human lineage (20 breakpoints relative to the ancestral genome) but little rearrangement in mouse (only three rearrangements relative to the ancestor). The occurrence of the human breakpoints near segmental duplications supports a mechanistic link between these two features that may explain the unusual history of this genomic region in the primate lineage. Together with our recent analysis of chromosome 15, this analysis sheds light on the forces that shape the large-scale structure of genomes and, in the process, create both evolutionary variation and the chromosomal fragility underlying human disease.
METHODS
The finished sequence of human chromosome 17 was generated almost entirely (>99%) at the Broad Institute of MIT and Harvard (formerly the Whitehead Institute/MIT Center for Genome Research) (Supplementary Table S6). Chromosomal positions in this paper refer to NCBI human build 35. The finished sequence of mouse chromosome 11 was generated almost entirely (>99%) at the Sanger Institute (Supplementary Table S6). Chromosomal positions in this paper refer to NCBI mouse build 34. Methods for clone mapping, sequencing and tiling-path validation are described in the Supplementary Information. The gene catalogue and annotation for human chromosome 17 were produced as previously published16.
Syntenic maps and segmental duplications
Syntenic maps and segmental duplications were constructed as previously described15,16,20.
Duplication class clustering
Pairwise duplications were clustered by physical overlap of ≥150 bp and at least 5% of the smaller element. We extended this by transitive closure to build maximally linked sets (see Supplementary Information).
Identification of core elements of duplicons
For each duplicon class, aligned coverage of each base in the duplicated region (the union of all duplicons) was recorded. Core elements were chosen by visual inspection to identify the longest contiguous sequence in the most deeply covered region. For each duplicon class, the longest core element sequence was aligned to the entire genome with PatternHunter27, and all full-length alignments were identified as core element copies.
Construction of core element phylogeny
Construction of core element phylogeny was performed as previously described20.
Acknowledgements
Special thanks are due to L. Gaffney for help with figures and tables. We thank T. Furey for help with lists of genetic markers and placement of RefSeqs, and K. Lindblad-Toh for sharing data on the opossum genome. We thank A. Kong for providing updated genetic map data, T. Hudson for technical advice, and the members of the Baylor College of Medicine Human Genome Sequencing Center, the J. Craig Venter Institute Joint Technology Center, and the Washington University Genome Sequencing Center for generation and early release of the genome assembly of the rhesus macaque. We also acknowledge the HUGO Gene Nomenclature Committee (S. Povey (chair), E. A. Bruford, R. C. Lovering, M. J. Lush, K. M. B. Sneddon, T. P. Sneddon, C. C. Talbot Jr and M. W. Wright) for assigning official gene symbols for human chromosome 17. We are grateful to all the members, present and past, of the Broad (and Whitehead) and Wellcome Trust Sanger sequencing platforms for their dedication and the consistent high quality of their data. The sequencing of human chromosome 17 was supported by a grant to the Whitehead Institute Center for Genome Research (now the Broad Institute) from the National Human Genome Research Institute (NHGRI). The sequencing of mouse chromosome 11 was supported by a grant to the Sanger Institute by the Wellcome Trust.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information Accession numbers for all clones contributing to the finished sequence of human chromosome 17 can be found in Supplementary Table S2, and for mouse chromosome 11 in Supplementary Table S5. The updated human chromosome 17 sequence can be accessed through GenBank accession number NC_000017. The updated mouse chromosome 11 sequence can be accessed through the accession numbers listed in Supplementary Table S5.
Reprints and permissions information is available at npg.nature.com/reprintsandpermissions.
The authors declare no competing financial interests.
References
- 1.Debry RW, Seldin MF. Human/mouse homology relationships. Genomics. 1996;33:337–351. doi: 10.1006/geno.1996.0209. [DOI] [PubMed] [Google Scholar]
- 2.International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 3.Grimwood J, et al. The DNA sequence and biology of human chromosome 19. Nature. 2004;428:529–535. doi: 10.1038/nature02399. [DOI] [PubMed] [Google Scholar]
- 4.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 5.International Chicken Genome Sequencing Consortium Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 6.Bourque G, Zdobnov EM, Bork P, Pevzner PA, Tesler G. Comparative architectures of mammalian and chicken genomes reveal highly variable rates of genomic rearrangements across different lineages. Genome Res. 2005;15:98–110. doi: 10.1101/gr.3002305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chance PF, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 8.Park SS, et al. Structure and evolution of the Smith–Magenis syndrome repeat gene clusters, SMS-REPs. Genome Res. 2002;12:729–738. doi: 10.1101/gr.82802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen KS, et al. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nature Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- 10.Inoue K, et al. The 1.4-Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes. Genome Res. 2001;11:1018–1033. doi: 10.1101/gr.180401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lupski JR, et al. DNA duplication associated with Charcot–Marie–Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 12.Lupski JR, Garcia CA. In: The Metabolic and Molecular Basis of Inherited Diseases. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. McGraw-Hill; New York: 2001. pp. 5759–5788. [Google Scholar]
- 13.Potocki L, et al. Molecular mechanism for duplication. 17p11.2—the homologous recombination reciprocal of the Smith–Magenis microdeletion. Nature Genet. 2000;24:84–87. doi: 10.1038/71743. [DOI] [PubMed] [Google Scholar]
- 14.Barbouti A, et al. The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am. J. Hum. Genet. 2004;74:1–10. doi: 10.1086/380648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey JA, et al. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nusbaum C, et al. DNA sequence and analysis of human chromosome 18. Nature. 2005;437:551–555. doi: 10.1038/nature03983. [DOI] [PubMed] [Google Scholar]
- 17.Nusbaum C, et al. DNA sequence and analysis of human chromosome 8. Nature. 2006;439:331–335. doi: 10.1038/nature04406. [DOI] [PubMed] [Google Scholar]
- 18.Hillier LW, et al. Generation and annotation of the DNA sequences of human chromosomes 2 and 4. Nature. 2005;434:724–731. doi: 10.1038/nature03466. [DOI] [PubMed] [Google Scholar]
- 19.Deloukas P, et al. The DNA sequence and comparative analysis of human chromosome 10. Nature. 2004;429:375–381. doi: 10.1038/nature02462. [DOI] [PubMed] [Google Scholar]
- 20.Zody MC, et al. Analysis of the DNA sequence and duplication history of human chromosome 15. Nature. 2006;440:671–675. doi: 10.1038/nature04601. [DOI] [PubMed] [Google Scholar]
- 21.Paulding CA, Ruvolo M, Haber DA. The Tre2 (USP6) oncogene is a hominoid-specific gene. Proc. Natl Acad. Sci. USA. 2003;100:2507–2511. doi: 10.1073/pnas.0437015100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yunis JJ, Prakash O. The origin of man: a chromosomal pictorial legacy. Science. 1982;215:1525–1530. doi: 10.1126/science.7063861. [DOI] [PubMed] [Google Scholar]
- 23.Stefansson H, et al. A common inversion under selection in Europeans. Nature Genet. 2005;37:129–137. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 24.Stankiewicz P, Park SS, Inoue K, Lupski JR. The evolutionary chromosome translocation 4;19 in Gorilla gorilla is associated with microduplication of the chromosome fragment syntenic to sequences surrounding the human proximal CMT1A-REP. Genome Res. 2001;11:1205–1210. doi: 10.1101/gr.181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eichler EE. Recent duplication, domain accretion and the dynamic mutation of the human genome. Trends Genet. 2001;17:661–669. doi: 10.1016/s0168-9525(01)02492-1. [DOI] [PubMed] [Google Scholar]
- 26.Stankiewicz P, Shaw CJ, Withers M, Inoue K, Lupski JR. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res. 2004;14:2209–2220. doi: 10.1101/gr.2746604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma B, Tromp J, Li M. PatternHunter: faster and more sensitive homology search. Bioinformatics. 2002;18:440–445. doi: 10.1093/bioinformatics/18.3.440. [DOI] [PubMed] [Google Scholar]