

Abstract
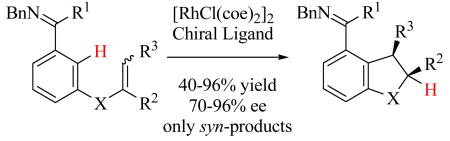
Highly enantioselective catalytic intramolecular ortho-alkylation of aromatic imines containing alkenyl groups tethered at the meta position relative to the imine directing group has been achieved using [RhCl(coe)2]2 and chiral phosphoramidite ligands. Cyclization of substrates containing 1,1- and 1,2-disubstituted as well as trisubstituted alkenes were achieved with enantioselectivities >90% ee for each substrate class. Cyclization of substrates with Z-alkene isomers proceeded much more efficiently than substrates with E-alkene isomers. This further enabled the highly stereoselective intramolecular alkylation of certain substrates containing Z/E-alkene mixtures via a Rh-catalyzed alkene isomerization with preferential cyclization of the Z-isomer.
Introduction
Transition metal catalyzed carbon–hydrogen (C–H) bond activation1 is one of the most powerful strategies for carbon–carbon (C–C) bond formation. Because the C–H bond is ubiquitous in organic substances, this method has broad potential. Moreover, direct conversion of C–H bonds into C–C bonds without intermediate pre-activation should shorten reaction sequences and reduce waste resulting in environmental benefits. Finally, catalytic C–H activation should enable novel bond connections to rapidly give rise to structurally complex molecules. In spite of these attractions, challenges in both the reactivity and selectivity for these transformations remain.
Following Murai’s ground-breaking discovery of Ru-catalyzed chelation-assisted alkylation of aromatic ketones,2 Jun reported that the scope of the reaction could be extended to isomerizable alkenes by the use of an imine directing group and Rh-catalyst.3 Previous work in our group extended this chemistry to intramolecular reactions, which showed significantly broadened reaction scope over intermolecular coupling reactions.4 For example, substrates with di- and even trisubstituted alkenes underwent cyclization, and aromatic aldimines and ketimines with heteroatom tethers such as vinyl ethers, allylic ethers, and allylic amines, also reacted cleanly. The synthetic utility of this transformation was demonstrated in its application to the synthesis of potent biologically active natural products and drug candidates.5
Examples of effective enantioselective catalytic C–H bond functionalizations are rare despite the high degree of activity in this area.6 Murai reported a chelation-assisted enantioselective alkylation utilizing the pyridine directing group in the intramolecular coupling of olefinic C–H bonds with alkenes; however, this transformation proceeds with only modest ee’s.7 While the cyclization of the analogous imidazolyl-1,5-diene gave significantly higher enantioselectivities, the reaction is limited in generality.7 The atropselective alkylation of 2-arylpyridines by transition metal-catalyzed C–H functionalization has also been reported although the ee did not exceed 49%.8 More recently Widenhoefer has reported the enantioselective platinum-catalyzed intramolecular alkylation of indoles tethered to unactivated terminal alkenes with selectivities up to 90% ee.9 On the other hand, high stereoselectivity and broader substrate scope have been obtained in intramolecular hydroacylation as initially reported for the cyclization of 4-pentanals into β-substituted cyclopentanones.10 In addition, Yu has very recently also reported on the Pd-catalyzed enantioselective alkylation of diphenyl(2-pyridyl)methane with alkylboronic acids using monoprotected amino acids as the chiral ligands.11
We previously communicated the catalytic enantioselective alkylation of aromatic ketimines with tethered 1,1-disubstituted alkenes (eq 1),12 which represented one of the first highly enantioselective catalytic reactions involving aromatic C H bond activation. Herein, we disclose the full details of this work and further report on ligand optimization that has enabled greatly expanded substrate scope, including the intramolecular hydroarylation of 1,2-disubstituted and 1,1,2-trisubstituted alkene substrates. Cyclization of substrates with Z-alkene isomers proceeded much more efficiently than substrates with E-alkene isomers. This further enabled the highly stereoselective intramolecular alkylation of certain substrates containing Z/E-alkene mixtures via a Rh-catalyzed alkene isomerization with preferential cyclization of the Z-isomer.
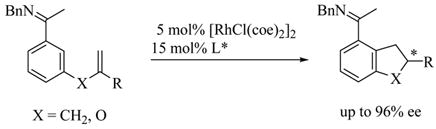 |
(1) |
Results and Discussion
A. Enantioselective Cyclization of 1,1-Disubstituted Alkenes
A.1. Ligand Screening
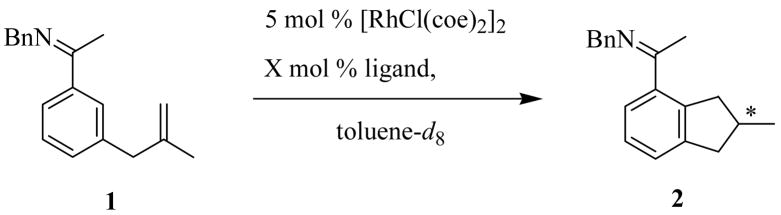
We initiated our study by testing the cyclization of alkene 1 using Rh complexes with various ligands in order to identify the optimal ligand for this reaction. Our efforts focused on chiral monodentate ligands13 because catalysts derived from chelating phosphines were inefficient for this reaction.4 Of the range of chiral monodentate phosphines screened (Chart 1), only phosphoramidites gave acceptable enantioselectivities. A brief summary of results for chiral phosphoramidites and the structurally related TADDOL-based ligands is given in Table 1.
Chart 1.

Chiral Monodentate Phosphorus Ligands
Table 1.
Asymmetric Cyclization of Alkene 1 Using Various Chiral Monophosphine Ligands
 | ||||||
|---|---|---|---|---|---|---|
| entry | ligand | ligand (mol%) | temp (°C) | time (h) | %yielda | % eeb |
| 1 | L1 | 15 | 75 | 20 | 93c | 17 (R) |
| 2 | L2 | 15 | 75 | 20 | 94c | 9 (R) |
| 3 | L3 | 15 | 75 | 20 | 91c | 38 (R) |
| 4 | L4 | 15 | 125 | 20 | 34c | 0 |
| 5 | L5 | 15 | 125 | 20 | 6c | n.t. |
| 6 | L6 | 15 | 125 | 2.5 | 15c | 19 (S) |
| 7 | L7 | 15 | 125 | 2.5 | 14c | 0 |
| 8 | L8 | 15 | 125 | 2.5 | 52c | 58 (S) |
| 9 | L9 | 5 | 125 | 1 | 96d | 83 (S) |
| 10 | L9 | 10 | 125 | 1 | 98d | 83 (S) |
| 11 | L9 | 15 | 125 | <2 | 100 | 83 (S) |
| 12 | L9 | 20 | 125 | 6 | 0 | -- |
| 13 | L9 | 30 | 125 | 6 | 0 | -- |
| 14 | L10 | 15 | 125 | <2 | 100 | 88 (S) |
| 15 | L11 | 15 | 125 | <2 | 99 | 87 (S) |
Yields based on 1H-NMR integration relative to 2,6-dimethoxytoluene internal standard.
Ee’s determined after hydrolysis of 2 with 1N HCl (aq) using chiral GC or HPLC. Sense of induction is indicated in parentheses. n.t.: not tested.
Remainder of mass balance is unreacted starting material.
Remainder of mass balance is the double bond isomer of 1.
High enantioselectivities and complete conversions were obtained with the (S)-binol-derived phosphoramidites with a bulky amine substituent (L9 – 11, entries 11, 14, and 15). Alkene 1 cyclized quantitatively in the presence of 5 mol% [RhCl(coe)2]2 and 15 mol% L10 to give 2 in 88% ee within 2 hours at 125 °C. In contrast, (−)-TADDOL-based phosphites L1 – 3 (entries 1 – 3), (−)-TADDOL-based phosphonite L4 (entry 4), (S)-binol-derived phosphites L5 (entry 5), and phosphoramidites L6 – 8 (entries 6 – 8) that incorporate unhindered secondary amines all proved to be ineffective catalysts, giving either poor conversions, poor ee’s, or both. Both diastereomeric ligands L10 and L11 afforded the same enantiomer in the same yield and with comparable enantioselectivities (entries 14 and 15). These data, combined with the result that L9, bearing an achiral amine substituent, also gave the same sense of induction (entry 11), indicates that the chirality of the binaphthyl moiety is primarily responsible for the asymmetric induction and that the sterics of the amine substituent, rather than its chirality, contribute to the overall induction and catalyst efficiency.
A.2. Further Optimization of Reaction Parameters
The ratio of ligand to Rh had a significant effect on the reaction efficiency. The optimal phosphoramidite to Rh ratio for the cyclization of 1 was found to be between 1:1 and 1.5:1. Increasing this ratio dramatically inhibited the reaction (Table 1, entries 9 – 13). Similar results have been observed in Rh-catalyzed hydrogenation using phosphoramidite ligands.14
To further enhance the enantioselectivity, the reaction temperature was lowered using ligands L9, L10, and L11, which all gave quantitative cyclizations of 1 at 125 °C (Table 1). Indeed, at 50 °C, 1 cyclized using L10 in 95% ee and 94% yield in only 9 hours (Table 2, entry 2). Increases in enantioselectivities were also obtained using ligands L9 and L11 at lower temperatures, although the reactions were significantly slower (Table 2, entries 1 and 3).
Table 2.
| substrate | product | Entry | ligand | temp (°C) | time (h) | %yieldc | % eed |
|---|---|---|---|---|---|---|---|
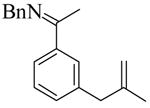
1 |
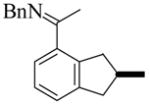
(S)-2 |
1e | L9 | 50 | 48 | 65f | 93 |
| 2 | L10 | 50 | 9 | 94 | 95 | ||
| 3e | L11 | 50 | 48 | 92 | 95 | ||
|
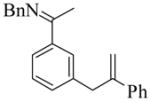
| |||||||
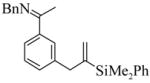
3 |
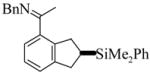
(S)-4 |
4 | L9 | 125 | 0.3 | 91 | 70 |
| 5 | L10 | 50 | 20 | 75 | 25 | ||
| 6 | L11 | 125 | 0.25 | 100 | 27 | ||
| 7 | L11 | 50 | 20 | 96 | 42 | ||
|
| |||||||
5 |
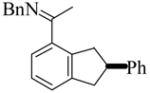
(S)-6 |
8 | L9 | 75 | 4 | 100 | 83 |
| 9 | L10 | 75 | 3 | 96 | 90 | ||
| 10 | L11 | 75 | 3.5 | 98 | 90 | ||
|
| |||||||
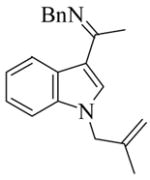
7 |
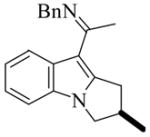
(R)-8 |
11e | L9 | 125 | 12 | 78 | 63 |
| 12e | L10 | 125 | 1 | 90 | 70 | ||
| 13e | L11 | 125 | 1 | 99 | 68 | ||
|
| |||||||
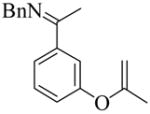
9 |
(R)-10 |
14 | L9 | 50 | >200 | 69 | 89 |
| 15 | L10 | 50 | 1.5 | 99 | 93 | ||
| 16 | L10 | rt | 23 | 95 | 96 | ||
| 17 | L11 | 50 | 1.5 | 99 | 95 | ||
| 18 | L11 | rt | 100 | 99 | 96 | ||
Reactions performed using 5 mol % [RhCl(coe)2]2 and 15 mol % ligand in toluene-d8.
The absolute configurations of (S)-6 and (R)-10 were assigned by chemical derivatization and X-ray structure determination (see reference 12). The absolute configurations of (S)-2, (S)-4, and (R)-8 were assigned by analogy.
Yields based on 1H-NMR integration relative to 2,6-dimethoxytoluene internal standard.
Ee’s determined after hydrolysis of the imine product using chiral GC or HPLC.
Performed using 10 mol% ligand.
Yield is low due to unreacted material (9%) and formation of double bond isomer (20%).
A.3. Substrate Scope
To explore the scope of this enantioselective cyclization, substrates 3, 5, 7, and 9 were evaluated using the optimal ligands L9, L10, and L11 (Table 2). These substrates cyclized in nearly quantitative yields under the optimal conditions for each substrate. Ligands L10 and L11 consistently gave faster reaction rates than L9. Ligands L10 and L11 also showed higher enantioselectivities for all substrates except the sterically encumbered silyl substrate 3, for which the least hindered ligand L9 gave the optimal result (70% ee, entry 4).
Vinyl ether 9 exhibited the most efficient reaction of all the substrates explored. Even at room temperature the reaction proceeded cleanly with ligand L10 giving the desired product (R)-10 in high yield and with 96% ee (entry 16). Ligand L11 afforded a less efficient catalyst but provided the same high level of selectivity as that obtained with diastereomer L10 (entry 18).
A4. Applications of Enantioselective Cyclizations of 1,1-Disubstituted Alkene Substrates
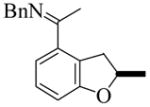
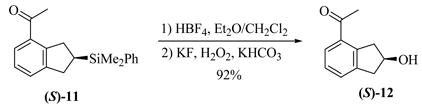
The substrate examples shown above demonstrate the utility of this methodology for the synthesis of a diversity of chiral products including chiral indanes, dihydropyrroloindoles, and dihydrobenzofurans. In addition, the cyclization of alkene 3 enables the introduction of a hydroxyl group, as the SiMe2Ph functionality can be oxidized with retention of configuration using the conditions developed by Fleming15 and Tamao.16 Phenylsilane (S)-11 was converted to the fluorosilane, which was cleanly oxidized using mild hydrogen peroxide conditions to give (S)-12 in 92% overall yield (eq. 2).12 Furthermore, we also applied the enantioselective cyclization methodology to the asymmetric synthesis of the potent protein kinase C inhibitor tricyclic indole 15 (eq. 3).5b In the course of the synthesis, a key intermediate 14 was prepared in 61% yield and 90% ee by the cyclization of alkene 13. This synthesis is noteworthy not only because it provided much more efficient entry to inhibitor 15, but also because it represents the first example of the enantioselective catalytic cyclization of an aldimine rather than a ketimine substrate.
 |
(2) |
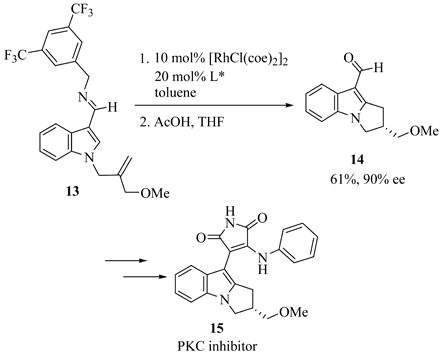 |
(3) |
B. Enantioselective Cyclization of 1,2-Disubstituted Alkenes
Next, our study focused on 1,2-disubstituted alkenes, which were more difficult substrates for enantioselective cyclization because Z/E-alkenes isomerization occurs at rates that are competitive with cyclization.
B.1. Initial Evaluation of Olefin Isomers
We began our study on 1,2-disubstituted alkene substrates by examining which of three isomeric alkenes, 16, 17, and 18, provided the 2,3-dihydro-3-methylbenzofuran product 19 with the greatest efficiency and selectivity (Table 3). We also monitored alkene isomerization as the reactions proceeded. Z-Alkene 18 was the best substrate with respect to both yield and enantioselectivity (entry 3). Alkene 18 cyclized in the presence of 10 mol% [RhCl(coe)2]2 and 20 mol% L10 to give (R)-19 in 82% yield and 85% ee in 96 hours at 50 °C. None of the benzopyran that might be produced upon isomerization of the alkenyl group from vinyl to allyl was seen. Much lower yield and enantioselectivity were observed in the reaction of allyl substrate 16 (entry 1). Reaction of E-alkene 17 was slower than that of the Z-isomer 18 (entry 2). However, both isomers gave the same stereoselectivity, and Z/E isomerization of the substrate was observed during the cyclizations of both 17 and 18 (entries 2 and 3). It is therefore likely that regardless of the Z or E stereochemistry of the starting material, cyclization proceeded through the Z-alkene.
Table 3.
Comparison of Isomeric Substrates for Asymmetric Cyclization
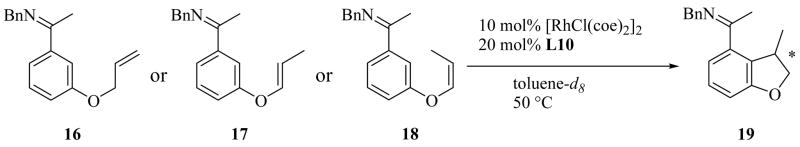 | ||||||
|---|---|---|---|---|---|---|
| Entry | substrate | time (h) | 17, %a | 18, %a | %yielda | % eeb |
| 1 | 16 | 96 | -- | -- | 19 | < 22 (S)c |
|
| ||||||
| 2 | 17 | 0 | 83 | n.d. | n.d. | -- |
| 6 | 53 | 25 | 6 | -- | ||
| 20 | 35 | 29 | 25 | -- | ||
| 44 | 26 | 24 | 39 | -- | ||
| 72 | 23 | 19 | 47 | -- | ||
| 96 | 21 | 16 | 51 | 84 (R) | ||
|
| ||||||
| 3 | 18 | 0 | n.d. | 90 | n.d. | -- |
| 6 | 12 | 53 | 32 | -- | ||
| 21 | 10 | 24 | 57 | -- | ||
| 45 | 12 | 12 | 75 | -- | ||
| 74 | 10 | 7 | 80 | -- | ||
| 96 | 10 | 4 | 82 | 85 (R) | ||
Amount of 17 and 18, and yields based on 1H NMR integration relative to 2,6-dimethoxytoluene internal standard. n.d.: not detected.
Ee’s determined after hydrolysis of 19 with silica gel using chiral HPLC. Sense of induction is indicated in parentheses.
Approximate value due to peak overlapping in HPLC analysis.
B.2. Ligand Screening
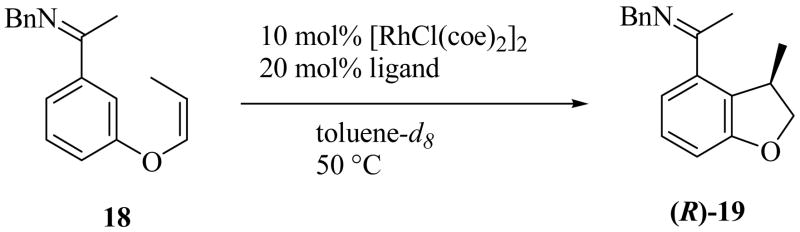
We next screened various phosphoramidite ligands (Chart 1)13 for enhancing the enantioselectivity of the reaction using Z-alkene 18 as a substrate (Table 4). N,N- Diisopropylphosphoramidite L9 provided high yield and the highest enantioselectivity among the (S)-binaphthyl type of ligands (76% yield, 87% ee, entry 2). N,N-Dicyclohexylphosphoramidite L12 showed lower selectivity (77% ee, entry 5) compared to L9. Increased steric hindrance as exemplified by the N-tert-butyl, N-isopropylphosphoramidite L13 (entry 6) resulted in a dramatic decrease in yield as did the less hindered N,N-dibenzylphosphoramidite L8 (entry 1). Ligand L11, the diastereomer of L10, provided lower yield but the same stereoselectivity as L9 and L10 (61% yield, 85% ee, entry 4). These results indicate that the stereoselectivity is predominantly controlled by the diol moiety of the phosphoramidite ligands, in analogy to what was observed in the reaction of 1,1-disubstituted alkenes. Accordingly, the chiral diol was varied, keeping the diisopropylamino group intact. (S)-Octahydrobinaphthol-based ligand L18 gave the best result (82% yield and 91 % ee, entry 11). (S)-Dimethylbinaphthol-based ligand L14 also resulted in increases in enantioselectivity (90% ee, entry 7), and the (S)-VANOL-based ligand L16 (entry 9) and the (S)-VAPOL-based ligand L17 (entry 10) accelerated the cyclization; however, none of these were better than L18 overall. The hindered (S)-biphenol-, (S)-octahydrobinaphthol-, and (−)-TADDOL-based ligands, L15 (entry 8), L20 (entry 13), and L21 (entry 14), respectively, all gave dramatically reduced yields.
Table 4.
Asymmetric Cyclization of Alkene 18 Using Various Chiral Phosphoramidite Ligands
 | ||||
|---|---|---|---|---|
| entry | ligand | time (h) | %yielda | % eeb |
| 1 | L8 | 44 | 7 | n.t. |
|
| ||||
| 2 | L9 | 20 | 44 | -- |
| 96 | 76 | 87 | ||
|
| ||||
| 3 | L10 | 45 | 75 | -- |
| 96 | 82 | 85 | ||
|
| ||||
| 4 | L11 | 96 | 61 | 85 |
| 5 | L12 | 96 | 71 | 77 |
| 6 | L13 | 20 | 2 | n.t. |
| 7 | L14 | 96 | 64 | 90 |
| 8 | L15 | 95 | 27 | 80 |
| 9 | L16 | 21 | 81 | 86 |
| 10 | L17 | 43 | 76 | 88 |
| 11 | L18 | 96 | 82 | 91 |
| 12 | L19 | 68 | 79 | 87 |
| 13 | L20 | 96 | 20 | 78 |
| 14 | L21 | 95 | 6 | 5 |
Yields based on 1H NMR integration relative to 2,6-dimethoxytoluene internal standard.
Ee’s determined after hydrolysis of (R)-19 with silica gel using chiral HPLC. n.t.: not tested.
B.3. Further Optimization of Reaction Parameters
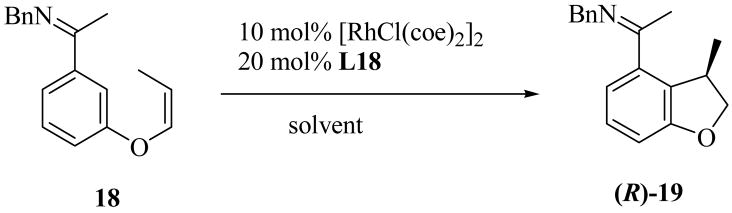
In the interest of exploring reaction efficiency, we further investigated the reaction conditions using 18 as a substrate and L18 as a ligand (Table 5). Increasing the temperature from 50 to 75 °C accelerated the reaction and resulted in only a modest reduction in selectivity (entry 2). Although high enantioselectivities were obtained in all solvents that were investigated (entries 1, 3, and 4), 1,4-dioxane provided both the fastest reaction and the highest conversion (entry 4).
Table 5.
Solvent Effects in Asymmetric Cyclization of Alkene 18
 | |||||
|---|---|---|---|---|---|
| entry | solvent | temp (°C) | time (h) | %yielda | % eeb |
| 1 | toluene-d8 | 50 | 45 | 65 | -- |
| 96 | 82 | 91 | |||
| 2 | toluene-d8 | 75 | 20 | 93 | 89 |
| 3 | THF-d8 | 50 | 47 | 74 | 91 |
| 4 | dioxane-d8 | 50 | 46 | 82 | 90 |
Yields based on 1H NMR integration relative to 2,6-dimethoxytoluene internal standard.
Ee’s determined after hydrolysis of (R)-19 with silica gel using chiral HPLC.
B.4. Substrate Scope
We also studied the scope of 1,2-disubstituted alkenes in this enantioselective cyclization reaction using the optimal ligands (Table 6). The ethyl-, i-butyl-, and phenyl-substituted substrates 20, 22, and 24 all cyclized with high enantioselectivities (entries 2 – 4). In the reactions of aldimine substrate 26, enantioselectivities were high, but the yields were much lower than for the corresponding ketimine substrate 18 regardless of ligand used (entries 5 – 7). In addition, L19 rather than L18 provided the highest enantioselectivity for the cyclization of aldimine substrate 26 (87–89% ee, entry 7).
Table 6.
Asymmetric Cyclization of 1,2-Disubstituted and 1,1,2-Trisubstituted Alkenesa
| substrate | product | entry | ligand | temp (°C) | time (h) | %yieldb | % eec |
|---|---|---|---|---|---|---|---|
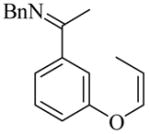
18 |
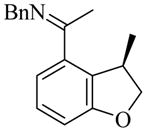
(R)-19 |
1 | L18 | 50 | 46 (46–48)d | 82 (65–71)e | 90 |
|
| |||||||
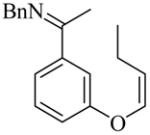
20 |
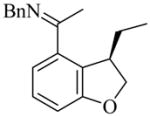
(R)-21 |
2 | L18 | 50 | 96 | 76 (53)e | 91–92 |
|
| |||||||
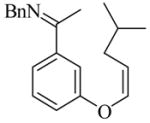
22 |
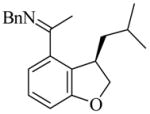
(R)-23 |
3 | L18 | 75 | 67 | 69 (48)e | 90 |
|
| |||||||
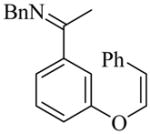
24 |
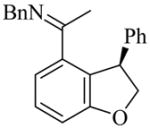
(R)-25 |
4 | L18 | 75 | 21 (22)d | 93 (65)e | 87 |
|
| |||||||
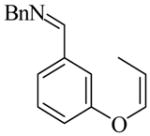
26 |
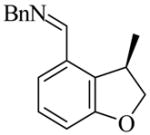
(R)-27 |
5 | L16 | 75 | 72 | 53 | 82 |
| 6f | L18 | 75 | 96 | 25 | 84 | ||
| 7 | L19 | 75 | 72 | 40 (27)e | 87–89 | ||
|
| |||||||
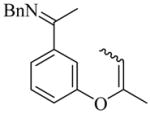
29(Z/E = 4/1) |
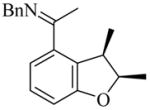
(2R,3R)-30 |
8 | L19 | 50 | 72 | 80 (59)e | 91–93 |
|
| |||||||
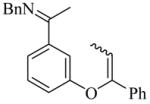
31(Z/E = 9/1) |
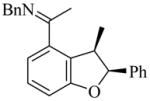
(2S,3R)-32 |
9g | L19 | 75 | 92 | 50 (45)e | 89–90 |
Reactions performed using 10 mol % [RhCl(coe)2]2 and 20 mol % ligand in 1,4-dioxane-d8 or 1,4-
dioxane.
Yield of N-benzylimine product determined by 1H NMR using 2,6-dimethoxytoluene as an
internal standard.
Ee’s determined after hydrolysis of products with silica gel or HCl/H2O-dioxane using chiral HPLC.
Figures in parentheses are for the experiments for ketone product isolation.
Isolated yield obtained after hydrolysis with HCl/H2O–dioxane, which hydrolyzed not only the N-
benzylimine but also any unreacted vinyl ether starting material.
Reaction carried out in toluene-d8. gIn the NMR-experiment, the reaction solution was heated at 50 °C for 6 h before increasing the temperature to 75 °C.
C. Stereoselective Cyclization of 1,1,2-Trisubstituted Alkenes
Finally, we explored the challenging stereoselective cyclization of 1,1,2-trisubstituted alkenes, in which two stereocenters would be set. We began our investigation with olefin isomers 28 and 29. We again monitored not only conversion to product but also alkene isomerization (Table 7). Reaction of the E-alkene isomer 28 using L16 in toluene at 75 °C for 68 hours gave 30 in modest yield and with low enantioselectivity (40% yield and 31% ee, entry 1). In contrast, a faster reaction that resulted in a higher yield and enantioselectivity was observed for substrate 29, which was a 4:1 mixture of Z and E isomers (81% yield and 69% ee, entry 2). The enantioselectivity of the reaction was increased by lowering the reaction temperature and by using 1,4-dioxane as the solvent (80% ee, entry 3). Ligands L18 and L19 resulted in additional increases in enantioselectivity, although the reaction efficiency was lower than that observed for L16 (entries 4 and 5). The highest selectivity was achieved with L19 (91% ee, entry 5), and with this ligand, the E-isomer 28 was completely unreactive to either cyclization or alkene isomerization. Additionally, trisubstituted alkene substrate 31 also cyclized to yield (2S,3R)-32 with high enantioselectivity, although a higher reaction temperature was required in this case (Table 6, entry 9). Interestingly, cyclization of the 1,1,2-trisubstituted alkene substrates 29 and 31, which were Z/E mixtures, gave only the syn-isomer products as determined by NMR and X-ray structural analysis (see Supporting Information and Section D), further indicating that when Z/E mixtures of trisubstituted alkene substrates are used only the Z-alkene isomer, which is expected to give the syn-product, cyclizes.
Table 7.
Asymmetric Cyclization of Alkene 28 and 29
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | Substrate | ligand | temp (°C) solvent | time (h) | E-alkene (%) | Z-alkene (%) | %yielda | % eeb |
| 1 | 28 | L16 | 75 | 0 | 84 | n.d. | n.d. | -- |
| toluene-d8 | 6 | 55 | n.d. | 21 | -- | |||
| 20 | 41 | 2 | 32 | -- | ||||
| 47 | 33 | 3 | 38 | -- | ||||
| 68 | 31 | 5 | 40 | 31 | ||||
|
| ||||||||
| 2 | 29 | L16 | 75 | 0 | 18 | 69 | n.d. | -- |
| toluene-d8 | 6 | 10 | 14 | 66 | -- | |||
| 20 | 4 | 4 | 81 | 69 | ||||
|
| ||||||||
| 3 | 29 | L16 | 50 | 0 | 19 | 75 | n.d. | -- |
| dioxane-d8 | 6 | 15 | 10 | 72 | -- | |||
| 22 | 13 | n.d. | 81 | 80 | ||||
|
| ||||||||
| 4 | 29 | L18 | 50 | 0 | 17 | 70 | n.d | -- |
| dioxane-d8 | 6.5 | 16 | 45 | 30 | -- | |||
| 22 | 16 | 20 | 60 | -- | ||||
| 46 | 14 | 7 | 69 | -- | ||||
| 72 | 13 | 2 | 74 | 88 | ||||
|
| ||||||||
| 5 | 29 | L19 | 50 | 0 | 21 | 78 | n.d. | -- |
| dioxane-d8 | 6 | 21 | 49 | 29 | -- | |||
| 21 | 20 | 22 | 59 | -- | ||||
| 48 | 20 | <10c | 74 | -- | ||||
| 72 | 20 | <7c | 80 | 91 | ||||
Amount of olefin isomers and yields based on 1H NMR integration relative to 2,6-dimethoxytoluene internal standard. n.d.: not detected.
Ee’s determined after hydrolysis of (2R,3R)-30 with silica gel using chiral HPLC.
Approximate value due to peak overlapping in 1H NMR.
D. Determination of Absolute Configuration for Cyclization Products
The absolute configuration of the two cyclization products, (R)-19 and (2R,3R)-30, were determined by X-ray crystallography after their derivatization. The N-benzylimine (R)-19 was hydrolyzed under acidic conditions and the resulting ketone was converted to the N-sulfinyl imine (SS,R)-33 by condensation with (S)-2,4,6-trimethylbenzenesulfinamide (Figure 1). (2R,3R)-30 was similarly derivatized to hydrazone (2R,3R)-34 (Figure 2). CIF files for the X-ray crystal structures of (SS,R)-33 and (2R,3R)-34 are provided in the Supporting Information. The absolute stereochemistry of the representative cyclized products was thus determined to be (R)-19 and (2R,3R)-30. The absolute configurations of (R)-21, (R)-23, (R)-25, (R)-27, and (2S,3R)-32 were assigned by analogy to the absolute configurations of (R)-19 and (2R,3R)-30.
Figure 1.
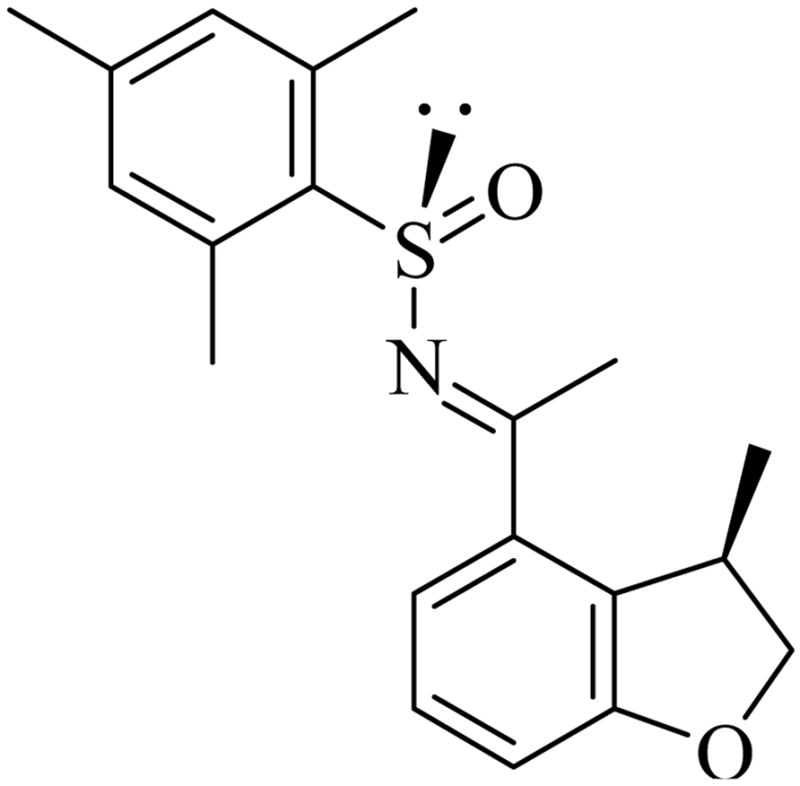
Absolute configuration determined by X-ray structure obtained of sulfinyl imine (Ss,R)-33
Figure 2.
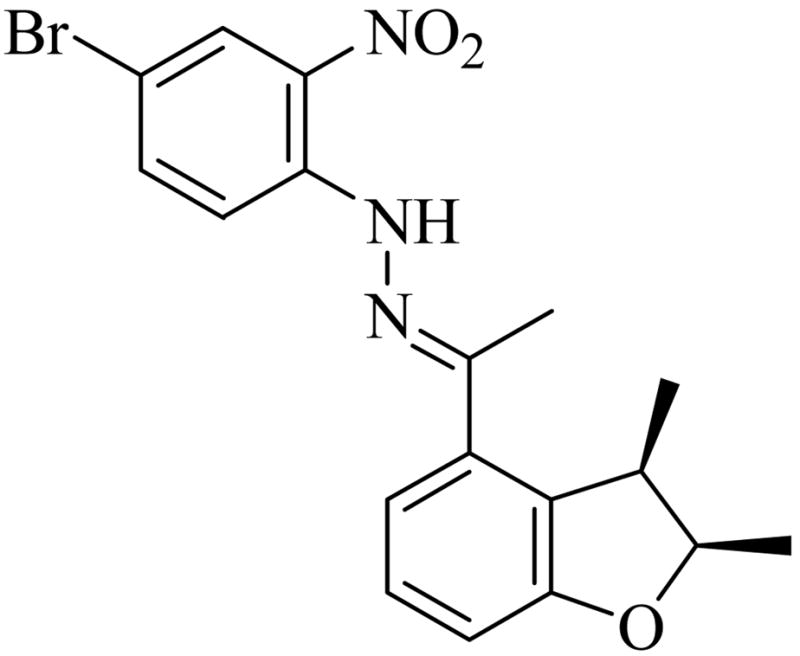
Absolute configuration determined by X-ray structure obtained hydrazone (2R,3R)-34
E. Mechanistic Discussion
Phosphoramidites have recently gained prominence as ligands for asymmetric catalysis due to their remarkable effects in catalytic asymmetric conjugate additions17 and Rh-catalyzed hydrogenation reactions.14 The exceptional rates and stereoselectivities observed may be attributed to the unique binding properties of the phosphoramidite ligands, which include σ donation to the metal center and enhanced π acceptor ability compared to phosphines.
The results presented in Table 1 (entries 9 – 13), which display the effect of the ligand-to-Rh ratio, strongly suggest that only one ligand is bound to one Rh center throughout the catalytic cycle. Excess ligand most likely generates an inactive Rh species having more than one phosphoramidite ligand bound. This conclusion is also supported by the fact that phosphoramidites bind very strongly to Rh as a result of their π acceptor properties.14 Thus, it seems likely that multiple phosphoramidite ligands on a Rh center could render it inactive.
Based on our results, and in accordance with the catalytic cycle described by Jun18 for the analogous intermolecular reaction, a possible catalytic cycle is given in Scheme 1. Pre-coordination of the imine and successive C–H oxidative addition to the Rh center would generate a Rh–H complex 35. Coordination of the alkenyl group to the metal, followed by migratory insertion of the double bond into the Rh–H bond, would provide metallacycle 36, which can then undergo reductive elimination to afford the product. Deuterium labeling studies performed by Jun and coworkers on the analogous intermolecular reaction indicate that the reductive elimination step is rate-determining.18
Scheme 1.

The stereoselectivities are presumably due to highly diastereoselective migratory insertion. The fact that both Z- and E-isomers of 1,2-disubstituted alkenes give the same selectivity (Table 3, entries 2 and 3) can be rationalized by the observation that the E-olefin isomer was converted to the Z-isomer during the reaction, which could then react. The trisubstituted E-alkene substrate 28 reacted much more slowly and with lower selectivity than that of the corresponding substrate 29 with a 4:1 Z- to E-isomer ratio (Table 7, entries 1 and 2). This lower selectivity can perhaps be explained by isomerization of the double bond from vinyl to allyl, which then cyclized with decreased selectivity, similar to that observed in the reaction of 16 (Table 3, entry 1).
Conclusions
In summary, we have developed a highly stereoselective intramolecular hydroarylation of alkenes via directed C H bond activation using a Rh/chiral phosphoramidite catalyst system, which represents a very rare example of an enantioselective catalytic reaction involving aromatic C H bond activation.
Moreover, the identified catalyst system enables the intramolecular alkylation reaction to proceed at low temperatures, leading to increased selectivity. Finally, good substrate scope was achieved with 1,1- and 1,2-disubstituted as well as 1,1,2-trisubstituted alkenes all serving as effective substrates. For the cyclization of the 1,2-disubstituted and 1,1,2-trisubstituted alkenes, the Z-alkene isomers were much more effective substrates than the corresponding E-isomers. This stereoselective catalytic transformation provides access to a range of chiral indanes, dihydrobenzofurans, and dihydropyrroloindoles with different substitution patterns and therefore should be applicable to the asymmetric synthesis of a range of biologically active compounds.
Experimental Section
General procedure for 1H NMR experiments
In a glovebox, to a medium-walled NMR tube was added a mixture of [RhCl(coe)2]2 (0.005 mmol, 10 mol%), phosphoramidite ligand (0.010 mmol, 20 mol%) in 0.40 mL of solvent, and a solution of imine (0.050 mmol) and 2,6-dimethoxytoluene internal standard (0.010 mmol) in 0.10 mL of solvent. The tube was fitted with a Cajon adapter, the mixture was frozen using liquid N2, and then the tube was flame sealed under vacuum. The NMR tube was then placed in oil bath heated to the appropriate temperature and the progress of the reaction was monitored periodically by 1H NMR spectroscopy. After the indicated reaction time, the sealed tube was opened and the mixture was concentrated. The residue was dissolved in a small amount of methylene chloride, silica gel was added, and the mixture was concentrated to dryness. The residue was subjected to silica gel column chromatography and eluted with a 1:20 mixture of ethyl acetate and hexanes for chiral HPLC analysis. Racemates for HPLC analysis were prepared as crude material by using PCy3 or FcPCy2 as a ligand instead of a chiral phosphoramidite.
(R)-1-(3-Methyl-2,3-dihydrobenzofuran-4-yl)ethanone ((R)-48) [Table 6, entry 1]
In a glovebox, to a medium-walled NMR tube was added a mixture of [RhCl(coe)2]2 (3.6 mg, 0.0050 mmol) and (S)-diisopropyl-(8,9,10,11,12,13,14,15-octahydro-3,5-dioxa-4-phosphacyclohepta[2,1-a;3,4-a′]dinaphthalen-4-yl)amine (4.2 mg, 0.0099 mmol) in 1,4-dioxane (0.40 mL) and a solution of benzyl-[1-{3-[((Z)-propenyl)oxy]phenyl}ethylidene]amine (13.4 mg, 0.0505 mmol) in 1,4-dioxane (0.10 mL). The tube was fitted with a Cajon adapter, the mixture was frozen, and then the tube was flame sealed under vacuum. The NMR tube was then placed in oil bath heated to 50 °C for 48 h. After the reaction, the sealed tube was opened and the mixture was concentrated. To the residue were added 1,4-dioxane (0.50 mL) and concentrated HCl/H2O (1/1) (0.50 mL). The mixture was stirred at room temperature for 3 h and then extracted with diethyl ether four times. The combined organic layer was concentrated and the residue was purified by silica gel column chromatography (silica gel: 15 mL, eluted with 20:1 hexanes/ethyl acetate) to give the title compound as a colorless oil (6.3 mg, 71% yield). IR (ZnSe, thin film) νmax (cm−1): 1680, 1584, 1442, 1355, 1256, 1236. 1H NMR (400 MHz, CDCl3): δ 7.38 (d, J = 8.0 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 4.54 (t, J = 8.6 Hz, 1H), 4.29 (dd, J = 2.8, 8.6 Hz, 1H), 4.04–3.96 (m, 1H), 2.60 (s, 3H), 1.24 (d, J = 6.8 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 199.0, 160.5, 133.8, 133.6, 128.2, 122.1, 114.1, 79.0, 37.0, 28.1, 20.1. HRMS (EI): m/z calcd. for C11H12O2 (M+): 176.08373; found: 176.08366. Chiral HPLC (Chiralcel AS column, 1% iPrOH/hexanes, 1mL/min): major, 6.41 min; minor, 5.90 min; 90% ee. [α]D25 +135.35 (c 0.99, CHCl3). Maximum value based upon sample enantiomeric purity: [α]D25 +150.39 (c 0.99, CHCl3).
By a similar procedure starting from benzyl-[1-{3-[((Z)-propenyl)oxy]phenyl}ethylidene]amine (132.8 mg, 0.5005 mmol), [RhCl(coe)2]2 (36.0 mg, 0.0502 mmol), and (S)-diisopropyl-(8,9,10,11,12,13,14,15-octahydro-3,5-dioxa-4-phosphacyclohepta[2,1-a;3,4-a′]dinaphthalen-4-yl)amine (42.5 mg, 0.100 mmol), the title compound was also obtained as a colorless oil in 57.3 mg (65% yield) and 90% ee.
(2R,3R)-1-(2,3-Dimethyl-2,3-dihydrobenzofuran-4-yl)ethanone ((2R,3R)-53) [Table 6, entry 8]
In a glovebox, to a medium-walled NMR tube was added a mixture of [RhCl(coe)2]2 (3.5 mg, 0.0049 mmol) and (S)-(8,9,10,11,12,13,14,15-octahydro-3,5-dioxa-4-phosphacyclohepta[2,1-a;3,4-a′]dinaphthalen-4-yl)-bis((R)-1-phenylethyl)amine (5.5 mg, 0.010 mmol) in 1,4-dioxane (0.40 mL) and a solution of benzyl-[1-[3-(1-methylpropenyloxy)phenyl]ethylidene]amine (Z/E = 4/1 for olefin) (13.9 mg, 0.0497 mmol) in 1,4-dioxane (0.10 mL). The tube was fitted with a Cajon adapter, the mixture was frozen, and then the tube was flame sealed under vacuum. The NMR tube was then placed in oil bath heated to 50 °C for 72 h. After the reaction, the sealed tube was opened and the mixture was concentrated. To the residue was added 1,4-dioxane (0.50 mL) and concentrated HCl/H2O (1/1) (0.50 mL). The mixture was stirred at room temperature for 3 h and then was extracted with diethyl ether four times. The combined organic layer was concentrated and the residue was purified by silica gel column chromatography (silica gel: 15 mL, eluted with 20:1 hexanes/ethyl acetate) to give the title compound as a colorless oil (5.6 mg, 59% yield). IR (ZnSe, thin film) νmax (cm−1): 1680, 1583, 1442, 1355, 1263, 1231. 1H NMR (400 MHz, CDCl3): δ 7.38 (d, J = 8.0 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H), 6.96 (d, J = 8.0 Hz, 1H), 4.75 (quint, J = 6.8 Hz, 1H), 3.83 (quint, J = 6.8 Hz, 1H), 2.60 (s, 3H), 1.49 (d, J = 6.8 Hz, 3H)1.06 (d, J = 6.8 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 199.0, 160.1, 135.4, 133.6, 128.0, 122.3, 114.0, 83.3, 40.0, 28.1, 15.0, 13.8. HRMS (EI): m/z calcd. for C12H14O2 (M+): 190.0994, found: 190.0993. Chiral HPLC (Chiralcel AS column, 0.5% iPrOH/hexanes, 1mL/min): major, 13.5 min; minor, 12.7 min; 93% ee. CD (c = 4 × 10−5 M, MeOH): λmax (Δε): 251 (+6.20). A 1H-1H NOESY spectrum of (2R,3R)-53 indicated that the geometry of the two protons on the dihydrofuran ring was cis.
Supplementary Material
Experimental details, including synthetic procedures, characterization, and X-ray crystallographic data of (SS,R)-33 and (2R,3R)-34 (PDF, CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by NIH Grant GM069559 (to J.A.E.) and the Director and Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, U.S. Department of Energy, under Contract DE-AC03-76SF00098 (to R.G.B.). Support for H.H. by Eisai Co., Ltd. is also gratefully acknowledged. We thank Dr. Frederick J. Hollander of the UC Berkeley CHEXray facility for solving the X-ray crystal structures of the N-sulfinyl imine (SS,R)-33 and hydrazone (2R,3R)-34 used to determine the absolute configurations of (R)-19 and (2R,3R)-30, respectively.
References
- 1.For recent reviews of C–H activation, see: Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174–238. doi: 10.1021/cr0509760.Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731.Kakiuchi F, Chatani N. Top Organomet Chem. 2004;11:45–79.Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861–2903. doi: 10.1021/cr0200217.Ritleng V, Sirlin C, Pfeffer M. Chem Rev. 2002;102:1731–1769. doi: 10.1021/cr0104330.Jia C, Kitamura T, Fujiwara Y. Acc Chem Res. 2001;34:633–639. doi: 10.1021/ar000209h.Miura M, Satoh T. Top Organomet Chem. 2005;14:55–83.Campeau LC, Fagnou K. Chem Soc Rev. 2007;37:1058–1068. doi: 10.1039/b616082d.
- 2.Murai S, Kakiuchi F, Sekine S, Tanaka Y, Kamatani A, Sonoda M, Chatani N. Nature. 1993;366:529–531. [Google Scholar]
- 3.(a) Jun C-H, Hong J-B, Kim Y-H, Chung K-Y. Angew Chem Int Ed. 2000;39:3440–3442. doi: 10.1002/1521-3773(20001002)39:19<3440::aid-anie3440>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]; (b) Lim SG, Ahn JA, Jun CH. Org Lett. 2004;6:4687–4690. doi: 10.1021/ol048095n. [DOI] [PubMed] [Google Scholar]
- 4.(a) Thalji RK, Ahrendt KA, Bergman RG, Ellman JA. J Am Chem Soc. 2001;123:9692–9693. doi: 10.1021/ja016642j. [DOI] [PubMed] [Google Scholar]; (b) Ahrendt KA, Bergman RG, Ellman JA. Org Lett. 2003;5:1301–1303. doi: 10.1021/ol034228d. [DOI] [PubMed] [Google Scholar]; (c) Thalji RK, Ahrendt KA, Bergman RG, Ellman JA. J Org Chem. 2005;70:6775–6781. doi: 10.1021/jo050757e. [DOI] [PubMed] [Google Scholar]
- 5.(a) O’Malley SJ, Tan KL, Watzke A, Bergman RG, Ellman JA. J Am Chem Soc. 2005;127:13496–13497. doi: 10.1021/ja052680h. [DOI] [PubMed] [Google Scholar]; (b) Wilson RM, Thalji RK, Bergman RG, Ellman JA. Org Lett. 2006;8:1745–1747. doi: 10.1021/ol060485h. [DOI] [PubMed] [Google Scholar]; (c) Watzke A, Wilson RM, O’Malley SJ, Bergman RG, Ellman JA. Synlett. 2007:2383–2389. [Google Scholar]
- 6.For a leading reference on enantioselective carbenoid and nitrenoid insertions see: Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485.
- 7.Fujii N, Kakiuchi F, Yamada A, Chatani N, Murai S. Chem Lett. 1997;26:425–426. [Google Scholar]
- 8.Kakiuchi F, Le Gendre P, Yamada A, Ohtaki H, Murai S. Tetrahedron Asymmetry. 2000;11:2647–2651. [Google Scholar]
- 9.Han X, Widenhoefer RA. Org Lett. 2006;8:3801–3804. doi: 10.1021/ol061441b. [DOI] [PubMed] [Google Scholar]
- 10.Taura Y, Tanaka M, Wu X-M, Funakoshi K, Sakai K. Tetrahedron. 1991;47:4879–4888.Barnhart RW, Wang X, Noheda P, Bergens SH, Whelan J, Bosnich B. J Am Chem Soc. 1994;116:1821–1830.Barnhart RW, McMorran DA, Bosnich B. Chem Commun. 1997:589–590.For an innovative strategy for the intramolecular hydroacylation of alkynes for the catalytic asymmetric synthesis of cyclopentenones via kinetic resolution see: Tanaka K, Fu GC. J Am Chem Soc. 2002;124:10296–10297. doi: 10.1021/ja0266161.Kundu K, McCullagh JV, Morehead AT., Jr J Am Chem Soc. 2005;127:16042–16043. doi: 10.1021/ja0564416.
- 11.Shi BF, Maugel N, Zhang YH, Yu JQ. Angew Chem Int Ed. 2008;47 doi: 10.1002/anie.200801030. EarlyView. [DOI] [PubMed] [Google Scholar]
- 12.Thalji RK, Bergman RG, Ellman JA. J Am Chem Soc. 2004;126:7192–7193. doi: 10.1021/ja0394986. [DOI] [PubMed] [Google Scholar]
- 13.(a) Arnold LA, Imbos R, Mandoli A, de Vries AHM, Naasz R, Feringa BL. Tetrahedron. 2000;56:2865–2878. [Google Scholar]; (b) Watanabe T, Knöpfel TF, Carreira EM. Org Lett. 2003;5:4557–4558. doi: 10.1021/ol035575q. [DOI] [PubMed] [Google Scholar]; (c) Hua Z, Vassar VC, Choi H, Ojima I. Proc Nat Acad Sci. 2004;101:5411–5416. doi: 10.1073/pnas.0307101101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang FY, Chan ASC. Tetrahedron Asymmetry. 1998;9:1179–1182. [Google Scholar]; (e) Duursma A, Minnaard AJ, Feringa BL. Tetrahedron. 2002;58:5773–5778. [Google Scholar]; (f) Alexakis A, Burton J, Vastra J, Benheim C, Fournioux X, van den Heuvel A, Levêque J-M, Mazé F, Rosset S. Eur J Org Chem. 2000:4011–4027. [Google Scholar]
- 14.van den Berg M, Minnaard AJ, Haak RM, Leeman M, Schudde EP, Meetsma A, Feringa BL, de Vries AHM, Malijaars CEP, Willans CE, Hyett D, Boogers JAF, Henderickx HJW, de Vries JG. Adv Synth Catal. 2003;345(1 2):308–323. [Google Scholar]
- 15.(a) Fleming I, Henning R, Parker DC, Plaut HE, Sanderson PEJ. J Chem Soc, Perkin Trans 1. 1995;4:317. [Google Scholar]; (b) Fleming I. Chemtracts: Org Chem. 1996;9:1. [Google Scholar]
- 16.Tamao K. Adv Silicon Chem. 1996;3:1. [Google Scholar]
- 17.Feringa BL. Acc Chem Res. 2000;33:346–353. doi: 10.1021/ar990084k. [DOI] [PubMed] [Google Scholar]
- 18.Jun C-H, Moon CW, Hong J-B, Lim S-G, Chung K-Y, Kim Y-H. Chem Eur J. 2002;8:485–492. doi: 10.1002/1521-3765(20020118)8:2<485::AID-CHEM485>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details, including synthetic procedures, characterization, and X-ray crystallographic data of (SS,R)-33 and (2R,3R)-34 (PDF, CIF). This material is available free of charge via the Internet at http://pubs.acs.org.