

The pyrrole-imidazole alkaloids constitute a family of more than 100 natural products,[1,2] among which palau'amine (1, Scheme 1) exists as one of the more structurally complex members. This polycyclic guanidine alkaloid, which was isolated from Stylotella aurantium, was found to possess potent cytotoxic, antibiotic, and immunosuppressive activities.[3] Despite the fact that its acute toxicity is relatively low (LD50 = 13 mg kg−1 in mice), palau'amine exhibited significant cytotoxicity in a variety of cancer cell lines, including P-388 (IC50 = 0.1 μg mL−1) and A-549 (IC50 = 0.2 μg mL−1). Moreover, 1 exhibits remarkable immunosuppressive responses in the mixed lymphocyte assay (IC50< 18 ngmL−1). Elegant strategies for the synthesis of advanced cyclopentane fragments related to 1 have been reported.[4] These include, inter alia, a tethered Pauson–Khand approach,[5] Diels–Alder chlorination/ring contraction,[6,7] Diels–Alder oxidative ring contraction,[8,9] spirocyclization of alkylidene glycolamidines and alkylidene hydantoins,[10,11] 1,3-dipolar cycloaddition of thiosemicarbazide derivatives and alkene/enamide metathesis,[12,13] Diels–Alder cyclopropane fragmentation,[14] oxidative tandem radical cyclization,[15] and most recently intramolecular guanidine-conjugate addition[16,17] in a total synthesis of the related axinellamine alkaloids. Additionally, synthetic efforts toward the phakellin heterocyclic core of 1 have also been reported.[18–25]
Scheme 1.
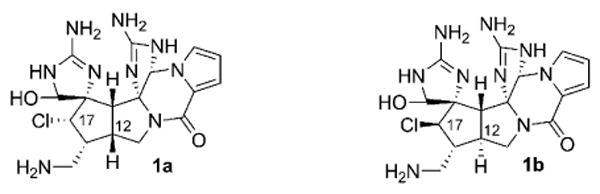
Originally proposed (1a) and revised (1b) structures of palau'amine.
The construction of the central chlorinated cyclopentane skeleton within 1, with control over the relative stereochemical configuration of all of its substituents, emerges as a demanding challenge in this synthetic target. This synthetic hurdle is further complicated by the recent discovery highlighting the stereochemical revision of palau'amine[26–29] based on NMR spectroscopy, molecular calculations, and comparison to related natural products. These data suggest that the endo-disposed C17 chloro group and the C12 cis-fused ring junction in the originally proposed structure 1a instead exists as its C12,C17 diastereomeric counterpart 1b, which incorporates a rare trans-fused [3.3.0]-bicyclic skeleton.
In focusing on the elaborately substituted cyclopentane scaffold 2 (Scheme 2) of 1a and 1b, retrosynthetic disconnection of the C18–R4 and C16–[N] substituents ([N] = nitrogen functional group) would furnish the [2.2.1]-bicyclo substrate 3. The resulting norbornene-like intermediate 3 would possess all of the diverse latent functionalities present in the cyclopentane core, including the C17-chloro substituent. However, direct application of a Diels–Alder transform to 3 is likely to be challenging. Indeed, selectively functionalized cyclopentadienes such as 4 are not trivial to access, and diene isomerization is virtually unavoidable if one of the substituents at C17 is a proton, as in the natural product.
Scheme 2.
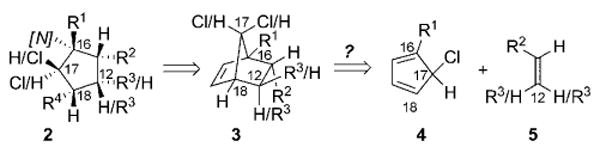
Retrosynthetic analysis. [N] = nitrogen functional group.
However, control over the configuration at C17, along with that of the other substituents on the cyclopentane core, could be established indirectly through the [3,3]-sigmatropic rearrangement of bridged tricyclodecadienes. The classic finding by Woodward and Katz[30] which involves the preparation of the bridged allylic alcohol 6 (Scheme 3), detailed its susceptibility to equilibration through [3,3]-sigmatropic rearrangement to form homoallylic alcohol 7. Mapping the stereochemical course of the Woodward–Katz reaction of the C* center in 6/7 (Scheme 3) to that of C17 in 3 (Scheme 2) bodes well for the preparation of a relatively simple Diels–Alder adduct that can be used to establish the stereochemistry of the cyclopentane cores of 1a and 1b.
Scheme 3.
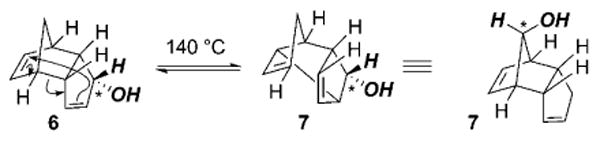
Tricyclodecadiene equilibration through [3,3]-sigmatropic rearrangement.
The strategy was initially validated in a sequence directed toward the synthesis of the originally proposed structure 1a. This synthetic sequence commenced with the dihydroquinone 8 (Scheme 4), which is readily available from the Diels–Alder reaction of benzoquinone with cyclopentadiene. Nucleophilic epoxidation of the electron-deficient alkene (95% yield) allowed for ethoxide-mediated Favorskii-like rearrangement of the resultant keto-epoxide to generate the cyclopentenone 9 (60% yield).[31] Subsequent α alkylation of the ketone enolate of 9 with BOMCl provided the α-benzyloxymethyl ketone 10 (53% yield),[32] which served as a versatile intermediate with which to execute the rearrangement strategy. Indeed, 10 underwent spontaneous [3,3]-sigmatropic rearrangement to furnish a significant quantity of the enoate 11 in a 72:28 thermodynamic ratio (10:11).[33] The cyclopentenone group of 10 was then subjected to conjugate addition with benzeneselenol in an endo-selective manner.[34] This operation served to avoid premature sigmatropic rearrangement through selective disruption of the 1,5-diene system in 10 through “protection” of the alkene moiety, and it capitalized on a Curtin–Hammett energy profile to obviate the production of enoate 11. This conjugate addition allowed for subsequent introduction of the C17-chloro substituent through an initial exo-selective hydride addition to the C17 ketone (70% yield), followed by activation of the alcohol and displacement with inversion using a chloride nucleophile, to afford the exo-chloride substituent at C17 (12; 82% yield). Selenide oxidation and elimination regenerated the 1,5-diene system (13), which spontaneously underwent [3,3]-sigmatropic rearrangement to form the 1,5-diene isomer 14 (92% yield). This product incorporates the chloro substituent at C17 in an orientation proximal to the norbornene π functionality, and was unambiguously verified by NOE signals between the C17 hydrogen and the C12 angular proton.
Scheme 4.
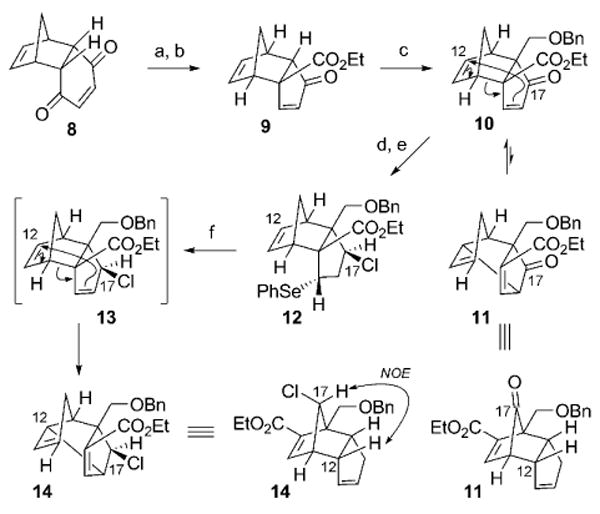
Reagents and conditions: a) H2O2, Na2CO3, EtOH, 0 °C, 95%; b) NaOH, EtOH, 23 °C, 60%; c) LiN(TMS)2, −78 °C, THF; BOMCl, −78 °C→−20 °C, 53%; d) PhSeH, AcOH, EtOH, 0 °C; NaBH4, 0 °C, 70%; e) PBu3, CCl4, 90 °C, 82%; f) mCPBA, −20 °C, CH2Cl2; Et3N, 90 °C, 92%. BOMCl = benzyl chloromethyl ether, Bn = benzyl, mCPBA = meta-chloroperbenzoic acid, TMS = trimethylsilyl.
The stereoselective synthesis of tricyclo[5.2.1.0]decadiene 14 establishes the C17-chloro relative stereochemical configuration in the originally proposed structure of palau'amine 1a, together with all of the remaining substituents on the central cyclopentane core in latent form. Advancement of the enoate 14 (Scheme 5) involved installation of the nitrogen functionality at the C16 quaternary carbon center; this process was initially envisioned to proceed through oxidative cleavage of the enoate group. However, the steric demands associated with such a reaction with 14 proved insurmountable, and led to the implementation of a Beckmann ring expansion strategy. Accordingly, hydrolysis of the ethyl ester in 14 allowed for subsequent acyl azide formation and Curtius rearrangement to afford ketone 15 (87% yield, over 3 steps) following hydrolysis under acidic conditions. After conversion of the ketone into the corresponding oxime (91% yield) by condensation with hydroxylamine,[35] thionyl-chloride-mediated Beckmann rearrangement ensued (54% yield).[36] The process was completely regioselective, effecting the net insertion of a nitrogen atom at C16 to form 16, as was verified by a significant NOE signal between its lactam NH group and the C20 methylene protons. Functional group manipulation of the cyclopentene substructure within 16 was then initiated by alkene ozonolysis followed by a reductive work up (NaBH4)[11] to afford the corresponding diol. Protection of the hydroxy groups provided the bis(acetate ester) 17 (77% yield, over 2 steps), thus allowing for N acylation of the lactam with Boc2O to furnish the bicyclic imide 18 (99% yield). Further elaboration to a less-strained advanced cyclopentane intermediate, with differentiation of C13 and C6 oxygen functionalities, can also be accomplished by saponification of both acetate esters, which is accompanied by hydrolysis of the cyclic imide. Treatment of the resultant diol carboxylic acid under acidic conditions effected an intramolecular ester formation to afford lactone 19 (96% yield, over 2 steps). The structure of 19 was verified by extensive NOE analysis, from which the relative stereochemical arrangement of its synthetic precursors could also be inferred with confidence. Both the bridged (i.e., 18) and non-bridged (i.e., 19) cyclopentane cores map directly onto the originally proposed structure of palau'amine (1a). Indeed, both have the potential to serve as useful advanced precursors for future elaboration of the nitrogen-rich periphery of 1a.
Scheme 5.
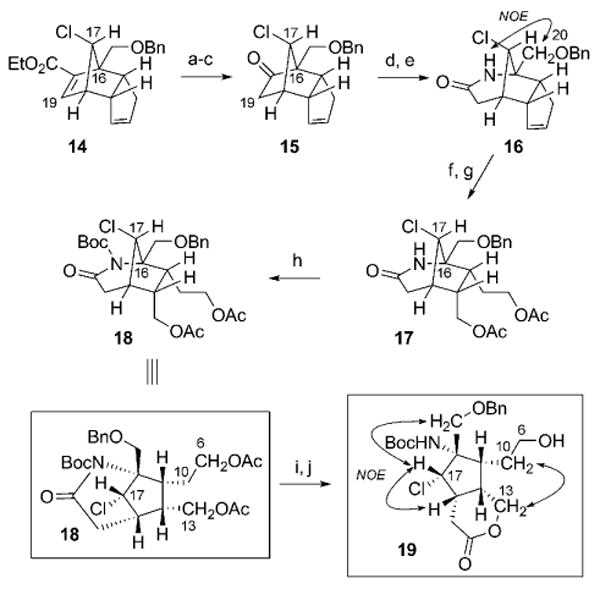
Reagents and conditions: a) NaOH, EtOH, 23 °C, 97%; b) (PhO)2P(O)N3, Et3N, 23 °C, toluene; MeOH, 80 °C; c) 12 n HCl, MeOH, 23 °C, 90% (2 steps); d) HONH2·HCl, pyridine, EtOH, H2O, 90 °C, 91%; e) SOCl2, Et2O, 23 °C, 54%; f) O3, MeOH, CH2Cl2, −78 °C; NaBH4; g) Ac2O, pyridine, 23 °C; 77% (over 2 steps); h) Boc2O, DMAP, THF, 23 °C→85 °C, 99%; i) K2CO3, MeOH, H2O, 23 °C; j) TsOH, CH2Cl2, 23 °C, 96% (over 2 steps). Boc = tert-butoxycarbonyl, DMAP = 4-dimethylaminopyridine, Ts = para-toluenesulfonyl.
However, the recent evidence supporting a revised structural assignment for palau'amine (1b) challenged the utility of this route in its ability to access the redefined target structure. Nevertheless, the above-described Diels–Alder/[3,3]-rearrangement strategy could also be conveniently modified to accommodate the stereochemical inversions at C12 and C17. This effort revisits the common early-stage enone intermediate 10 (Scheme 4), which was found to exist in a 72:28 isomeric ratio with its enoate counterpart 11. This dynamic 10/11 mixture was subjected to selective Meerwein–Verley–Pondorf reduction of the bridging ketone group in 11, giving the secondary alcohol 20 in high yield (>97%; Scheme 6). This process again capitalizes on the Curtin–Hammett energy profile of the system, only this time in the opposite direction to advance 10 through sigmatropic rearrangement and reduction. The hydroxy group in 20 underwent substitution with chloride resulting in net retention of configuration at C17, presumably as a result of π-bond anchimeric assistance,[37] to afford the chloride 21 (97% yield, over 2 steps), which constitutes the C17 epimer of 14 (see Scheme 4).
Scheme 6.
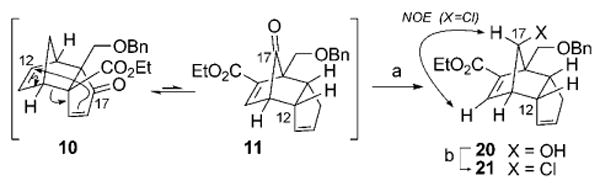
Reagents and conditions: a) Al(OiPr)3, iPrOH, reflux; b) SOCl2, CH2Cl2, 45 °C, 97% (over 2 steps).
Further derivatization of the tricyclo[5.2.1.0]decadiene 21 (Scheme 7) followed a similar strategy to that of its C17 epimer. Removal of the ester carbon moiety was accomplished through ester hydrolysis and Curtius rearrangement to afford ketone 22 (92% yield, over 3 steps). The C16-nitrogen functionality was introduced, as before (see Scheme 5), by regioselective Beckmann rearrangement followed by Boc protection of the resultant lactam to obtain imide 23 (53% yield, over 3 steps). Upon cleavage of the alkene in 23 by Lemieux–Johnson oxidation (Scheme 7) and exposure to silica gel and Et3N, C12 epimerization ensued to provide the di-aldehyde 24 (86% yield) as the thermodynamically favored diastereomer. The successful preparation of 24 constitutes a near-functional equivalent to the bridged chlorocyclopentane 18 (see Scheme 5), with the key stereochemical differences residing at C12 and C17. Moreover, an alternate sequence by which to prepare an appropriately functionalized non-bridging form of the chlorocyclopentane core of 1b can be accomplished by direct ozonolysis of the cyclopentene in 23 (Scheme 7), followed by reduction with NaBH4 and exposure to TsOH. This process led to spontaneous intramolecular alcoholysis of the imide to form the C13–C19 lactone, whose C6 hydroxy group was protected as the silyl ether 25 (91% yield, over 2 steps). Removal of the C20 benzyl ether and tethering of the resultant hydroxy group with the C16 carbamate (26; 62% yield, over 2 steps) precluded unproductive aziridine formation at this juncture.[38,39] This strategy allowed for sequential lactone hydrolysis, methyl ester formation, and C12 oxidation/epimerization to afford aldehyde 27 (90% yield, over 3 steps), which is a near-functional equivalent of the C12,C17 diastereomeric counterpart to the chlorocyclopentane 19 (see Scheme 5). Thus, stereoselective access to the advanced intermediates 24 and 27 provides promising avenues towards preparation of the revised palau'amine structure 1b.
Scheme 7.
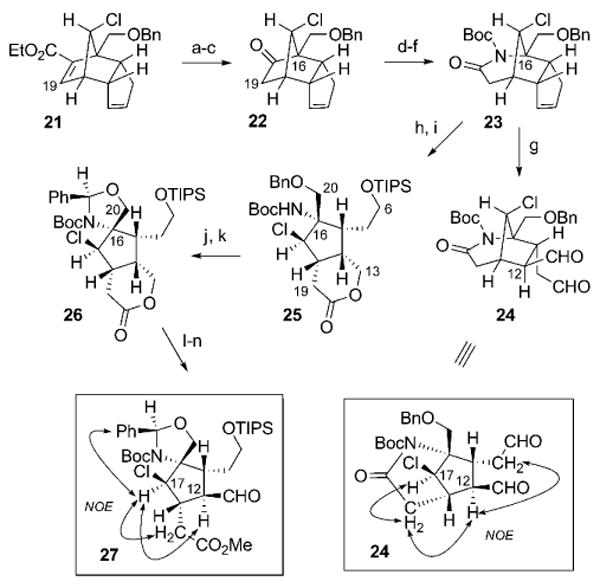
Reagents and conditions: a) NaOH, EtOH, 23 °C, 98%; b) (PhO)2P(O)N3, Et3N, toluene, 23 °C; EtOH, 80 °C; c) HCl, EtOH, 94% (over 2 steps); d) HONH2·HCl, pyridine, EtOH, H2O, 95 °C, 96%; e) SOCl2, Et2O, 23 °C, 59%; f) Boc2O, Et3N, DMAP, THF, reflux, 94%; g) OsO4, NaIO4, THF, H2O, 23 °C; SiO2, Et3N, EtOAc, 23 °C, 86%; h) O3, MeOH, CH2Cl2 −78 °C; NaBH4, 23 °C; i) TsOH·H2O, CH2Cl2, 23 °C; TIPSCl, imidazole, 23 °C, 91% (over 2 steps); j) Pd(OH)2/C, H2, THF, 23 °C; k) TsOH·H2O, PhCH(OMe)2, toluene, 23 °C, 62% (over 2 steps); l) NaOH, MeOH, 23 °C; m) TMSCHN2, benzene, MeOH, 23 °C; n) Dess–Martin periodinane, CH2Cl2, 23 °C; SiO2, Et3N, CH2Cl2, 23 °C; 90% (over 3 steps). TIPS = triisopropylsilyl.
In summary, the syntheses of the functionalized chlorocyclopentane cores of both the proposed original and revised structures of palau'amine have been accomplished. The essential features of the syntheses include a Diels–Alder/[3,3]-sigmatropic rearrangement sequence and Beckmann rearrangement, allowing for construction of the highly congested central cyclopentane with high stereocontrol.
Supplementary Material
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200801969.
Footnotes
This work is supported by the NIH-NIGMS (R01-GM67659) and Merck. A portion of this work was performed in the Department of Chemistry, University of Illinois, Urbana, IL 61801 (USA).
References
- 1.Al Mourabit A, Potier P. Eur J Org Chem. 2001:237–243. [Google Scholar]
- 2.Hoffmann H, Lindel T. Synthesis. 2003:1753–1783. [Google Scholar]
- 3.Kinnel RB, Gehrken HP, Scheuer PJ. J Am Chem Soc. 1993;115:3376–3377. [Google Scholar]
- 4.a Jacquot DEN, Lindel T. Curr Org Chem. 2005;9:1551–1565. [Google Scholar]; b Arndt HD, Riedrich M. Angew Chem Int Ed. 2008;47:4785–4788. doi: 10.1002/anie.200801793. [DOI] [PubMed] [Google Scholar]
- 5.Koenig SG, Miller SM, Leonard KA, Löwe RS, Chen BC, Austin DJ. Org Lett. 2003;5:2203–2206. doi: 10.1021/ol0344063. [DOI] [PubMed] [Google Scholar]
- 6.Dilley AS, Romo D. Org Lett. 2001;3:1535–1538. doi: 10.1021/ol015864j. [DOI] [PubMed] [Google Scholar]
- 7.Dransfield PJ, Wang SH, Dilley A, Romo D. Org Lett. 2005;7:1679–1682. doi: 10.1021/ol0473602. [DOI] [PubMed] [Google Scholar]
- 8.Lovely CJ, Du HW, He Y, Dias HVR. Org Lett. 2004;6:735–738. doi: 10.1021/ol036403w. [DOI] [PubMed] [Google Scholar]
- 9.Sivappa R, Hernandez NM, He Y, Lovely CJ. Org Lett. 2007;9:3861–3864. doi: 10.1021/ol0711568. [DOI] [PubMed] [Google Scholar]
- 10.Garrido-Hernandez H, Nakadai M, Vimolratana M, Li QY, Doundoulakis T, Harran PG. Angew Chem. 2005;117:775–779. doi: 10.1002/anie.200462069. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:765–769. doi: 10.1002/anie.200462069. [DOI] [PubMed] [Google Scholar]
- 11.Cernak TA, Gleason JL. J Org Chem. 2008;73:102–110. doi: 10.1021/jo701866g. [DOI] [PubMed] [Google Scholar]
- 12.Katz JD, Overman LE. Tetrahedron. 2004;60:9559–9568. [Google Scholar]
- 13.Lanman BA, Overman LE, Paulini R, White NS. J Am Chem Soc. 2007;129:12896–12900. doi: 10.1021/ja074939x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starr JT, Koch G, Carreira EM. J Am Chem Soc. 2000;122:8793–8794. [Google Scholar]
- 15.Tan XH, Chen C. Angew Chem. 2006;118:4451–4454. [Google Scholar]; Angew Chem Int Ed. 2006;45:4345–4348. doi: 10.1002/anie.200601208. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi J, Seiple IB, Young IS, O'Malley DP, Maue M, Baran PS. Angew Chem. 2008;120:3634–3636. doi: 10.1002/anie.200705913. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:3578–3580. doi: 10.1002/anie.200705913. [DOI] [PubMed] [Google Scholar]
- 17.O'Malley DP, Yamaguchi J, Young IS, Seiple IB, Baran PS. Angew Chem. 2008;120:3637–3639. [Google Scholar]; Angew Chem Int Ed. 2008;47:3581–3583. doi: 10.1002/anie.200801138. [DOI] [PubMed] [Google Scholar]
- 18.Foley LH, Buchi G. J Am Chem Soc. 1982;104:1776–1777. [Google Scholar]
- 19.Wiese KJ, Yakushijin K, Horne DA. Tetrahedron Lett. 2002;43:5135–5136. [Google Scholar]
- 20.Chung R, Yu ES, Incarvito CD, Austin DJ. Org Lett. 2004;6:3881–3884. doi: 10.1021/ol0490532. [DOI] [PubMed] [Google Scholar]
- 21.Feldman KS, Skoumbourdis AP. Org Lett. 2005;7:929–931. doi: 10.1021/ol0500113. [DOI] [PubMed] [Google Scholar]
- 22.Schroif-Gregoire C, Travert N, Zaparucha A, Al-Mourabit A. Org Lett. 2006;8:2961–2964. doi: 10.1021/ol0608451. [DOI] [PubMed] [Google Scholar]
- 23.Zöllinger M, Mayer P, Lindel T. J Org Chem. 2006;71:9431–9439. doi: 10.1021/jo061813u. [DOI] [PubMed] [Google Scholar]
- 24.Vergne C, Appenzeller J, Ratinaud C, Martin MT, Debitus C, Zaparucha A, Al-Mourabit A. Org Lett. 2008;10:493–496. doi: 10.1021/ol702866m. [DOI] [PubMed] [Google Scholar]
- 25.Wang SH, Romo D. Angew Chem. 2008;120:1304–1306. [Google Scholar]; Angew Chem Int Ed. 2008;47:1284–1286. doi: 10.1002/anie.200703998. [DOI] [PubMed] [Google Scholar]
- 26.Buchanan MS, Carroll AR, Addepalli R, Avery VM, Hooper JNA, Quinn RJ. J Org Chem. 2007;72:2309–2317. doi: 10.1021/jo062007q. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi H, Kitamura K, Nagai K, Nakao Y, Fusetani N, van Soest RWM, Matsunaga S. Tetrahedron Lett. 2007;48:2127–2129. [Google Scholar]
- 28.Grube A, Köck M. Angew Chem. 2007;119:2372–2376. [Google Scholar]; Angew Chem Int Ed. 2007;46:2320–2324. doi: 10.1002/anie.200604076. [DOI] [PubMed] [Google Scholar]
- 29.Köck M, Grube A, Seiple IB, Baran PS. Angew Chem. 2007;119:6706–6714. doi: 10.1002/anie.200701798. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:6586–6594. doi: 10.1002/anie.200701798. [DOI] [PubMed] [Google Scholar]
- 30.Woodward RB, Katz TJ. Tetrahedron. 1959;5:70–89. [Google Scholar]
- 31.Tanaka H, Kozuki Y, Ogasawara K. Tetrahedron Lett. 2002;43:4175–4178. [Google Scholar]
- 32.Lange JHM, Klunder AJH, Zwanenburg B. Tetrahedron. 1991;47:1495–1508. [Google Scholar]
- 33.Equilibration is achieved upon standing in solution at ambient temperature for 3 days or at 70 °C for 50 min.
- 34.Miyashita M, Yoshikoshi A. Synthesis. 1980:664–666. [Google Scholar]
- 35.Martínez AG, Vilar ET, Fraile AG, de la Mayo Cerero S, Maroto BL. Tetrahedron. 2004;60:9447–9451. [Google Scholar]
- 36.Kovacic P, Field KW, Wnuk TA. J Chem Eng Data. 1971;16:141–142. [Google Scholar]
- 37.Story PR. J Org Chem. 1961;26:287–290. [Google Scholar]
- 38.Wang SH, Dilley AS, Poullennec KG, Romo D. Tetrahedron. 2006;62:7155–7161. [Google Scholar]
- 39.Grube A, Immel S, Baran PS, Köck M. Angew Chem. 2007;119:6842–6845. doi: 10.1002/anie.200701935. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:6721–6724. doi: 10.1002/anie.200701935. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200801969.