

Abstract
The ApbC protein has been shown previously to bind and rapidly transfer iron-sulfur ([Fe-S]) clusters to an apoprotein (Boyd, J. M., Pierik, A. J., Netz, D. J., Lill, R., and Downs, D. M. (2008) Biochemistry 47, 8195–8202. This study utilized both in vivo and in vitro assays to examine the function of variant ApbC proteins. The in vivo assays assessed the ability of ApbC proteins to function in pathways with low and high demand for [Fe-S] cluster proteins. Variant ApbC proteins were purified and assayed for the ability to hydrolyze ATP, bind [Fe-S] cluster, and transfer [Fe-S] cluster. This study details the first kinetic analysis of ATP hydrolysis for a member of the ParA subfamily of “deviant” Walker A proteins. Moreover, this study details the first functional analysis of mutant variants of the ever expanding family of ApbC/Nbp35 [Fe-S] cluster biosynthetic proteins. The results herein show that ApbC protein needs ATPase activity and the ability to bind and rapidly transfer [Fe-S] clusters for in vivo function.
Proteins containing iron-sulfur ([Fe-S]) clusters are employed in a wide array of metabolic functions (reviewed in Ref. 1). Research addressing the biosynthesis of the iron-molybdenum cofactor of nitrogenase in Azotobacter vinelandii led to the discovery of an operon (iscAnifnifUSVcysE1) involved in the biosynthesis of [Fe-S] clusters (reviewed in Ref. 2). Subsequent experiments led to the finding of two more systems involved in the de novo biosynthesis of [Fe-S] clusters, the isc and the suf systems (3, 4). Like Escherichia coli, the genome of Salmonella enterica serovar Typhimurium encodes for the isc and suf [Fe-S] cluster biosynthesis machinery.
Recent studies have identified a number of additional or non-isc/-suf-encoded proteins that are involved in bacterial [Fe-S] cluster biosynthesis and repair. Examples include the following: CyaY, an iron-binding protein believed to be involved in iron trafficking and iron delivery (5–7); YggX, an Fe2+-binding protein that protects the cell from oxidative stress (8, 9); ErpA, an alternate A-type [Fe-S] cluster scaffolding protein (10); NfuA, a proposed intermediate [Fe-S] delivery protein (11–13); YtfE, a protein proposed to be involved in [Fe-S] cluster repair (14, 15); and CsdA-CsdE, an alternative cysteine desulferase (16).
Analysis of the metabolic network anchored to thiamine biosynthesis in S. enterica identified lesions in three non-isc or -suf loci that compromise Fe-S metabolism as follows: apbC, apbE, and rseC (17–21). This metabolic system was subsequently used to dissect a role for cyaY and gshA in [Fe-S] cluster metabolism (6, 22, 23). Of these, the apbC (mrp in E. coli) locus was identified as the predominant site of lesions that altered thiamine synthesis by disrupting [Fe-S] cluster metabolism (17, 18).
ApbC is a member of the ParA subfamily of proteins that have a wide array of functions, including electron transfer (24), initiation of cell division (25), and DNA segregation (26, 27). Importantly, ATP hydrolysis is required for function of all well characterized members of this subfamily, and all members contain a “deviant” Walker A motif, which contains two lysine residues instead of one (GKXXXGK(S/T)) (28). ApbC has been shown to hydrolyze ATP (17).
Recently, five proteins with a high degree of identity to ApbC have been shown to be involved in [Fe-S] cluster metabolism in eukaryotes. The sequence alignments of the central portion of these proteins and bacterial ApbC are shown in Fig. 1. HCF101 was demonstrated to be involved in chloroplast [Fe-S] cluster metabolism (29, 30). The CFD1, Npb35, and huNbp35 (formally Nubp1) proteins were demonstrated to be involved in cytoplasmic [Fe-S] cluster metabolism (31, 32). Ind1 was demonstrated to be involved in the maturation of [Fe-S] clusters in the mitochondrial enzyme NADH:ubiquinone oxidoreductase (33). There is currently no report of any of these proteins hydrolyzing ATP.
FIGURE 1.

Protein sequence alignments of members of the ApbC/Nbp35 subfamily of ParA family of proteins. Protein alignments were assembled using the Clustal_W method in the Lasergene® software and show only the central portion of the proteins, which have the highest sequence conservation. The three boxed areas highlight the Walker A box, conserved Ser residue, and CXXC motif. Proteins listed are as follows: ApbC (S. enterica serovar Typhimurium LT2), CFD1 (S. cerevisiae), Nbp35 (S. cerevisiae), HCF101 (Arabidopsis thaliana), huNpb35 (formally Nubp1) (Homo sapiens), and Ind1 (Candida albicans).
Biochemical analysis of ApbC indicated that it could bind and transfer [Fe-S] clusters to Saccharomyces cerevisiae apo-isopropylmalate isomerase (34). Additional genetic studies indicated that ApbC has a degree of functional redundancy with IscU, a known [Fe-S] cluster scaffolding protein (35, 36).
In this study we investigate the correlation between the biochemical properties of ApbC (i.e. ATPase activity, [Fe-S] cluster binding, and [Fe-S] cluster transfer rates) and the in vivo function of this protein. This is the first detailed kinetic analysis of ATP hydrolysis for a member of the ParA subfamily of deviant Walker A proteins and the first functional analysis of a member of the ever expanding family of ApbC/Nbp35 proteins. Data presented indicate that noncomplementing variants have distinct biochemical properties that place them in three distinct classes.
EXPERIMENTAL PROCEDURES
Materials—FeCl3 (ACS grade), thiamine (>99%), Li2S (>98%), Fe(NH4)2(SO4)2 (>99%), 3-(2-pyridyl)-5,6-di(2-furyl)-1,2,4-triazine-5′,5″-disulfonic acid (>99%), ascorbic acid (>99%), (C2H3O2)2Zn·2H2O (reagent grade), N,N-dimethyl-p-phenylenediamine sulfate (98%), and l-cysteine (>98%) were purchased from Sigma. dl-threo-3-Isopropylmalic acid (96%) was purchased from Wako Pure Chemical Co Osaka, Japan. All other chemicals were of the highest purity available. The BCA protein assay kit and bovine serum albumin were purchased from Pierce. E. coli BL21 (AI*) cells were purchased from Novagen.
Bacterial Strains, Culture Media, and Growth Conditions—Bacterial strains were derivatives of S. enterica serovar Typhimurium strain LT2 (specifically DM1) whose genotypes are shown in Table 1. Plasmids carrying the desired allele of apbC fused to a C-terminal His tag in pET20b were constructed as described (17) using XhoI instead of NcoI. All constructs used were verified by sequencing at the University of Wisconsin Biotechnology Center. Plasmids were moved between strains via electroporation. Overnight cultures were grown in Difco nutrient broth with appropriate antibiotic for strains harboring a plasmid. Test cultures were grown at 37 °C shaking at an approximate 45° angle to increase aeration. The absorbance of cultures was taken at 650 nm on a Bausch and Lomb Spectronic 20.
TABLE 1.
Strains and plasmids
Unless indicated otherwise, strains were constructed for this study. The yggX::Gm allele has been described (77). Tn 10d refers to the transposition-defective mini-Tn10 (Tn10D-16 D-17) (78).
|
Strain
|
Genotype
|
||
| DM5104 | DM1 wild type | ||
| DM5986 | apbC55:Tn10d(Tc) yggX::Gma | ||
|
DM9450
|
purF2085 apbC55::Tn10d(Tc)
|
||
| Plasmid | Vector and insert | Insert | |
| pET20b | pET20b(Ap) | None | |
| pES1 | pET20b(Ap) | apbCb | |
| pES2 | pET20b(Ap) | apbC (K121A)b | |
| pJB1 | pET20b(Ap) | apbC (S122A) | |
| pJB2 | pET20b(Ap) | apbC (C70A) | |
| pJB3 | pET20b(Ap) | apbC (C283A) | |
| pJB4 | pET20b(Ap) | apbC (C286A) | |
| pJB5 | pET20b(Ap) | apbC (S182A) | |
| pJB6 | pET20b(Ap) | apbC (S116A) | |
| pJB7 | pET20b(Ap) | apbC (C283A, C286A) | |
Resistances are indicated as follows: Tc, tetracycline; Ap, ampicillin; Gm, gentamycin.
Plasmids were constructed for a previous study (17).
Nutritional Analysis—Complementation analyses were performed in strains where plasmids encoded the only copy of apbC present in the cell. Complementation was assessed using 5-ml cultures of no carbon E (NCE) medium supplemented with 1 mm MgSO4 and trace minerals. Cultures were started with a 1:100 inoculum. When present in the media, the following were used at the specified final concentration: adenine, 0.4 mm; thiamine, 100 nm; glucose, 11 mm; tricarballylate, 20 mm; gluconate, 11 mm; and ampicillin, 30 μg/ml.
Three growth assays, using two genetic backgrounds, were used to examine the ability of ApbC proteins to function in vivo. The two genetic backgrounds used were apbC yggX and purF apbC. In vivo function was assayed by growth on tricarballylate as a carbon and energy source or growth in the absence of exogenous thiamine on either glucose or gluconate, respectively. The requirement for ApbC in these growth conditions has been described previously (17, 18, 23, 35, 37).
Anaerobic Work—Anaerobic work was performed using a Coy anaerobic glove box (Grass Lake, MI) or vacuum manifold. Before placement inside the anaerobic chamber, solutions were made anoxic by repeated evacuation and flushing with N2 gas passed over a heated copper column for removal of O2. Outside of the glove box, all solutions were added to anaerobic cuvettes using gas-tight Hamilton syringes.
ApbC and Leu-1 Protein Purification—Purification of ApbC and Leu-1 has been described previously (34).
Protein Concentration Determination—Protein concentration was determined using a colorimetric assay or an empirically determined extinction coefficient (apo-ApbC-His280 = 43.1 mm–1 cm–1). The colorimetric assay was copper-based and used a reagent containing bicinchoninic acid to detect of the cupreous ion (Pierce). Bovine serum albumin (2 mg/ml) was used as a standard.
[Fe-S] Cluster Reconstitution—Iron-sulfur cluster reconstitution was described previously (34). Briefly, ApbC protein (2.1 mg/ml; 52 μm) suspended in buffer A (50 mm Tris, pH 8.0, 150 mm NaCl) was incubated for 1 h in an anoxic environment in the presence of 5 mm DTT.2 A 5-fold excess of FeCl3 was added to the protein followed by a 5-fold excess Li2S. Proteins were incubated for 1 h, and excess S2–, Fe3+, and DTT was removed by passing the reaction mixture over a PD-10 column (GE Healthcare). One mm DTT was added to protein post desalting.
Leu-1 Activation Assays—Leu-1 activation assays have been described previously (34). Briefly, apo-isopropylmalate isomerase (apo-Leu-1) (25 μm) was pre-reduced with 5 mm DTT in the anaerobic glove box for at least 1 h prior to assay initiation. The Leu-1 activation assays contained 3.5 μm Leu-1, 5 mm DTT, 50 mm Tris-HCl, pH 8.0, and 150 mm NaCl in a total volume of 500 μl. Assays were initiated by the addition of 4 μm ApbC protein. Ten-μl aliquots of the assay mixture were removed at time points, and Leu-1 was assayed for the ability to convert 3-isopropylmalate to dimethylcitraconate acid spectrophotometrically (dimethylcitraconate ε235 = 4.35 mm–1 cm–1) (38).
The second-order rate constant for ApbC dependent Leu-1 activation was determined by linearization of the data and fitting the data using linear regression. The ApbC-dependent activation of Leu-1 is summarized by Reaction 1,
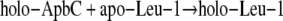 |
REACTION 1 |
The amount of holo-ApbC protein present (4 μm initial) in the assay mixture at a given time point was determined using Equation 1,
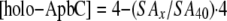 |
(Eq. 1) |
where SAx is the specific activity of Leu-1 at a fixed time point, and SA40 is the specific activity of Leu-1 after the reaction had proceeded for 40 min. The second-order rate constant of cluster transfer was determined by fitting the 1/[holo-ApbC protein] versus time post the addition of holo-ApbC protein to apo-Leu-1 data using linear regression, where k is the slope of the fit.
Spectroscopic Techniques—The UV-visible absorption spectra were recorded with a Lambda Bio 40 spectrophotometer (PerkinElmer Life Sciences) using 1.5-ml Sterna Cell cuvettes that can be anaerobically sealed (Atascadero, CA).
Metal Analysis—Inductively coupled plasma-mass spectrometry was conducted by the Soil and Plant Analysis Laboratory at the University of Wisconsin-Madison. The concentration of non-heme iron and acid-labile sulfide was also determined as described elsewhere (39).
Assay to Monitor Inorganic Phosphate—The direct colorimetric malachite green/molybdate/polyvinyl alcohol assay as described by Chan et al. (40) was used to monitor inorganic phosphate (Pi) release from nucleotide hydrolysis. Assays contained 0–0.34 mg of purified wild-type ApbC, 0 or 5 mm MgCl2, 0 or 200 mm KCl, 0 or 5 mm nucleotide triphosphate, and 50 mm Tris, pH 8.0. The assays were initiated by addition of ApbC. The mixture was allowed to react at room temperature for 30 min before 20 μl of the reaction mixture was removed, diluted to 250 μl with buffer A, and quickly added to 1 ml of the malachite green/molybdate/polyvinyl alcohol assay mixture. The mixture was incubated for 15 min at room temperature before sample absorbance was monitored at 630 nm in a Molecular Devices Spectra-MAX Plus microplate reader. Because of daily variation of absorbance, a standard curve containing 0–15 nmol of Pi was constructed for each set of assays.
Coupled Spectrophotometric Assay for ApbC ATPase Activity—A continuous spectrophotometric assay was developed that couples ADP production with the oxidation of NADH to NAD+ (41) as shown in Reactions 2 and 3.
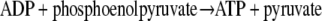 |
REACTION 2 |
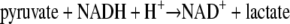 |
REACTION 3 |
The assays were initiated when purified ApbC protein (10–350 μg) was added to an assay mixture containing 0.2 mm NADH, 4 mm phosphoenolpyruvate, 0–12 mm MgCl2, 200 mm KCl, 20 units of lactate dehydrogenase, 20 units of pyruvate kinase, 0.01–10 mm ATP (stocks prepared in 1 m Tris, pH 8.0), and 50 mm Tris, pH 8.0. The assay volume was 1 ml, and assays were conducted anaerobically in 1.5 ml of Sterna Cell cuvettes (Atascadero, CA) that had a screw cap top and rubber septum. After the addition of ApbC, the oxidation of NADH was monitored by following the absorbance change at A340 from 2 to 4 min (linear region).
The linear steady-state portion of the A340 versus time plot was used to determine the Hill coefficient by fitting initial velocity data to Equation 2 (R2 ≥ 0.99),
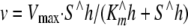 |
(Eq. 2) |
where v is velocity; S is the concentration of varied substrate; Vmax is the velocity at saturating substrate; Km is the Michaelis constant for the varied substrate, and h is the Hill coefficient (labeled nH in Table 4). The kinetic parameters Vmax and Km were determined by fitting the v versus [ATP] initial velocity data to Equation 3 (R2 ≥ 0.99),
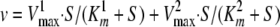 |
(Eq. 3) |
The velocity data as plotted in the double-reciprocal form was fit by Equation 4,
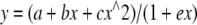 |
(Eq. 4) |
where the initial slope is defined as b – ae; the slope of the asymptote is c/e; the y intercept is a, and the asymptote intercept is (be – c)/e^2.
TABLE 4.
Nucleotide specificity of ApbC
Inorganic phosphate release was detected after a 30-min incubation using the malachite green/molybdate/polyvinyl alcohol assay as described under “Experimental Procedures.” Assays were done in triplicate, and the error is listed as standard deviation. Unless indicated, assays contained 0–0.34 mg of purified ApbC, 6 mm MgCl2, 200 mm KCl, 5 mm nucleotide mono-, di-, or triphosphate, and 50 mm Tris, pH 8.0.
| Nucleotide (5 mm) | Pi release |
|---|---|
| nmol mg-1 | |
| ATP | 518 ± 18 |
| ATP - Mg2+ | NDa |
| ATP - K+ | 63 ± 27 |
| ATP + AMP | 429 ± 36 |
| ATP + ADP | ND |
| ADP | ND |
| AMP | ND |
| dATP | 261 ± 30 |
| CTP | 42 ± 21 |
| GTP | 75 ± 15 |
| TTP | ND |
| UTP | ND |
No phosphate release was detected.
Computational Analysis—Nonlinear and linear regression analyses and curve fitting to first-order rate laws were performed using the software SigmaPlot (version 9.0).
Bioinformatic Analysis—Protein sequences were acquired from the NCBI website in FASTA format. The accession numbers for sequences used are as follows: Ind1 EAK91699; Nbp35 CAA96797.1; Cfd1, AAS56623; Hcf101, AAR97892.1, ApbC (mrp), NP_461098; and Nubp1, NP_002475.2.
Sequence alignments were performed using the Clustal_W function of the Lasergene software by DNAstar, Madison, WI. Default settings were used, including the Gonnet Series for Protein Weight Matrix, gap penalty of 10, and a gap length penalty of 0.2. Selected regions of sequences were used to give the best alignment of the similar regions. The protein sequences for the proteins used in Fig. 1 were acquired in FASTA format from the NCBI Protein Sequence Data Base.
The ApbC amino acid sequence was used as the input for the three-dimensional Jigsaw version 2.0 Comparative Modeling Server by Cancer Research UK using the automatic mode (42–44). The resulting text file alignment was viewed using the PyMOL Viewer from Delano Scientific, LLC, for protein structures. Regions of the proteins that do not have significant similarity are excluded from the structures. The accession numbers of the structures used to thread the ApbC sequence are 2bej_A for Soj and 2afk_H for NifH and can be found on the NCBI website.
RESULTS
Protein Sequence Alignments Show Significant Conservation of Protein Sequence within the ApbC/Nbp35 Subfamily of ParA Homologs—The ApbC/Nbp35 subfamily proteins show a high degree of conservation in their central and C-terminal domains. Representative members of the ApbC/Nbp35 family are shown in Fig. 1. The N-terminal 100 amino acids of bacterial ApbC are sparsely conserved in the plant homolog HCF101, yeast Nbp35, human Nubp1, and mitochondrial Ind1 and therefore are not included in the alignments. The N-terminal extension is missing in yeast Cfd1. The N-terminal region of ApbC showed no similarity to characterized functional domains of the N-terminal extension found in ParA and ParA homologs (25). The central and C-terminal domains of these proteins contain two highly conserved domains as follows: the Walker A box for ATP binding/hydrolysis, and a CXXC motif in the C-terminal third of the protein. In Fig. 1 the Walker A box, conserved Cys residues, and conserved Ser are boxed for clarity. All six of these proteins are thought to be involved in [Fe-S] cluster metabolism (29–35).
In Vivo Function of ApbC Is Abolished by Mutations in the Walker A Motif, CXXC Motif, and a Conserved Serine—A number of mutant alleles of apbC were created using plasmid pES1, which encodes the C-terminal hexahistidine-tagged ApbC protein (17). Directed mutations substituted single residues to alanine in three regions of the protein (see Fig. 1) as follows: 1) deviant Walker A motif necessary for ATP binding/hydrolysis (ApbCK116A, ApbCK121A, and ApbCS122A) (28); 2) conserved cysteine residues with spacing consistent with a metal-binding/thioredoxin motif (ApbCC283A and ApbCC286A) (45); and 3) conserved serine (ApbCS182A) and nonconserved cysteine (ApbCC70A).
To probe the in vivo function of ApbC variants, complementation assays were conducted examining three growth defects using two genetic backgrounds. Complementation of two of the growth defects required restoration of the low flux thiamine biosynthesis pathway (the amount of thiamine pyrophosphate in wild-type S. enterica grown in minimal glucose medium is 41.4 pmol/mg dry weight (46)). Stains lacking apbC and yggX (or purF) are thiamine auxotrophs likely because of poor cluster occupancy of ThiH/ThiC (47, 48, 80). Complementation of a third growth defect required function of the high flux tricarballylate catabolic metabolic pathway (35, 37, 49, 50). Strains lacking yggX and apbC are unable to use tricarballylate as a carbon and energy source, and presumably, this is from poor [Fe-S] cluster occupancy of TcuB (35).
Plasmids carrying alleles of apbC were introduced into two genetic backgrounds (apbC yggX and apbC purF) and provided the only source of apbC in the resulting strains. Each of the seven mutant alleles was tested for the ability to complement growth in both the low and high flux demand situations. The data in Table 2 show that substitutions in the Walker A (ApbCK116A, ApbCK121A, and ApbCS122A) and CXXC (ApbCC283A and ApbCC286A) motifs resulted in proteins unable to complement any of the three growth conditions. Changing the nonconserved Cys-70 to Ala had no detectable effect on in vivo function of the protein. In all cases, the empty vector and wild-type control plasmids displayed the growth patterns anticipated by past experiments (17, 35).
TABLE 2.
Growth allowed by ApbC variants
Doubling times were calculated using the formulas μ = ln(X/X0)/T, where μ is the growth rate; X and Xo are absorbance at 650 nm; T(h) is the time between absorbance readings X and Xo; and doubling time (g) = (ln 2)/μ (79). Numbers represent the average of three independent cultures.
|
Relevant chromosomal genotype
| |||||
|---|---|---|---|---|---|
|
ApbC variant
|
apbC yggX
|
purF apbC
|
|||
| Glua | Glu + Thi | TC + Thi | Glcn + Adn | Glcn + Adn + Thi | |
| Vector | NG (0.33)b | 2.5 ± 0.32 | NG (0.21) | NG (0.16) | 1.5 ± 0.07 |
| Wild type | 1.6 ± 0.03 | 1.7 ± 0.16 | 2.7 ± 0.26 | 2.4 ± 0.28 | 1.6 ± 0.08 |
| ApbC(C70A) | 1.7 ± 0.17 | 1.6 ± 0.06 | 3.1 ± 0.54 | 2.0 ± 0.02 | 1.4 ± 0.04 |
| ApbC(K116A) | NG (0.25) | 1.8 ± 0.12 | NG (0.24) | NG (0.18) | 1.7 ± 0.26 |
| ApbC(K121A) | NG (0.31) | 2.2 ± 0.10 | NG (0.19) | NG (0.10) | 2.1 ± 0.24 |
| ApbC(S122A) | NG (0.27) | 2.3 ± 0.16 | NG (0.15) | NG (0.12) | 1.9 ± 0.09 |
| ApbC(S182A) | 2.7 ± 0.09 | 1.7 ± 0.08 | NG (0.28) | 2.4 ± 0.15 | 1.5 ± 0.24 |
| ApbC(C283A) | NG (0.27) | 2.5 ± 0.12 | NG (0.19) | NG (0.15) | 1.6 ± 0.16 |
| ApbC(C286A) | NG (0.23) | 2.4 ± 0.22 | NG (0.20) | NG (0.18) | 1.6 ± 0.04 |
Abbreviations used are as follows: Glu, glucose; Thi, thiamine; TC, tricarballylate, Glcn, gluconate; Adn, adenine.
NG, no growth was determined when strains showed doubling times of greater than 6 h and/or a final A650 less than 0.35. Average final A650 is indicated in parentheses for strains determined as no growth.
The ApbCS182A variant qualitatively restored thiamine synthesis, although there was a 2–4-h lag before exponential growth initiated (data not shown). In addition, the doubling times of the yggX apbC or purF apbC strains carrying ApbCS182A, on glucose or gluconate in the absence of thiamine, were ∼1.6-fold higher than with the wild-type protein. The ApbCS182A variant was unique in its ability to restore growth under the low demand growth condition monitoring thiamine biosynthesis but not under the high demand condition monitoring tricarballylate utilization.
All ApbC variant proteins accumulated to qualitatively similar levels in yggX apbC cells when expressed off a plasmid and detected by Western blot analysis (data not shown). Thus, the phenotypes seen were not because of differential protein stability.
Purification and Metal Analysis of ApbC Proteins—ApbC proteins were expressed in E. coli BL21 AI* using the pET20 system and purified as C-terminal hexahistidine-tagged fusion proteins via Ni2+-affinity chromatography. The single column purification yielded proteins that were >95% pure. The behavior of all ApbC variants resembled that of the wild-type protein during purification except for ApbCC286A and ApbCC283A, which had lower yields and precipitated upon long dialysis or high concentration (>10 mg/ml).
To examine the possibility that variant ApbC proteins could form intermolecular disulfide bonds, the electrophoretic mobility of purified ApbC proteins was examined using nonreducing SDS-PAGE. Fig. 2 shows the separation of ApbC proteins into monomers, dimers, and hexamers using a nonreducing SDS-polyacrylamide gel. Covalent linkage of ApbC monomers was seen with the ApbCC283A and ApbCC286A proteins. A small but reproducible amount of dimer was also seen with the wild-type protein. All other ApbC proteins migrated as monomers. Upon the addition of 1 mm β-mercaptoethanol to the sample buffers, all of the protein samples migrated exclusively as monomers (data not shown). These data suggest that ApbCC283A and ApbCC286A can form covalent disulfide linkages spanning the dimer interface.
FIGURE 2.

Nonreducing SDS-PAGE analysis of purified ApbC proteins. The ApbC protein in each lane is labeled at the bottom of the gel, and the migration of protein as a monomer, dimer, and hexamer is labeled on the left-hand side of the gel. Three μg of each ApbC protein was loaded onto the gel.
Metal Binding by Variant ApbC Proteins—As was seen with the wild type, none of the ApbC proteins examined co-purified with a detectable concentration of iron or sulfur atoms. ApbC did bind two iron and two sulfur atoms per monomer when chemically reconstituted with ferrous iron and sulfide under reducing conditions (34).
All the ApbC variant proteins, except ApbCC283A,C286A, bound approximately two iron and two acid-labile sulfide atoms per ApbC monomer as has been reported previously with wild-type ApbC (Table 3) (34). The ApbCC283A,C286A variant protein bound approximately one atom of each iron and sulfide and was not further characterized here.
TABLE 3.
Variant ApbC proteins bind and transfer iron-sulfur clusters
ApbC proteins were reconstituted as described under “Experimental Procedures.” Iron and sulfide were quantified as described elsewhere (39). Metal binding data are the average of two independent reconstitutions.
| ApbC variant | Iron | Sulfur | Rate of cluster transfer to Leu-1a |
|---|---|---|---|
| m-1 min-1 | |||
| ApbC | 2.2 ± 0.2 | 2.1 ± 0.1 | 39,000 ± 2,000 |
| ApbCK16A | 2.0 ± 0.2 | 1.9 ± 0.0 | 25,000 ± 1,000 |
| ApbCK21A | 1.9 ± 0.1 | 1.9 ± 0.1 | 27,000 ± 1,000 |
| ApbCS22A | 1.9 ± 0.2 | 1.8 ± 0.1 | 43,000 ± 1,000 |
| ApbCS182A | 1.7 ± 0.2 | 1.8 ± 0.0 | 45,000 ± 1,000 |
| ApbCC283A | 1.8 ± 0.1 | 1.7 ± 0.2 | 45,000 ± 1,000 |
| ApbCC286A | 1.7 ± 0.2 | 1.9 ± 0.1 | 44,000 ± 5,000 |
| ApbCC283A,C286A | 0.9 ± 0.2 | 1.0 ± 0.0 | NDb |
Leu-1 activation assays were conducted as described previously (34). The second-order rate constant of cluster transfer was determined by a linear transformation of the data and fitting the data using linear regression as described under “Experimental Procedures.”
ND means not determined.
ApbC Variant Proteins Can Transfer [Fe-S] Clusters—The previously described model system using isopropylmalate isomerase protein Leu-1 from S. cerevisiae was used to assay the ability of variant ApbC proteins to transfer their respective [Fe-S] clusters (34, 51). Previous results showed that 2 mol of holo-ApbC are required for full activation of 1 mol of apo-Leu-1 (34). The assays described here had 3.5 μm Leu-1 protein and 4 μm ApbC protein; therefore, apo-Leu-1 was present in a 1.75-fold excess of holo-ApbC dimer.
All ApbC variants were capable of activating apo-Leu-1, but the data show ApbC protein variants can be divided into two classes (Table 3). One class of ApbC proteins, consisting of wild type, ApbCS122A, ApbCS182A, ApbCC283A, and ApbCC286A, activated Leu-1 rapidly and effectively with a second-order rate constant of ∼40,000 m–1 min–1. The second class of ApbC variant proteins, consisting of ApbCK116A and ApbCK121A, activated Leu-1 at ∼60% of the rate of the first class. The data distinguishing the two classes in cluster transfer are shown in Fig. 3.
FIGURE 3.

ApbC proteins activate Leu-1 at variable rates. A, time course of ApbC- (•) or ApbCK121A (○)-dependent Leu-1 activation. Leu-1 activity was monitored at fixed time points by the formation of isopropylmaleate, which was monitored at A235. Assays contained 4 μm reconstituted ApbC and 3.5 μm apo-Leu-1. The reaction was started by adding ApbC to the reaction vessel. B, plot of 1/[holo-ApbC protein] versus time post ApbC protein addition. The second-order rate constant of cluster transfer was determined by fitting the data using linear regression. The concentration of holo-ApbC protein at a given time point was estimated using Equation 1.
Kinetic Analysis of ApbC ATP Hydrolysis Activity—A discontinuous direct colorimetric assay designed to detect and quantify inorganic phosphate (Pi) production, indicative of nucleotide hydrolysis, was used to examine the requirements for ApbC ATPase activity and nucleotide specificity. As shown in Table 4, Pi release was dependent upon the presence of both nucleotide and Mg2+. An 8-fold increase in Pi release was seen upon the addition of K+ to the reaction mix. Although ApbC was unable to hydrolyze the α-β phosphodiester bond of ADP, the inclusion of equal amounts of ADP and ATP in the reaction mix resulted in complete inhibition of ApbC ATPase activity. A 1.2-fold decrease in the amount of Pi release was seen with the inclusion of both AMP and ATP. ApbC was capable of hydrolyzing dATP, CTP, and GTP, but the amount of Pi production was significantly lower than the amount of Pi production from ATP hydrolysis.
A continuous spectrophotometric assay coupling ATP hydrolysis to NADH oxidation using the enzymes pyruvate kinase and lactic dehydrogenase (Reactions 2 and 3) was used to define the kinetic parameters for ATPase activity of ApbC proteins. High protein concentrations and anaerobic reducing conditions (to prevent NADH oxidation) were used to compensate for the slow rate of ATP hydrolysis catalyzed by ApbC.
Kinetic parameters were determined from initial velocity data for the purified enzymes as described under “Experimental Procedures” (Table 5). The specific activity for wild-type ApbC was determined to be 45.7 nmol min–1 mg–1 (0.210 mg of ApbC, 5.1 mm MgCl2, 5 mm ATP, and 200 mm KCl, 50 mm Tris, pH 8.0). Thus, the activity of ApbC was found to be 3-fold higher using the coupled assay than that calculated from the Pi release data in Table 4. This difference may reflect inhibition of ApbC by the accumulating ADP in the discontinuous assay. The coupled assay has an ATP-regenerating system, and therefore very little ADP was present in the reaction mix upon initiation of the reaction by ApbC addition or during ApbC-catalyzed ATP hydrolysis.
TABLE 5.
Kinetic analysis of variant ApbC proteins
ATP hydrolysis was monitored using a spectrophotometric assay coupling the hydrolysis of ATP to the oxidation of NADH using the enzymes pyruvate kinase and lactic dehydrogenase. Assays contained the following: 0.2 mm NADH, 4 mm phosphoenolpyruvate, 20 units of each pyruvate kinase and lactic dehydrogenase, 0–12 mm MgCl2, 200 mm KCI. Km, Vmax, and nH (Hill coefficient) values were determined by fitting the v versus [S] curves to modified forms of either the Michaelis-Menten-Henri equation (Equation 3) or Hill equation (Equation 2). Assays were done in triplicate, and the error is listed as standard deviation.
| Km1 | Km2 | kcat | nHa | |
|---|---|---|---|---|
| μm | μm | min-1 | ||
| Wild type | 1.6 ± 1.1 | 300 ± 43 | 1.92 | 0.48 ± 0.05 |
| K116A | 5.7 ± 2.9 | 1260 ± 170 | 3.40 | 0.49 ± 0.06 |
| K121A | 6.4 ± 2.7 | 930 ± 330 | 1.05 | 0.35 ± 0.04 |
| C283A | 15.0 ± 6.0 | 3670 ± 1300 | 0.49 | 0.29 ± 0.05 |
| C286A | 11.0 ± 8.0 | 1380 ± 600 | 0.84 | 0.32 ± 0.06 |
Abbreviation used is as follows: nH, Hill coefficient.
The relative velocities at various ATP concentrations for ApbC are shown in Fig. 4. The v versus [ATP] plots (Fig. 4A) were best fit by modified forms of either the Hill equation (Equation 2) or the Michaelis-Menten-Henri equation (Equation 3) (Equation 3 as shown). The double-reciprocal plots (Fig. 4B) show data that are nonlinear and have a rising trend. This is characteristic of enzymes displaying negative cooperativity (52). To verify negative cooperativity, Hill plots were constructed for the data. The Hill plot for the wild-type enzyme is shown in Fig. 4C. The plot shows data that have two separate linear regions on the graph, each having a distinct slope. One of the slopes (n = 0.4) corresponds to the Hill coefficient (nH = 0.48; Table 5) derived from fitting the v versus [ATP] data to Equation 2.
FIGURE 4.

Kinetic results demonstrate cooperative behavior for S. enterica ApbC protein when ATP is varied (wild-type ApbC shown). The plots illustrate the velocity of ADP production while varying their concentration of ATP. Plots are as follows: A, v versus [substrate] fit by Equation 3; B, double-reciprocal plot of the data in plot 1 fit by Equation 4; C, Hill plot fit using a linear regression (n = slope). Datum points are the average of three assays. v is defined as nmol/min/mg.
The kinetic parameters for the wild-type and variant S. enterica enzymes are displayed in Table 5. Fitting the data to Equation 4 gives two Km values; one Km value for each hypothesized ATP-binding site. The wild-type enzyme had one low (1.6 μm) and one high (300 μm) Km value for ATP, as would be expected of an enzyme that displays negative kinetic cooperativity for a substrate.
With the exception of ApbCS122A and ApbCS182A, all of the variant ApbC proteins hydrolyzed ATP. The kinetic parameters vary slightly for each variant, and no unifying theme was noted. The kinetic differences observed for the variant proteins were manifest in turnover number, the second-site Km, and Hill coefficient.
Effect of an [Fe-S] Cluster on ATPase Activity—The ATPase activity of holo- and apo-ApbC was examined. The presence of [Fe-S] cluster had no effect on the kinetic parameters of ATP hydrolysis (data not shown). In addition, it was previously shown that the addition of 10 mm Mg·ATP had no effect on the rate that ApbC activated Leu-1 (34).
DISCUSSION
ApbC is a cytosolic protein whose absence impacts [Fe-S] cluster metabolism in S. enterica. Previous work in our laboratory has described two biochemical activities for ApbC. ApbC protein can hydrolyze ATP and bind and transfer an [Fe-S] cluster (17, 34). This study examined the two biochemical activities of a number of variant proteins that are not able to complement an apbC null mutation. The results herein show that both of these biochemical activities are required for in vivo function, and furthermore, they can be separated by mutation.
Previous data showed that the presence of ATP was not necessary the for [Fe-S] cluster transfer activity of ApbC (34). The data here confirm that the ability to hydrolyze ATP is not necessary for [Fe-S] cluster transfer. Based on these data we envision a general model in which ATP binding and hydrolysis are required to generate holo-ApbC in vivo and not for the subsequent transfer of the [Fe-S] cluster.
In this study we targeted residues in three locations of the protein for directed mutagenesis as follows: the Walker A motif, a conserved Ser, and the Cys residues present in a CXXC motif. Walker A motifs have been examined in detail and are used to bind and to hydrolyze ATP and transfer the message of ATP binding/hydrolysis to a second biochemical activity elsewhere in the protein (reviewed in Ref. 28). Motifs consisting of two Cys residues separated by two residues (CXXC) are common in proteins that bind [Fe-S] clusters.
The noncomplementing variant proteins examined herein were defined by three distinct classes. As seen previously, wild-type ApbC complemented an apbC null mutant, hydrolyzed ATP, bound [Fe-S] cluster, and rapidly transferred the cluster to apo-Leu-1; each class of variant ApbC proteins was defective in one or more of these four areas. The classification of these proteins is summarized in Table 6.
TABLE 6.
Noncomplementing ApbC variants can be divided into three classes
Growth results are as shown in Table 2.
The first class of ApbC proteins consisting of ApbCK116A and ApbCK121A hydrolyzed ATP with wild-type kinetics and were capable of binding an [Fe-S] cluster. However, cluster transfer from ApbC to Leu-1 was compromised in these variants.
Both reconstituted ApbCK116A and reconstituted ApbCK121A are dimers (data not shown) and display some degree of negative kinetic cooperativity (Table 5). One interpretation of these data suggest that variant proteins can still communicate ATP binding to adjacent monomers, but may be lacking the ability to communicate ATP binding and/or hydrolysis with the second function of the protein, which is necessary for in vivo function.
Other members of this ATPase superfamily of proteins that have mutations directed to the Walker motif have also been shown to hydrolyze ATP (general (28, 53), Lys-116 (54–57), Ser-122 (58), and Lys-121 (59–61)). In some proteins within this family such as NifH, MinD, ArsA, UvrB, and the F1-ATPase, making directed amino acid substitutions in the Walker A motif resulted in the absence of a signal between the ATPase domain and elsewhere within the protein (24, 25, 53, 59–63). In these cases this signal is necessary for the second function of the protein such as electron transfer or protein interaction. These parallels suggest the exciting possibility that further analysis of these ApbC variants will provide clues as to the signals causing cluster transfer from this protein in vivo.
Variants of two other bacterial proteins have been shown to be defective in [Fe-S] cluster transfer. The variants IscUD39A and Nfu-1D39A were shown to be compromised in [Fe-S] cluster transfer to apo-aconitase and apo-dinitrogen reductase, respectively (64, 65). These protein variants have been useful in dissecting the mechanism of [Fe-S] cluster biosynthesis and transfer (36, 66–69). There is no evidence to suggest that either of these proteins bind or hydrolyze ATP.
The second class of variant ApbC proteins consisting of ApbCS122A and ApbCS182A had no detectable ATPase activity, but could bind and rapidly transfer [Fe-S] cluster to Leu-1. The Ser-182 residue is conserved in all known ApbC homologs, and the Ser-122 residue resides in the Walker A motif. One explanation for the behavior of these variants is that the ATPase activity is required for loading the cluster in vivo, and thus, the in vitro system of [Fe-S] cluster reconstitution bypassed the need for this activity.
Interestingly, the ApbCS182A variant was able to complement under growth conditions in which the demand for [Fe-S] cluster-containing proteins was low (thiamine biosynthesis), but not when the demand for [Fe-S] proteins was high (tricarballylate utilization). It is plausible that ApbCS182A has a low rate of ATP hydrolysis that was out of the sensitivity range of the coupled assay, yet sufficient for the low demand of thiamine synthesis. Alternatively, the protein is not capable of hydrolyzing ATP, but the ApbCS182A variant is locked into a conformation that allows for a single turnover, which may fulfill the ApbC requirement in the low flux thiamine requirement condition.
The third class of variant ApbC proteins, consisting of ApbCC283A and ApbCC286A, could hydrolyze ATP and transfer an [Fe-S] cluster that was reconstituted on the proteins in vitro. Our hypothesis is that ApbC binds an [Fe-S] cluster that bridges the dimer interface ligated by four Cys residues, two cysteine ligands from each ApbC monomer. Therefore, we anticipated that these cysteines would be required for in vivo ApbC function, as they were. Similarly, studies by others have shown that the Cys residues that are proposed [Fe-S] cluster ligands in cluster scaffolding proteins, the proposed intermediate [Fe-S] cluster carrier NfuA, and eukaryotic ApbC homologs are essential for in vivo function (11, 31, 68, 70–73). Therefore, it was surprising that these ApbC variant proteins could bind an [Fe-S] cluster when artificially reconstituted in vitro. To the best of our knowledge there is no report of whether similar variants of the other proteins can bind an [Fe-S] cluster in vitro.
Previous work by Golbeck and co-workers (74) showed that externally supplied organic molecules such as 2-mercaptoethanol could act as an artificial ligand to a [4Fe-4S] cluster. Similar bio-inorganic studies have examined the ability of a 16-amino acid ferredoxin maquette peptide containing four Cys residues to bind a [4Fe-4S] cluster. The authors found that when one or more of these Cys residues were changed to Ala, the ferredoxin maquettes could still bind an [4Fe-4S] cluster, although the yield of [4Fe-4S] cluster binding was lower (75, 76). DTT was present in all in vitro ApbC reconstitution assays, making it feasible that the thiolate moieties of DTT were providing ligands for an [Fe-S] cluster that was bound by ApbC variants within an advantageous proteinaceous cavity. In this case, [Fe-S] cluster binding in the Cys → Ala variant ApbC proteins would be an artifact of the in vitro reconstitution.
A strength of this study lies in the combination of in vivo and in vitro approaches. This approach has allowed us to dissect the biochemical properties of ApbC within the context of biological activity. The resulting data showed that ApbC has two biochemical activities, and both activities are required for function in vivo. Identification of protein variants with no defect in the biochemical assays, but nonfunctional in vivo, indicates that some aspect of the in vitro assays is not physiologically relevant. We suggest that the chemical reconstitution of a cluster on ApbC does not reflect the process that occurs to generate holo-ApbC in vivo. The results described here with variant proteins provide a framework for continuing experiments intended to determine the function of ApbC in the network of [Fe-S] cluster metabolism.
Acknowledgments
We acknowledge W. W. Cleland (University of Wisconsin-Madison) for insightful discussions about enzyme kinetics, and Elizabeth Skovran for construction of some of the ApbC variants used in this study.
This work was supported, in whole or in part, by National Institutes of Health Grant GM47296 (to D. M. D.) and a National Institutes of Health Ruth Kirschstein postdoctoral training fellowship (to J. M. B.). This work was also supported by a 21st Century Scientist Scholars Award from the J. M. McDonnell Foundation (to D. M. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviation used is: DTT, dithiothreitol.
References
- 1.Beinert, H. (2000) J. Biol. Inorg. Chem. 5 2–15 [DOI] [PubMed] [Google Scholar]
- 2.Frazzon, J., and Dean, D. R. (2003) Curr. Opin. Chem. Biol. 7 166–173 [DOI] [PubMed] [Google Scholar]
- 3.Zheng, L., Cash, V. L., Flint, D. H., and Dean, D. R. (1998) J. Biol. Chem. 273 13264–13272 [DOI] [PubMed] [Google Scholar]
- 4.Takahashi, Y., and Tokumoto, U. (2002) J. Biol. Chem. 277 28380–28393 [DOI] [PubMed] [Google Scholar]
- 5.Layer, G., Ollagnier-de Choudens, S., Sanakis, Y., and Fontecave, M. (2006) J. Biol. Chem. 281 16256–16263 [DOI] [PubMed] [Google Scholar]
- 6.Vivas, E., Skovran, E., and Downs, D. M. (2006) J. Bacteriol. 188 1175–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bou-Abdallah, F., Adinolfi, S., Pastore, A., Laue, T. M., and Dennis Chasteen, N. (2004) J. Mol. Biol. 341 605–615 [DOI] [PubMed] [Google Scholar]
- 8.Cui, Q., Thorgersen, M. P., Westler, W. M., Markley, J. L., and Downs, D. M. (2006) Proteins Struct. Funct. Bioinformat. 62 578–586 [DOI] [PubMed] [Google Scholar]
- 9.Gralnick, J. A., and Downs, D. M. (2003) J. Biol. Chem. 278 20708–20715 [DOI] [PubMed] [Google Scholar]
- 10.Loiseau, L., Gerez, C., Bekker, M., Ollagnier-de Choudens, S., Py, B., Sanakis, Y., Teixeira de Mattos, J., Fontecave, M., and Barras, F. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 13626–13631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bandyopadhyay, S., Naik, S. G., O'Carroll, I. P., Huynh, B. H., Dean, D. R., Johnson, M. K., and Dos Santos, P. C. (2008) J. Biol. Chem. 283 14092–14099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angelini, S., Gerez, C., Ollagnier-de Choudens, S., Sanakis, Y., Fontecave, M., Barras, F., and Py, B. (2008) J. Biol. Chem. 283 14084–14091 [DOI] [PubMed] [Google Scholar]
- 13.Jin, Z., Heinnickel, M., Krebs, C., Shen, G., Golbeck, J. H., and Bryant, D. A. (2008) J. Biol. Chem. 283 28426–28435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Justino, M. C., Almeida, C. C., Goncalves, V. L., Teixeira, M., and Saraiva, L. M. (2006) FEMS Microbiol. Lett. 257 278–284 [DOI] [PubMed] [Google Scholar]
- 15.Justino, M. C., Almeida, C. C., Teixeira, M., and Saraiva, L. M. (2007) J. Biol. Chem. 282 10352–10359 [DOI] [PubMed] [Google Scholar]
- 16.Loiseau, L., Ollagnier-de Choudens, S., Lascoux, D., Forest, E., Fontecave, M., and Barras, F. (2005) J. Biol. Chem. 280 26760–26769 [DOI] [PubMed] [Google Scholar]
- 17.Skovran, E., and Downs, D. M. (2003) J. Bacteriol. 185 98–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petersen, L., and Downs, D. M. (1996) J. Bacteriol. 178 5676–5682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koo, M. S., Lee, J. H., Rah, S. Y., Yeo, W. S., Lee, J. W., Lee, K. L., Koh, Y. S., Kang, S. O., and Roe, J. H. (2003) EMBO J. 22 2614–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beck, B. J., Connolly, L. E., De Las Penas, A., and Downs, D. M. (1997) J. Bacteriol. 179 6504–6508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck, B. J., and Downs, D. M. (1998) J. Bacteriol. 180 885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gralnick, J., Webb, E., Beck, B., and Downs, D. (2000) J. Bacteriol. 182 5180–5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skovran, E., Lauhon, C. T., and Downs, D. M. (2004) J. Bacteriol. 186 7626–7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Igarashi, R. Y., and Seefeldt, L. C. (2003) Crit. Rev. Biochem. Mol. Biol. 38 351–384 [DOI] [PubMed] [Google Scholar]
- 25.Lutkenhaus, J., and Sundaramoorthy, M. (2003) Mol. Microbiol. 48 295–303 [DOI] [PubMed] [Google Scholar]
- 26.Leonard, T. A., Butler, P. J., and Lowe, J. (2005) EMBO J. 24 270–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedman, S. A., and Austin, S. J. (1988) Plasmid 19 103–112 [DOI] [PubMed] [Google Scholar]
- 28.Koonin, E. V. (1993) J. Mol. Biol. 229 1165–1174 [DOI] [PubMed] [Google Scholar]
- 29.Lezhneva, L., Amann, K., and Meurer, J. (2004) Plant J. 37 174–185 [DOI] [PubMed] [Google Scholar]
- 30.Stockel, J., and Oelmuller, R. (2004) J. Biol. Chem. 279 10243–10251 [DOI] [PubMed] [Google Scholar]
- 31.Roy, A., Solodovnikova, N., Nicholson, T., Antholine, W., and Walden, W. E. (2003) EMBO J. 22 4826–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hausmann, A., Aguilar Netz, D. J., Balk, J., Pierik, A. J., Muhlenhoff, U., and Lill, R. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 3266–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bych, K., Kerscher, S., Netz, D. J., Pierik, A. J., Zwicker, K., Huynen, M. A., Lill, R., Brandt, U., and Balk, J. (2008) EMBO J. 27 1736–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyd, J. M., Pierik, A. J., Netz, D. J., Lill, R., and Downs, D. M. (2008) Biochemistry 47 8195–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyd, J. M., Lewis, J. A., Escalante-Semerena, J. C., and Downs, D. M. (2008) J. Bacteriol. 190 4596–4602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raulfs, E. C., O'Carroll, I. P., Dos Santos, P. C., Unciuleac, M. C., and Dean, D. R. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 8591–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis, J. A., Horswill, A. R., Schwem, B. E., and Escalante-Semerena, J. C. (2004) J. Bacteriol. 186 1629–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gross, S. R., Burns, R. O., and Umbarger, H. E. (1963) Biochemistry 2 1046–1052 [DOI] [PubMed] [Google Scholar]
- 39.Balk, J., Pierik, A. J., Netz, D. J., Muhlenhoff, U., and Lill, R. (2004) EMBO J. 23 2105–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan, K. M., Delfert, D., and Junger, K. D. (1986) Anal. Biochem. 157 375–380 [DOI] [PubMed] [Google Scholar]
- 41.Kreuzer, K. N., and Jongeneel, C. V. (1983) Methods Enzymol. 100 144–160 [DOI] [PubMed] [Google Scholar]
- 42.Bates, P. A., Kelley, L. A., MacCallum, R. M., and Sternberg, M. J. (2001) Proteins 5 S39–S46 [DOI] [PubMed] [Google Scholar]
- 43.Bates, P. A., and Sternberg, M. J. (1999) Proteins 3 S47–S54 [Google Scholar]
- 44.Contreras-Moreira, B., and Bates, P. A. (2002) Bioinformatics (Oxf.) 18 1141–1142 [DOI] [PubMed] [Google Scholar]
- 45.Holmgren, A. (1985) Annu. Rev. Biochem. 54 237–271 [DOI] [PubMed] [Google Scholar]
- 46.Enos-Berlage, J. L., and Downs, D. M. (1999) J. Bacteriol. 181 841–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leonardi, R., Fairhurst, S. A., Kriek, M., Lowe, D. J., and Roach, P. L. (2003) FEBS Lett. 539 95–99 [DOI] [PubMed] [Google Scholar]
- 48.Martinez-Gomez, N. C., Robers, M., and Downs, D. M. (2004) J. Biol. Chem. 279 40505–40510 [DOI] [PubMed] [Google Scholar]
- 49.Lewis, J. A., and Escalante-Semerena, J. C. (2006) J. Bacteriol. 188 5479–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lewis, J. A., and Escalante-Semerena, J. C. (2007) Biochemistry 46 9107–9115 [DOI] [PubMed] [Google Scholar]
- 51.Netz, D. J., Pierik, A. J., Stumpfig, M., Muhlenhoff, U., and Lill, R. (2007) Nat. Chem. Biol. 3 278–286 [DOI] [PubMed] [Google Scholar]
- 52.Segel, I. H. (1975) Enzyme Kinetics, John Wiley & Sons, Inc., New York
- 53.Karkaria, C. E., Chen, C. M., and Rosen, B. P. (1990) J. Biol. Chem. 265 7832–7836 [PubMed] [Google Scholar]
- 54.Seefeldt, L. C., Morgan, T. V., Dean, D. R., and Mortenson, L. E. (1992) J. Biol. Chem. 267 6680–6688 [PubMed] [Google Scholar]
- 55.Tian, G. C., Yan, H. G., Jiang, R. T., Kishi, F., Nakazawa, A., and Tsai, M. D. (1990) Biochemistry 29 4296–4304 [DOI] [PubMed] [Google Scholar]
- 56.Reinstein, J., Schlichting, I., and Wittinghofer, A. (1990) Biochemistry 29 7451–7459 [DOI] [PubMed] [Google Scholar]
- 57.Sigal, I. S., Gibbs, J. B., D'Alonzo, J. S., Temeles, G. L., Wolanski, B. S., Socher, S. H., and Scolnick, E. M. (1986) Proc. Natl. Acad. Sci. U. S. A. 83 952–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seefeldt, L. C., and Mortenson, L. E. (1993) Protein Sci. 2 93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou, H., Schulze, R., Cox, S., Saez, C., Hu, Z., and Lutkenhaus, J. (2005) J. Bacteriol. 187 629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seeley, T. W., and Grossman, L. (1989) Proc. Natl. Acad. Sci. U. S. A. 86 6577–6581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parsonage, D., Al-Shawi, M. K., and Senior, A. E. (1988) J. Biol. Chem. 263 4740–4744 [PubMed] [Google Scholar]
- 62.Ryle, M. J., and Seefeldt, L. C. (1996) Biochemistry 35 4766–4775 [DOI] [PubMed] [Google Scholar]
- 63.Ryle, M. J., Lanzilotta, W. N., Mortenson, L. E., Watt, G. D., and Seefeldt, L. C. (1995) J. Biol. Chem. 270 13112–13117 [DOI] [PubMed] [Google Scholar]
- 64.Agar, J. N., Krebs, C., Frazzon, J., Huynh, B. H., Dean, D. R., and Johnson, M. K. (2000) Biochemistry 39 7856–7862 [DOI] [PubMed] [Google Scholar]
- 65.Smith, A. D., Jameson, G. N., Dos Santos, P. C., Agar, J. N., Naik, S., Krebs, C., Frazzon, J., Dean, D. R., Huynh, B. H., and Johnson, M. K. (2005) Biochemistry 44 12955–12969 [DOI] [PubMed] [Google Scholar]
- 66.Shimomura, Y., Wada, K., Fukuyama, K., and Takahashi, Y. (2008) J. Mol. Biol. 383 133–143 [DOI] [PubMed] [Google Scholar]
- 67.Shimomura, Y., Kamikubo, H., Nishi, Y., Masako, T., Kataoka, M., Kobayashi, Y., Fukuyama, K., and Takahashi, Y. (2007) J. Biochem. (Tokyo) 142 577–586 [DOI] [PubMed] [Google Scholar]
- 68.Johnson, D. C., Unciuleac, M. C., and Dean, D. R. (2006) J. Bacteriol. 188 7551–7561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Foster, M., Mansy, S., Hwang, J., Penner-Hahn, J., Surerus, K. K., and Cowan, J. A. (2000) J. Am. Chem. Soc. 122 6805–6806 [Google Scholar]
- 70.Vitale, G., Fabre, E., and Hurt, E. C. (1996) Gene (Amst.) 178 97–106 [DOI] [PubMed] [Google Scholar]
- 71.Dos Santos, P. C., Smith, A. D., Frazzon, J., Cash, V. L., Johnson, M. K., and Dean, D. R. (2004) J. Biol. Chem. 279 19705–19711 [DOI] [PubMed] [Google Scholar]
- 72.Kaut, A., Lange, H., Diekert, K., Kispal, G., and Lill, R. (2000) J. Biol. Chem. 275 15955–15961 [DOI] [PubMed] [Google Scholar]
- 73.Lu, J., Yang, J., Tan, G., and Ding, H. (2008) Biochem. J. 409 535–543 [DOI] [PubMed] [Google Scholar]
- 74.Antonkine, M. L., Maes, E. M., Czernuszewicz, R. S., Breitenstein, C., Bill, E., Falzone, C. J., Balasubramanian, R., Lubner, C., Bryant, D. A., and Golbeck, J. H. (2007) Biochim. Biophys. Acta 1767 712–724 [DOI] [PubMed] [Google Scholar]
- 75.Gibney, B. R., Mulholland, S. E., Rabanal, F., and Dutton, P. L. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 15041–15046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mulholland, S. E., Gibney, B. R., Rabanal, F., and Dutton, P. L. (1998) J. Am. Chem. Soc. 120 10296–10302 [Google Scholar]
- 77.Gralnick, J., and Downs, D. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 8030–8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Way, J. C., Davis, M. A., Morisato, D., Roberts, D. E., and Kleckner, N. (1984) Gene (Amst.) 32 369–379 [DOI] [PubMed] [Google Scholar]
- 79.Neidhardt, F. C., Ingraham, J. L., and Schaechter, M. (1990) Physiology of the Bacterial Cell, pp. 198–199, Sinauer Associates, Inc., Sunderland, MA
- 80.Martinez-Gomez, N. C., and Downs, D. M. (2008) ThiC is an [Fe-S] Cluster protein that requires AdoMet to generate the 4-amino-5-hydroxymethyl-2-methylpyrimidine moiety in thiamin synthesis. Biochemistry 47 9054–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]