

Abstract
Cytochrome c, a mitochondrial electron transfer protein containing a hexacoordinated heme, is involved in other physiologically relevant events, such as the triggering of apoptosis, and the activation of a peroxidatic activity. The latter occurs secondary to interactions with cardiolipin and/or post-translational modifications, including tyrosine nitration by peroxynitrite and other nitric oxide-derived oxidants. The gain of peroxidatic activity in nitrated cytochrome c has been related to a heme site transition in the physiological pH region, which normally occurs at alkaline pH in the native protein. Herein, we report a spectroscopic characterization of two nitrated variants of horse heart cytochrome c by using optical spectroscopy studies and NMR. Highly pure nitrated cytochrome c species modified at solvent-exposed Tyr-74 or Tyr-97 were generated after treatment with a flux of peroxynitrite, separated, purified by preparative high pressure liquid chromatography, and characterized by mass spectrometry-based peptide mapping. It is shown that nitration of Tyr-74 elicits an early alkaline transition with a pKa = 7.2, resulting in the displacement of the sixth and axial iron ligand Met-80 and replacement by a weaker Lys ligand to yield an alternative low spin conformation. Based on the study of site-specific Tyr to Phe mutants in the four conserved Tyr residues, we also show that this transition is not due to deprotonation of nitro-Tyr-74, but instead we propose a destabilizing steric effect of the nitro group in the mobile Ω-loop of cytochrome c, which is transmitted to the iron center via the nearby Tyr-67. The key role of Tyr-67 in promoting the transition through interactions with Met-80 was further substantiated in the Y67F mutant. These results therefore provide new insights into how a remote post-translational modification in cytochrome c such as tyrosine nitration triggers profound structural changes in the heme ligation and microenvironment and impacts in protein function.
Cytochrome c is a small globular heme protein present at >1 mm concentration in the intermembrane space of mitochondria, where it participates as a redox partner in the mitochondrial electron transport chain (1–4). In addition, when released to the cytosol it is involved in the apoptotic pathway (5, 6). The iron ion at the heme moiety is hexacoordinated with His-18 and Met-80 as fifth and sixth heme ligands, respectively (7). Cytochrome c can switch between the ferric and ferrous redox forms, with a characteristically high Eo of +260 mV (8, 9). Spectroscopic studies have indicated that cytochrome c3+ (ferricytochrome c) can exist in at least five distinct conformations in the pH range of 1 to 12, resulting from changes in heme axial coordination and/or in protein folding (10–12). Conformational state III is the dominant species at near neutral and physiological pH, and its structure has been studied extensively at near atomic resolution (13–17). The transformation of the state III species (referred to the “neutral” species from now on) into state IV species (the “alkaline” species) upon pH increase is called the alkaline transition (which occurs with a pKa of ∼9.3) (13, 18–24). This conformational change is present in all known isoforms of type c cytochrome and implies the displacement of Met-80 from the heme and its substitution by another ligand, Lys-72 or Lys-79 (13, 18, 24, 25), keeping the iron in a low spin state.
Although initially this was not appreciated as a phenomenon of biological relevance, recent work has established that a transition from state III to alternative states can be triggered on a pH-independent fashion under biologically relevant conditions and plays a role in mitochondrial physiology and apoptotic signaling (26). In this regard, cytochrome c association to a pool of the mitochondrial phospholipid cardiolipin (27–29) and posttranslational modifications such as methionine oxidation and tyrosine nitration (30, 31), promotes the transition of cytochrome c to “alternative low spin conformations” (32), which in some cases correspond to state IV.
Indeed, when the neutral species converts into the alkaline form, a band in the visible spectrum centered at 695 nm, indicative of the iron-sulfur bond established between the heme iron and Met-80, weakens in intensity and is eventually lost (10); data obtained by magnetic circular dichroism and low temperature EPR have clearly shown that the heme is kept as low spin (27, 32). As an alternative to these spectroscopic methods, changes in heme electronic structure in cytochrome c3+ can be monitored by looking at the paramagnetically shifted resonances in 1H NMR spectra (33–35), as both species are low spin and give sharp, sensitive signals, with different chemical shifts under a slow exchange regime (13, 19). NMR not only allows the characterization of the different conformations but also assists in the determination of the pKa for the observed transition, which can be followed at the residue level (19, 35). Moreover, cytochrome c3+-cardiolipin interactions have been evaluated preliminarily by 1H-NMR (36).
Among the chemical modifications that seem to alter the conformational state of cytochrome c3+, and in particular the heme microenvironment, is the nitration of tyrosine residues. Indeed, mammalian cytochromes c contain four highly conserved tyrosines, two that are solvent-exposed (Tyr-74 and Tyr-97) and two buried internally (Tyr-67 and Tyr-48). Although the functional impact of the nitration of cytochrome c by tetranitromethane was studied by seminal early work (37–39), a renewed interest appeared recently (40, 41), because tyrosine nitration has been established as a post-translational modification occurring in vivo due to the reactions of nitric oxide (•NO)-derived oxidants (31) and that can influence protein structure and function (for a recent review see) (42). In this regard, we should note that addition of a nitro group (–NO2) to tyrosine residues reduces the pKa of the phenolic –OH group by 2 or 3 units and adds a bulky moiety of 45 Da. Nitrated cytochrome c has been detected in cellular and animal models of disease associated to nitroxidative stress (43–45).
We have established that cytochrome c tyrosine nitration can occur
via a variety of biologically relevant reaction mechanisms that include
peroxynitrite
(ONOO–/ONOOH)6
(40,
41), nitrite
(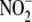 ) plus hydrogen peroxide
(H2O2)
(46), and • NO plus
H2O2
(47)-dependent reactions (for
a review see Ref. 48). Peptide
mapping studies have revealed that during peroxynitrite-mediated cytochrome
c nitration, the first nitrated tyrosine residue is either Tyr-74 or
Tyr-97, consistent with the fact that peroxynitrite-derived radicals (either
via homolytic cleavage to yield hydroxyl (•OH) and nitrogen
dioxide (•NO2) radicals or secondary to the reaction with
carbon dioxide, which yields carbonate radicals
(
) plus hydrogen peroxide
(H2O2)
(46), and • NO plus
H2O2
(47)-dependent reactions (for
a review see Ref. 48). Peptide
mapping studies have revealed that during peroxynitrite-mediated cytochrome
c nitration, the first nitrated tyrosine residue is either Tyr-74 or
Tyr-97, consistent with the fact that peroxynitrite-derived radicals (either
via homolytic cleavage to yield hydroxyl (•OH) and nitrogen
dioxide (•NO2) radicals or secondary to the reaction with
carbon dioxide, which yields carbonate radicals
(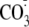 )
and •NO2), will preferentially react with solvent-exposed
tyrosine residues (40). Other
biochemical pathways of cytochrome c nitration have been reported
(46,
47), which may also involve
the nitration of Tyr-67 (48).
Nonetheless, we have observed that the nitro-Tyr-97 and nitro-Tyr-74
cytochrome c species undergo important functional changes that
include the induction of a peroxidatic activity (i.e. reduction of a
peroxide such as H2O2 with the concomitant oxidation of
a second substrate via a heme-dependent catalytic cycle)
(40,
41), the loss of electron
transport capabilities (40,
41), and the inability to
induce apoptosome activation
(48).
)
and •NO2), will preferentially react with solvent-exposed
tyrosine residues (40). Other
biochemical pathways of cytochrome c nitration have been reported
(46,
47), which may also involve
the nitration of Tyr-67 (48).
Nonetheless, we have observed that the nitro-Tyr-97 and nitro-Tyr-74
cytochrome c species undergo important functional changes that
include the induction of a peroxidatic activity (i.e. reduction of a
peroxide such as H2O2 with the concomitant oxidation of
a second substrate via a heme-dependent catalytic cycle)
(40,
41), the loss of electron
transport capabilities (40,
41), and the inability to
induce apoptosome activation
(48).
Although the functional effect, from none to either gain or loss of function (31, 42), of tyrosine nitration on a variety of proteins has been under active investigation over the last decade, structural studies are almost lacking. In particular, one crucial unanswered issue is how the site-specific nitration of one or more tyrosine residues in cytochrome c is able to decrease the pKa of the alkaline transition toward the neutral region and result in a conformational switch to render peroxidatic activity; the latter effect has been related to cardiolipin oxidation (29, 40, 56) and initiation of mitochondrial apoptotic signaling (26).
In this study, we have explored the effect of nitration at the solvent-exposed tyrosine residues (Tyr-97 and Tyr-74) of horse heart cytochrome c on the pKa of its alkaline transition and the changes in heme ligation and protein conformation by using a combination of NMR and optical spectroscopy studies. This work allowed us to provide a structural rationale for the remote effect of Tyr-74 nitration on the alkaline transition, which results in the gain of peroxidatic activity upon nitric oxide-derived oxidants reactions. Moreover, we propose a mechanism by which tyrosine nitration is transmitted conformationally within the protein framework. These data provide novel insights into the structural changes triggered by tyrosine nitration on cytochrome c and highlight the utility of NMR of paramagnetic systems for assessing the effects of post-translational modifications and interactions with protein and phospholipids ligands on the heme ligation and conformational states of cytochrome c3+.
EXPERIMENTAL PROCEDURES
Chemicals—Horse heart cytochrome c (C-7752) was obtained from Sigma. This cytochrome c preparation was selected because it is purified without the use of trichloroacetic acid, which otherwise leads to substantial amounts of deamidated and polymeric forms (57). The purity of the cytochrome c preparation used herein was >95% as determined by chromatographic, spectroscopic, electrophoretic, and mass spectrometric criteria. Sequencing grade modified trypsin was obtained from Promega. Hydrogen peroxide was obtained from Baker. D2O (99.8% 2H purity) was purchased from Sigma. All other reagents were analytical grade. HPLC7 solvents were of the highest available quality. Peroxynitrite, which was synthesized, quantitated, and handled as described previously (58), contained less than 20% residual nitrite.
Nitration and Purification of Modified Cytochrome c Species—A large scale nitration and purification procedure was adapted from previously described methods (40, 48) to obtain the amounts of mononitrated cytochrome c species required for the optical and NMR spectroscopy studies performed herein. Peroxynitrite was added as a continuous flow of 0.134 mm · min–1 for 30 min to cytochrome c3+ (200 μm) by using a motor-driven multi-syringe system (Sage Instruments, Boston), making three vials of 0.5 ml each time, under vigorous shaking condition in 200 mm potassium phosphate with 25 mm NaHCO3 and 100 μm diethylenetriaminepentaacetic acid at pH 7.0 and 25 °C. The final pH was always controlled and kept below 7.4. After reaction, salts were removed by a chromatography step in a HiTrap desalting column (Amersham Biosciences), and the mixture of nitrated cytochrome c forms was passed through a cation exchange sulfopropyl-TSK preparative column (21.5 mm (inner diameter) × 15.0 cm (length); Tosoh Biosep) in order to purify the mononitrated species. The flow rate was 3 ml · min–1. The column was equilibrated with 10 mm ammonium acetate buffer (pH 9.0) for 20 min, and then peaks were eluted using a linear gradient of 10–400 mm ammonium acetate (pH 9.0) for 20 to 80 min and kept up to 90 min at 400 mm ammonium acetate. Peaks 2 and 3 from the elution (Fig. 1A), containing nitrated cytochrome c (note that peak 1 corresponds to unmodified cytochrome c3+) were repurified in the same column (Fig. 1B) using the same solvent gradient, to avoid any cross-contamination. The high purity of different modified cytochrome c species was confirmed by mass spectrometry analysis of the whole protein and monitoring of the nitration of the tyrosine-containing peptides as described previously (40, 48). Mass spectrometry analysis was performed with a matrix-assisted laser desorption ionization time-of-flight mass spectrometer (model 4800 MALDI TOF/TOF Analyzer, Applied Biosystems, Framingham, MA) on the Analytical Biochemistry and Proteomics Core Facility of the Institut Pasteur de Montevideo. Tryptic digestion of HPLC peak 2 contained only the nitrated peptide 89TEREDLIAY(NO2)LK99 (theoretical m/z = 1395.72; detected m/z = 1395.69), corresponding to the nitration of Tyr-97. Similarly, the digestion of peak 3 only contained the nitrated peptide 74Y(NO2)IPGTK79 (theoretical m/z = 723.37; detected m/z = 723.30), corresponding to the nitration of Tyr-74 (40, 48).
FIGURE 1.

Purification of mononitrated cytochrome c species. Cytochrome c was nitrated by a flux of peroxynitrite as described under “Experimental Procedures.” A, the reaction mixture (60 mg of protein in 3 ml of 10 mm ammonium acetate) was separated by a preparative cation-exchange HPLC column, and three main peaks were found corresponding to unmodified (peak 1), nitro-Tyr-97 (peak 2)-, and nitro-Tyr-74 (peak 3)-cytochrome c, as confirmed by MALDI-TOF and peptide mapping (40). B, peaks 2 and 3 were repurified with the same HPLC column (2–5 mg of protein loaded), obtaining fractions of >99% purity, which were used for all subsequent NMR and optical spectroscopy studies.
Site-directed Mutagenesis, Expression, and Purification of Recombinant Cytochrome c Mutants—The plasmid pJRhrsN2 (kindly provided by Dr. Jon Rumbley, Chemistry Department, University of Minnesota, Duluth) was employed to produce wild type recombinant cytochrome c and the Y97F, Y74F, Y67F, and Y48F mutants. Plasmid pJRhrsN2 codes for horse heart cytochrome c, as described (59), carrying the two substitutions H26N and H33N. The four single Y to F mutants were obtained using the QuikChange II site-directed mutagenesis kit (Stratagene). The resultant pJRhrsN2-derived plasmids were transformed into Escherichia coli strain BL21 Star (DE3) (Invitrogen) for protein expression.
Recombinant proteins were expressed and purified according to Rumbley et al. (59) with minor modifications. Twenty-five ml of starter culture grown in LB medium and 100 μg/ml ampicillin was inoculated in 1 liter of Terrific Broth with the antibiotic. The culture was grown with vigorous shaking until A550 nm = 0.8. Expression of cytochrome c Y to F mutants was induced with 0.8 mm isopropyl 1-thio-β-d-galactopyranoside for 50 h at 303 K. The protein was purified using a CM-Sepharose fast-flow column (Amersham Biosciences). Fractions with a 410/280 nm absorbance ratio > 4.0 were considered pure, as supported by SDS-PAGE analysis.
1H NMR Spectroscopy—Lyophilized samples were dissolved in 100 mm phosphate buffer prepared in D2O at pH7. NMR spectra were acquired with a Bruker Avance II spectrometer operating at 600.13 MHz (1H frequency) with presaturation of the water signal during the relaxation delay. A spectral window of 70 ppm was used, and the total recycle time was 300 ms.
For pH titration studies, the pH was adjusted close to a desired value by the addition of 0.1–1 m NaOH or 0.1–1 m HCl dissolved in D2O, and a 1H spectrum was recorded at 298 and 318 K. After recording of each spectrum, the pH was measured again, and the mean value (before and after the experiment) was reported. The reported pH values were corrected for the deuterium isotope effect (60). The pKa values were determined by fitting the intensity of heme methyl 8, heme methyl 3, and Met-80 methyl signals of the neutral species as a function of pH to a Henderson-Hasselbalch equation.
Absorption and Circular Dichroism Spectroscopy Studies—Electronic absorption spectra were obtained in the 650–750 nm region to evaluate the band centered at 695 nm, indicative of the heme iron-Met-80 coordination (10), using a Shimadzu UV-2401 PC spectrophotometer in a 0.1-ml quartz cuvette with a 1-cm path. Because of the relatively weak absorption coefficient of this band (ε695 = 865 m–1 cm–1 for control cytochrome c3+ in the pH range of 4.5 to 7.0; this work, and see also Ref. 61), assays were performed with 0.5 mm cytochrome c3+ or nitrocytochrome c3+ in 50 mm phosphate buffer. The concentration of the different cytochrome c species was adjusted by quantitation of the Soret band absorption at 410 nm (ε410 = 106,000 m–1 cm–1), known to be nearly identical for control and nitrated cytochrome c3+ (41). For pH titration studies, the pH of the sample was adjusted to the reported values by the addition of 0.1–1 m NaOH or 0.1–1 m HCl, and spectra were collected at each pH point. Circular dichroism spectra were recorded using a Jasco J-810 Spectropolarimeter using thermostatted cells of 0.1-cm path length.
RESULTS
Preparation of Nitro-Tyr-97 and Nitro-Tyr-74 Cytochrome c Species—Horse heart cytochrome c contains 4 tyrosine residues. Tyr-67 and Tyr-48 are buried within the protein structure, whereas Tyr-97 and Tyr-74, instead, are solvent-accessible; hence both of them are preferentially nitrated by peroxynitrite-derived radicals (40, 48). Accordingly, in the preparative method for obtaining nitrated cytochrome c reported herein (see “Experimental Procedures”), Tyr-97 and Tyr-74 were the two major modified residues obtained when cytochrome c3+ was treated with a peroxynitrite flow during 30 min, as shown in Fig. 1A. Indeed, early reaction products, corresponding to peaks 2 and 3 in Fig. 1, were compatible with two mononitrated species, as evidenced by the 45-Da increase in molecular mass expected for the addition of a –NO2 group. Peptide mapping of tryptic fragments confirmed that peaks 2 and 3 corresponded to nitro-Tyr-97 and nitro-Tyr-74 cytochrome c, respectively, in agreement with previous data (40, 48). Repurification of the peaks through the same HPLC column (Fig. 1B) rendered pure forms of mononitrated cytochrome c (>99% purity) and with overall final yields at approximately 1 and 3% of initial cytochrome c for the nitro-Tyr-74 and nitro-Tyr-97 species, respectively. Longer incubation times (>30 min) of cytochrome c with the peroxynitrite flux induced the subsequent appearance of di- and trinitrated cytochrome c species eluting with earlier retention times (not shown).
Absorption and CD Spectroscopy of Nitrated Cytochrome c—The integrity of the sixth coordination position occupied by Met-80 in the native state of cytochrome c can be followed by inspecting the charge transfer band at 695 nm (62). The decrease or loss of the 695 nm band in mononitrated cytochrome c in either Tyr-97 or Tyr-74 at pH 7.4 suggested the possibility of an “early” alkaline transition (40) due to the decrease in pI (9.28 for native cytochrome c; (63)) determined by the addition of a –NO2 group to tyrosine, known to decrease the pKa of the phenolic –OH group from ∼10 to 7.5 (64). Herein, we performed a detailed spectrophotometric pH titration of native, nitro-Tyr-97, and nitro-Tyr-74 cytochrome c, which shows that this band is lost in all three cases, with pKa values of 9.4, 8.7, and 7.3, respectively (Fig. 2). The extinction coefficient values at 695 nm of the nitro-cytochrome c species were somewhat smaller than that of the control protein, even under acidic pH (Fig. 2, inset). These data indicate that nitration of Tyr-97 has a relatively mild effect in promoting the alkaline transition, whereas Tyr-74 nitration has a profound effect, lowering the pKa value by ∼2 pH units (Table 1).
FIGURE 2.

pH titrations of native and nitrated cytochrome c samples followed by electronic absorption at 695 nm. Electronic absorption spectra of 0.5 mm cytochrome c (▪), nitro-Tyr-97-cythocrome c (•), and nitro-Tyr-74-cytochrome c (▴). Spectra were recorded at 298 K in 50 mm phosphate buffer. Inset, absorption band centered at 695 nm for control and mononitrated cytochromes at pH 5.0. The data were fitted to the Henderson-Hasselbalch function to obtain the pKa values for each cytochrome c species. The correlation coefficient (r2) for the data was ∼0.99.
TABLE 1.
pKa values determined for the alkaline transition in control and nitro-Tyr-97- and nitro-Tyr-74-cytochrome c by NMR and optical spectroscopy
The data were fitted to the Henderson-Hasselbalch function to obtain the pKa values. NMR determinations were performed at 318 K and optical determinations at 298 K.
|
Sample
|
pKa values
|
|
|---|---|---|
| NMR | 695 nm band | |
| Control | 9.3 ± 0.1 | 9.4 ± 0.1 |
| Nitro-Tyr-97 | 8.6 ± 0.2 | 8.7 ± 0.2 |
| Nitro-Tyr-74 | 7.2 ± 0.1 | 7.3 ± 0.1 |
CD spectra were also recorded at different pH values in these samples. The native form of cytochrome c at pH 7 is characterized by a Cotton effect at 410 nm, corresponding to the maximum of the Soret band (Fig. 3A and Ref.65, 66). Instead, at pH 11, the spectrum displays a positive band centered at 405 nm. In the case of nitro-Tyr-74, this change is already noticeable at pH 9 (Fig. 3B), in agreement with the behavior of the 695 nm band in the absorption spectrum (32, 41).
FIGURE 3.

Circular dichroism spectra of cytochrome forms. CD spectra in the near UV and visible regions corresponding to 10 μm control (A), nitro-Tyr-74 (B), and Y67F (C) cytochrome c, recorded at pH 7 (solid line), pH 9 (dash-dot-dashed line), and pH 11 (dotted line).
1H NMR Spectroscopy of Nitrated Cytochrome c—1H NMR spectroscopy has been employed extensively to characterize the heme moiety and the axial iron ligands in a plethora of heme proteins, particularly cytochromes (33–35). We decided to follow the pH dependence of the nitrated cytochromes using this high resolution technique to unequivocally characterize the different species formed. The usual ligands to the cytochrome c heme iron in the neutral species are indicated in Fig. 4A. When the spectra were recorded under conditions tailored for the detection of fast relaxing nuclei (located close to the Fe(III) ion), several resonances were located outside the diamagnetic envelope (10–0 ppm). Fig. 4B shows the spectrum of native cytochrome c3+ at near neutral pH, where the resonances corresponding to two heme methyl groups and Met-80 are indicated. As already reported, spectra recorded at different pH values evidenced the transition from the native low spin form (III) at neutral pH to the alkaline low spin form (IV) (Fig. 4B) (13, 18, 19, 24). Several resonances change at high pH (such as those from methyl groups 3, 5, and 8) revealing an overall perturbation of the heme environment. Detachment of Met-80 from the heme iron was unequivocally evidenced by the loss of the intense resonance at –20 ppm, which corresponds to the ε-CH3 group of this residue. This titration allowed us for comparative purposes, to determine a reference pKa value of 9.3 at 318 K for the un-nitrated protein under our working conditions (Table 1). This value is in good agreement with those reported previously (13, 18–24), which vary between 8.9 and 9.5 pH units depending on the protein isoform, ionic strength, and temperature.
FIGURE 4.

pH titrations of native and nitrated cytochrome c samples followed by NMR spectroscopy. A, scheme showing the ligands of the heme iron atom. Groups that are referred in the text are labeled. Panel A was rendered with PyMol (DeLano Scientific, San Carlos, CA). B–D, pH titrations of native cytochrome c (B) and of cytochrome c nitrated at Tyr-97 (C) or Tyr-74 (D) monitored by NMR. Spectra were acquired at 318 K in 100 mm phosphate buffer prepared in 100% D2O. Signal labeling is indicated in B. Resonances corresponding to the alkaline species are labeled as 8-CH3′, 5-CH3′, and Lys′.
In Fig. 4, C and D, the pH titrations of nitro-Tyr-97 and nitro-Tyr-74 cytochrome c followed by 1H NMR spectroscopy are shown. In both cases, raising the pH resulted in spectral perturbations similar to those reported for the native protein: the heme methyl resonances are located at smaller chemical shift values (20–25 ppm), and (most important) the upfield signal corresponding to Met-80 was lost. This demonstrates that both mononitrated species experience a transition similar to the one found for the native cytochrome. However, the pKa values for the transition are perturbed in both cases, being shifted to lower pH values. Nitration at Tyr-97 lowers the pKa value from 9.3 to 8.6, whereas Tyr-74 nitration drastically lowers the pKa to 7.2 (Table 1).
A broad upfield resonance located at –9 ppm, which can be identified in the alkaline forms of both nitro-Tyr-97 and nitro-Tyr-74 cytochrome c, closely resembles a signal assigned to Lys-72 in the alkaline form of native cytochrome c (Fig. 4, B–D). This residue replaces Met-80 as the axial ligand, preserving the low spin character at the Fe(III) center. Finding this resonance in both of the nitrated forms further supports the similarity of the metal site structure in the alkaline form of the three samples.
Overall, NMR, absorption and CD spectroscopy studies revealed that nitration at both Tyr-97 and Tyr-74 shifts the alkaline transition to lower pH values, with the perturbation being substantially larger for Tyr-74. The NMR data demonstrate that Met-80 is detached from the heme iron and is replaced by a Lys residue, i.e. nitration does not alter the identity of the alkaline species but leads to an earlier transition. In the case of nitro-Tyr-74, this effect results in a significant population of the alternative low spin form at neutral pH.
Analysis of Tyr to Phe Mutants—Different mutations leading to Tyr to Phe substitution were obtained in order to analyze the perturbations in the same protein sites affected by nitration. Because of minor differences existing between the recombinant wild type protein and the commercial protein purified from horse heart (59), we determined by NMR the pKa value for the alkaline transition of the recombinant protein to be 9.1 ± 0.1 (similar to that of the commercial cytochrome, which is 9.3). The pKa values for the alkaline transition in the Y97F, Y74F, Y48F, and Y67F point mutants were determined by NMR titrations.
The NMR spectra of the Y97F, Y74F, and Y48F cytochrome c mutants recorded at different pH values (Fig. 5) showed that the resonances corresponding to the neutral and alkaline species are identical to those observed for the wild type protein, with relatively small changes in the pKa of this transition, which was lower in the three cases by 0.4–0.5 pH units (ca. 8.6–8.7). Optical studies of the 695 nm band further supported the hypothesis that the spectral properties are nearly identical to those of wild type cytochrome c at physiological pH (not shown).
FIGURE 5.

NMR studies of cytochrome c wild type and Tyr to Phe single mutants at different pH values. pH titration of recombinant wild type cytochrome c (A) and Y48F (B), Y74F (C), and Y97F (D) single mutants at 318 K. Spectra were acquired at 318 K in 100 mm phosphate buffer prepared in 100% D2O.
In the case of the Y67F mutant (Fig. 6A), the 1H NMR spectrum of the neutral form displays some perturbations compared with the spectrum of the wild type protein (Fig. 6B). This was not completely unexpected, based on the proximity of this residue to the heme site (Fig. 4A). The signal assignment was similar to that reported for the wild type protein as confirmed by COSY and NOESY experiments (not shown) (35). Regarding the alkaline transition, spectral changes were observed at higher pH values than in the native protein, resulting in a lower limit of 11.0 for the pKa (Fig. 6A). At alkaline pH, the intensity of the heme 8-CH3 and 3-CH3 groups decreased together with that of Met-80, again revealing a transition that implies detachment of this axial residue. The newly formed species displayed heme methyl signals around 20 ppm that resemble those of the alternative low spin form in the native protein. However, some additional resonances were also observed, revealing the presence of more species at high pH (see Figs. 5 and 6A).
FIGURE 6.

NMR studies on the Y67F cytochrome c mutant at different pH values. A, pH titration of the Y67F cytochrome c mutant. Spectra were acquired at 318 K in 100 mm phosphate buffer prepared in 100% D2O. B, NMR spectra of recombinant wild type cytochrome c at pD 7.1.
The CD spectra of Y67F cytochrome c recorded at pH 7 and 11 in the near UV and visible regions displayed a similar form but with quite different intensities. Because the NMR spectra recorded reveal a 60:40 ratio of the neutral and alkaline species at pH 11, the reduced negative elipticity at 410 nm compared with the spectrum at pH 7 may well correspond to the contribution of a positive Soret band, as observed for the pure high pH form in the native protein (Fig. 3C). The effect of the Y67F replacement is in agreement with that reported for a similar mutation in rat cytochrome c, which was evaluated following the 695 nm band in the absorption spectrum (62).
DISCUSSION
The alkaline transition of cytochrome c, as well as the influence of different chemical or physicochemical perturbations on it, has been studied extensively (13, 18, 20, 23, 24). It has been shown that the alkaline form of cytochrome c is stabilized by chemical modifications of amino acids, cleavage of certain parts of the polypeptide chain, perturbation of interactions inside the heme crevice, interactions with anionic phospholipids, increased temperature, and the presence of denaturants, resulting in a lower pKa (29, 41, 67–72). Such conditions are associated with local or global destabilization of the protein fold induced by the deprotonation of one or more residues. The solution structure of the alkaline form of the K79A variant of yeast cytochrome c revealed that most protein regions are well defined in the structure and are similar to the same regions in the native form, except for the segment encompassing residues Asn-70 to Ile-85, known as the Ω-loop, which becomes “floppy” in the alkaline form (13, 73).
Here, we have shown that nitration of Tyr-74 significantly perturbs the pKa of the alkaline transition down to the near neutral pH region (Table 1). The high pH form in this variant preserves the spectral features reported for the native protein at pH 11, such as the detachment of Met-80 from the heme iron and ligation of a Lys residue. In contrast, nitration of cytochrome c at Tyr-97 shows a slightly lower pKa compared with the native protein (i.e. by 0.5 pH units). Both Tyr-97 and Tyr-74 side chains are solvent-accessible, in line with the high degree of peroxynitrite-dependent nitration displayed by both residues, and both are located far from the heme site (the Fe-OH-Tyr distances are 13.8 and 14.7 Å, respectively). However, the impact of nitration on the alkaline transition is quite different in each case. Examination of the structure of native cytochrome c reveals that Tyr-74 is located in the Ω-loop, whereas Tyr-97 is located in the opposite side of the protein structure (15). Despite the similar solvent accessibility, Tyr-97 is located in a more rigid area of the protein fold, and its influence in promoting early conformational changes may be rather modest. Thus, it is tempting to speculate that the drastic effect induced by nitration of Tyr-74 is related to the destabilization of the Ω-loop. Accordingly, nitration at Tyr-74 is also the one that promotes the strongest gain of peroxidatic activity at physiological pH (40, 48).
Tyr nitration is expected to affect the pKa of the phenolic moiety. Indeed, the pKa of 3-nitrotyrosine in solution is 7.2, as compared with a value of 10.1 for Tyr (64). In a previous study, we speculated that nitration of Tyr-74 is responsible for decreasing its pKa value and that this change serves to shift the alkaline transition in the protein toward near neutral pH values (40). This assumption would imply that deprotonation of Tyr-74 is responsible for triggering the conformational change coupled to the alkaline transition. However, the present study shows that the Y74F mutant exhibits an alkaline transition similar to the wild type protein, allowing us to rule out this hypothesis. Moreover, the pKa change induced by the Tyr to Phe substitution is similar at positions 48, 74, and 97, confirming that deprotonation of Tyr-74 does not trigger the alkaline transition in cytochrome c. Thus, we are left with the possibility that nitration of Tyr-74 induces a destabilization of the Ω-loop. However, this hypothesis requires further elaboration to explain how this perturbation in a solvent-accessible residue is transmitted to the heme site and the fact that a relatively minor post-translational modification results in such a dramatic change of pKa.
Nitration of the aromatic ring of Tyr-74 is expected to present a steric strain with the Glu-66 side chain (i.e. the residue preceding Tyr-67) (Fig. 7). In turn, the phenolic proton of Tyr-67 is involved in an H-bond with the sulfur atom of Met-80 (15, 74), i.e. providing a direct connection between the nitrated residue and an iron ligand (Fig. 7). Our observation that the alkaline transition in the Y67F mutant (pKa ∼ 11.0) is shifted to a much higher pH value compared with the native protein may be due to removal of this H-bonding interaction. This hypothesis is further supported by the NMR structure of the alkaline form of yeast cytochrome c (13), where (in addition to the different conformation adopted by the Ω-loop) Tyr-67 and Met-80 are distant from the heme site and have lost the H-bond between them.
FIGURE 7.

Contact network proposed to lead to disruption of the Fe–S–Met-80 bond upon nitration at Tyr-74. Shown is a view of the CE–NO2–Tyr-74–COO–Glu-66–Glu-66–Tyr-67—O–Tyr-67—S–Met-80–Fe pathway proposed to transmit the effect of nitration at Tyr-74 to the heme ring. The figure was rendered with PyMol (DeLano Scientific) based on the PDB coordinates 1 hrc.
We therefore propose that Tyr-67 is involved in Met-80 detachment by wiring a conformational change in the Ω-loop to the metal site. Thus, Tyr-74 nitration perturbs the interaction with Glu-66; this perturbation is transmitted via the Tyr-74–Glu-66–Tyr-67–Met-80–Fe network (Fig. 7). The involvement of Glu-66 in this pathway is supported by the recent results obtained by Maity et al. (73), who measured the shift in the pKa of the alkaline transition upon mutations at several positions of the sequence. They found that the E66A replacement is one of the few mutations not located in the Ω-loop that shifts the pKa of the alkaline transition.
This analysis allows us to postulate the following scenario for the effect of Tyr nitration on cytochrome c: although both Tyr-97 and Tyr-74 are almost equally nitrated based on their similar solvent accessibility, nitration of Tyr-74 has a profound effect on the heme site structure and the protein flexibility at neutral pH. This phenomenon is due to the stabilization of the alkaline form, resulting in a lower pKa and therefore rendering a high concentration of cytochrome c molecules in which Met-80 has detached from the iron ion and has been replaced by a Lys residue. This ligand switch gives rise to an increase of the peroxidatic activity at pH 7 (41), as the replacement ligand, Lys-72, is more weakly bound to the heme and is readily displaceable by hydrogen peroxide and other peroxides, including peroxynitrite. Further, the gain of peroxidatic activity should favor the nitration of heme-adjacent Tyr-67 via iron-mediated reactions with either peroxynitrite or even hydrogen peroxide (in the presence of nitrite or nitric oxide) (40), supported by the detection of dinitrated cytochrome c species (i.e. in Tyr-74 and Tyr-67) during peroxynitrite exposure even under conditions of a large excess of remaining unmodified cytochrome c. This phenomenon indicates a preferential nitration of a second site (Tyr-67), after nitro-Tyr-74 cytochrome c formation, which most likely occurs secondary to a direct and fast iron-catalyzed reaction of the species present in the alternative low spin conformation. We also propose a mechanism by which the conformational change in nitro-Tyr-74 cytochrome c is triggered (Fig. 7).
The biological relevance of this study relies on the fact that nitrated mitochondrial proteins, including cytochrome c, have been identified under a variety of disease conditions, and some of them are likely to participate in the pathogenic process. In the case of cytochrome c, we envision that the conformational changes induced by tyrosine nitration reported herein can be tied to at least four processes8: 1) creating an intramitochondrial peroxidatic activity that can participate in oxidative sensing and signaling of apoptosis via oxidation of critical mitochondrial components including cardiolipin (26, 29, 56); 2) facilitating mitochondrial cytochrome c translocation in non-apoptotic cells to the cytosol and nucleus to exert yet to defined functions (but that may imply adaptive responses at the gene level)9; 3) affecting cytochrome c interactions with cytosolic partners such as Apaf-1, as nitrocytochrome c is not capable of apoptosome activation (48, 76) and can even interfere on unmodified cytochrome c-dependent apoptosome activation (interestingly, Tyr-97 is very close to Lys-7, which plays a critical role in Apaf-1 binding (77), and nitration seems to interfere with this process); and 4) potentially impeding tyrosine phosphorylation (78), recently reported to occur in vivo (77).
At the structural level, we have shown that NMR of the paramagnetic active site in a metalloprotein is sensitive enough to monitor remote changes; it could be further exploited to study different post-translational modifications, mutations, and physicochemical factors and interactions that may influence cytochrome c structure and function. This work may be extended to many other redox metalloproteins known to be modified by nitration in vitro and in vivo (42), thus setting the ground for future studies in this area.
Acknowledgments
The Bruker Avance II 600-MHz NMR spectrometer was purchased with funds from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) (PME2003-0026) and the Consejo Nacional de Investigaciones Científicas y Técnicas de Argentina (CONICET). We thank Dr. Carlos Batthyany and the Analytical Biochemistry and Proteomics Core Facility of the Institut Pasteur de Montevideo, Uruguay, for assistance on the HPLC-based methods. We also thank Lucía Bonilla (Departamento de Bioquímica, Facultad de Medicina, Universidad de la República, Uruguay) for participation in the preparation of nitrated cytochrome c species.
This work was supported, in whole or in part, by National Institutes of Health Grant R01-GM068682 (to A. J. V.). This work was also supported by grants from the Howard Hughes Medical Institute (to A. J. V. and R. R.), International Centre of Genetic Engineering and Biotechnology (to R. R.), and Comisión Sectorial de Investigación Científica (CSIC), Universidad de la República (to L. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Peroxynitrite is the product of the diffusion-controlled reaction
(k = 1.6 × 1010 m–1
s–1; (49))
between superoxide
(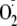 )
and •NO radicals. Cytochrome c3+ reacts at
moderate rates with
)
and •NO radicals. Cytochrome c3+ reacts at
moderate rates with
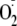 –(k
= 3 × 105 m–1
s–1 (50)) and
very slowly with •NO (k = 7.2 × 102
m–1 s–1
(51,
52)) and therefore can inhibit
peroxynitrite formation (53)
and decrease nitration yields. Nonetheless, cytochrome c3+
can still be nitrated by fluxes of
–(k
= 3 × 105 m–1
s–1 (50)) and
very slowly with •NO (k = 7.2 × 102
m–1 s–1
(51,
52)) and therefore can inhibit
peroxynitrite formation (53)
and decrease nitration yields. Nonetheless, cytochrome c3+
can still be nitrated by fluxes of
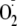 and •NO (A. Cassina, and R. Radi, unpublished data),
underscoring the concept that is not possible to completely block
peroxynitrite formation even by strong radical scavenging systems
(31,
54,
55).
and •NO (A. Cassina, and R. Radi, unpublished data),
underscoring the concept that is not possible to completely block
peroxynitrite formation even by strong radical scavenging systems
(31,
54,
55).
The abbreviations used are: HPLC, high pressure liquid chromatography; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight.
It is worth mentioning that in the moth/pigeon cytochrome c peptide model of antigen recognition (encompassing cytochrome c residues 88–103), a peptide containing nitro-Tyr-97 profoundly alters T-cell recognition (75) in a way that supports the proposal that nitrated epitopes of autologous proteins could participate in autoimmunological processes. In this regard, nitrocytochrome c may contribute to autoimmune responses by exposing a self-antigen. The structural aspects behind the aberrant interactions of the nitrated cytochrome c peptide at the T-cell and/or major histocompatibility complex level are unknown.
L. C. Godoy, C. Muñoz Pinedo, L. Castro, S. Cardaci, C. M. Schonoff, M. King, V. Tórtora, M. Marin, Q. Miao, J. Fei Jiangs, A. Kapralov, R. Jemmerson, G. G. Silkstone, J. N. Patel, J. Evans, M. T. Wilson, D. R. Green, V. E. Kagan, R. Radi, and J. B. Mannick, submitted for publication.
References
- 1.Margoliash, E., and Schejter, A. (1996) in Cytochrome C: A Multidisciplinary Approach (Scott, R. A., and Mauk, A. G., eds) p. vi, University Science Books, Sausalito, CA
- 2.Radi, R., Turrens, J. F., and Freeman, B. A. (1991) Arch. Biochem. Biophys. 288 118–125 [DOI] [PubMed] [Google Scholar]
- 3.Salamon, Z., and Tollin, G. (1997) J. Bioenerg. Biomembr. 29 211–221 [DOI] [PubMed] [Google Scholar]
- 4.Wilkstrom, M., and Sarastre, M. (1984) Bioenergetics: New Comprehensive Biochemistry, Vol. 9, pp. 49–94, Elsevier Science, New York [Google Scholar]
- 5.Jiang, X., and Wang, X. (2004) Annu. Rev. Biochem. 73 87–106 [DOI] [PubMed] [Google Scholar]
- 6.Skulachev, V. P. (1998) FEBS Lett. 423 275–280 [DOI] [PubMed] [Google Scholar]
- 7.Senn, H., and Wuthrich, K. (1985) Q. Rev. Biophys. 18 111–134 [DOI] [PubMed] [Google Scholar]
- 8.Heineman, W. R., Norris, B. J., and Goelz, J. F. (1975) Anal. Chem. 47 79–84 [DOI] [PubMed] [Google Scholar]
- 9.Liu, G., Shao, W., Zhu, S., and Tang, W. (1995) J. Inorg. Biochem. 60 123–131 [DOI] [PubMed] [Google Scholar]
- 10.Dickerson, R. E., and Timkovich, R. (1975) The Enzymes (Boyer, P. D., ed) 3rd Ed., Vol. 11, pp. 397–547, Academic Press, New York [Google Scholar]
- 11.Dopner, S., Hildebrandt, P., Rosell, F. I., and Mauk, A. G. (1998) J. Am. Chem. Soc. 120 11246–11255 [Google Scholar]
- 12.Scott, R. A., and Mauk, A. G. (eds) (1996) Cytochrome C: A Multidisciplinary Approach (Mauk, A. G., and Scott, R. A., eds) pp. 475–487, University Science Books, Sausalito, CA
- 13.Assfalg, M., Bertini, I., Dolfi, A., Turano, P., Mauk, A. G., Rosell, F. I., and Gray, H. B. (2003) J. Am. Chem. Soc. 125 2913–2922 [DOI] [PubMed] [Google Scholar]
- 14.Banci, L., Bertini, I., Gray, H. B., Luchinat, C., Reddig, T., Rosato, A., and Turano, P. (1997) Biochemistry 36 9867–9877 [DOI] [PubMed] [Google Scholar]
- 15.Bushnell, G. W., Louie, G. V., and Brayer, G. D. (1990) J. Mol. Biol. 214 585–595 [DOI] [PubMed] [Google Scholar]
- 16.Ochi, H., Hata, Y., Tanaka, N., Kakudo, M., Sakurai, T., Aihara, S., and Morita, Y. (1983) J. Mol. Biol. 166 407–418 [DOI] [PubMed] [Google Scholar]
- 17.Takano, T., and Dickerson, R. E. (1981) J. Mol. Biol. 153 95–115 [DOI] [PubMed] [Google Scholar]
- 18.Ferrer, J. C., Guillemette, J. G., Bogumil, R., Inglis, S. C., Smith, M., and Mauk, A. G. (1993) J. Am. Chem. Soc. 115 7507–7508 [Google Scholar]
- 19.Hong, X. L., and Dixon, D. W. (1989) FEBS Lett. 246 105–108 [DOI] [PubMed] [Google Scholar]
- 20.Moore, G. R., and Pettigrew, G. W. (1990) Cytochromes c: Evolutionary, Structural, and Physicochemical Aspects (Springer Series in Molecular Biology), Springer-Verlag, Berlin
- 21.Osheroff, N., Borden, D., Koppenol, W. H., and Margoliash, E. (1980) J. Biol. Chem. 255 1689–1697 [PubMed] [Google Scholar]
- 22.Pearce, L. L., Gartner, A. L., Smith, M., and Mauk, A. G. (1989) Biochemistry 28 3152–3156 [DOI] [PubMed] [Google Scholar]
- 23.Pettigrew, G. W., and Moore, G. R. (1987) Cytochromes c: Biological Aspects (Springer Series in Molecular Biology), Springer-Verlag, Berlin
- 24.Rosell, F. I., Ferrer, J. C., and Mauk, A. G. (1998) J. Am. Chem. Soc. 120 11234–11245 [Google Scholar]
- 25.Pollock, W. B., Rosell, F. I., Twitchett, M. B., Dumont, M. E., and Mauk, A. G. (1998) Biochemistry 37 6124–6131 [DOI] [PubMed] [Google Scholar]
- 26.Kagan, V. E., Tyurin, V. A., Jiang, J., Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A., Osipov, A. N., Belikova, N. A., Kapralov, A. A., Kini, V., Vlasova, I. I., Zhao, Q., Zou, M., Di, P., Svistunenko, D. A., Kurnikov, I. V., and Borisenko, G. G. (2005) Nat. Chem. Biol. 1 223–232 [DOI] [PubMed] [Google Scholar]
- 27.Nantes, I. L., Zucchi, M. R., Nascimento, O. R., and Faljoni-Alario, A. (2001) J. Biol. Chem. 276 153–158 [DOI] [PubMed] [Google Scholar]
- 28.Kawai, C., Prado, F. M., Nunes, G. L., Di Mascio, P., Carmona-Ribeiro, A. M., and Nantes, I. L. (2005) J. Biol. Chem. 280 34709–34717 [DOI] [PubMed] [Google Scholar]
- 29.Basova, L. V., Kurnikov, I. V., Wang, L., Ritov, V. B., Belikova, N. A., Vlasova, I. I., Pacheco, A. A., Winnica, D. E., Peterson, J., Bayir, H., Waldeck, D. H., and Kagan, V. E. (2007) Biochemistry 46 3423–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen, Y. R., Chen, C. L., Liu, X., Li, H., Zweier, J. L., and Mason, R. P. (2004) Free Radic. Biol. Med. 37 1591–1603 [DOI] [PubMed] [Google Scholar]
- 31.Radi, R. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4003–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mugnol, K. C., Ando, R. A., Nagayasu, R. Y., Faljoni-Alario, A., Brochsztain, S., Santos, P. S., Nascimento, O. R., and Nantes, I. L. (2008) Biophys. J. 94 4066–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertini, I., Turano, P., and Vila, A. J. (1993) Chem. Rev. 93 2833–2932 [Google Scholar]
- 34.Bren, K. L., Kellogg, J. A., Kaur, R., and Wen, X. (2004) Inorg. Chem. 43 7934–7944 [DOI] [PubMed] [Google Scholar]
- 35.Feng, Y. Q., Roder, H., and Englander, S. W. (1990) Biophys. J. 57 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soussi, B., Bylund-Fellenius, A. C., Schersten, T., and Angstrom, J. (1990) Biochem. J. 265 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schejter, A., Aviram, I., and Sokolovsky, M. (1970) Biochemistry 9 5118–5122 [DOI] [PubMed] [Google Scholar]
- 38.Sokolovsky, M., Aviram, I., and Schejter, A. (1970) Biochemistry 9 5113–5118 [DOI] [PubMed] [Google Scholar]
- 39.Skov, K., Hofmann, T., and Williams, G. R. (1969) Can J. Biochem. 47 750–752 [DOI] [PubMed] [Google Scholar]
- 40.Batthyany, C., Souza, J. M., Duran, R., Cassina, A., Cervenansky, C., and Radi, R. (2005) Biochemistry 44 8038–8046 [DOI] [PubMed] [Google Scholar]
- 41.Cassina, A. M., Hodara, R., Souza, J. M., Thomson, L., Castro, L., Ischiropoulos, H., Freeman, B. A., and Radi, R. (2000) J. Biol. Chem. 275 21409–21415 [DOI] [PubMed] [Google Scholar]
- 42.Souza, J. M., Peluffo, G., and Radi, R. (2008) Free Radic. Biol. Med. 45 357–366 [DOI] [PubMed] [Google Scholar]
- 43.Alonso, D., Encinas, J. M., Uttenthal, L. O., Bosca, L., Serrano, J., Fernandez, A. P., Castro-Blanco, S., Santacana, M., Bentura, M. L., Richart, A., Fernandez-Vizarra, P., and Rodrigo, J. (2002) Neuroscience 111 47–56 [DOI] [PubMed] [Google Scholar]
- 44.Cruthirds, D. L., Novak, L., Akhi, K. M., Sanders, P. W., Thompson, J. A., and MacMillan-Crow, L. A. (2003) Arch. Biochem. Biophys. 412 27–33 [DOI] [PubMed] [Google Scholar]
- 45.Peluffo, G., and Radi, R. (2007) Cardiovasc. Res. 75 291–302 [DOI] [PubMed] [Google Scholar]
- 46.Castro, L., Eiserich, J. P., Sweeney, S., Radi, R., and Freeman, B. A. (2004) Arch. Biochem. Biophys. 421 99–107 [DOI] [PubMed] [Google Scholar]
- 47.Chen, Y. R., Chen, C. L., Chen, W., Zweier, J. L., Augusto, O., Radi, R., and Mason, R. P. (2004) J. Biol. Chem. 279 18054–18062 [DOI] [PubMed] [Google Scholar]
- 48.Souza, J. M., Castro, L., Cassina, A. M., Batthyany, C., and Radi, R. (2008) Methods Enzymol. 441 197–215 [DOI] [PubMed] [Google Scholar]
- 49.Kissner, R., Nauser, T., Bugnon, P., Lye, P. G., and Koppenol, W. H. (1997) Chem. Res. Toxicol. 10 1285–1292 [DOI] [PubMed] [Google Scholar]
- 50.Butler, J., Koppenol, W. H., and Margoliash, E. (1982) J. Biol. Chem. 257 10747–10750 [PubMed] [Google Scholar]
- 51.Hoshino, M., Ozawa, K., Seki, H., and Ford, P. C. (1993) J. Am. Chem. Soc. 115 9568–9575 [Google Scholar]
- 52.Radi, R. (1996) Chem. Res. Toxicol. 9 828–835 [DOI] [PubMed] [Google Scholar]
- 53.Kelm, M., Dahmann, R., Wink, D., and Feelisch, M. (1997) J. Biol. Chem. 272 9922–9932 [DOI] [PubMed] [Google Scholar]
- 54.Demicheli, V., Quijano, C., Alvarez, B., and Radi, R. (2007) Free Radic. Biol. Med. 42 1359–1368 [DOI] [PubMed] [Google Scholar]
- 55.Quijano, C., Romero, N., and Radi, R. (2005) Free Radic. Biol. Med. 39 728–741 [DOI] [PubMed] [Google Scholar]
- 56.Belikova, N. A., Jiang, J., Tyurina, Y. Y., Zhao, Q., Epperly, M. W., Greenberger, J., and Kagan, V. E. (2007) Int. J. Radiat. Oncol. Biol. Phys. 69 176–186 [DOI] [PubMed] [Google Scholar]
- 57.Margoliash, E., and Lustgarten, J. (1962) J. Biol. Chem. 237 3397–3405 [PubMed] [Google Scholar]
- 58.Radi, R., Beckman, J. S., Bush, K. M., and Freeman, B. A. (1991) J. Biol. Chem. 266 4244–4250 [PubMed] [Google Scholar]
- 59.Rumbley, J. N., Hoang, L., and Englander, S. W. (2002) Biochemistry 41 13894–13901 [DOI] [PubMed] [Google Scholar]
- 60.Glasoe, P. K., and Long, F. A. (1960) J. Phys. Chem. 64 188–190 [Google Scholar]
- 61.Wilson, M. T., and Greenwood, C. (1971) Eur. J. Biochem. 22 11–18 [DOI] [PubMed] [Google Scholar]
- 62.Luntz, T. L., Schejter, A., Garber, E. A., and Margoliash, E. (1989) Proc. Natl. Acad. Sci. U. S. A. 86 3524–3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pepaj, M., Wilson, S. R., Novotna, K., Lundanes, E., and Greibrokk, T. (2006) J. Chromatogr. A 1120 132–141 [DOI] [PubMed] [Google Scholar]
- 64.Creighton, T. E. (1993) Proteins: Structures and Molecular Properties, 2nd Ed., pp. 14–17, W. H. Freeman, New York
- 65.Blauer, G., Sreerama, N., and Woody, R. W. (1993) Biochemistry 32 6674–6679 [DOI] [PubMed] [Google Scholar]
- 66.Santucci, R., and Ascoli, F. (1995) J. Inorg. Biochem. 170 3197–3292 [Google Scholar]
- 67.Adams, P. A. (1991) in Peroxidases in Chemistry and Biology (Everse, J., Everse, K. E., and Grisham, M. B., eds) Vol. 2, pp. 171–200, CRC Press, Boca Raton, FL [Google Scholar]
- 68.Banci, L., Bertini, I., Reddig, T., and Turano, P. (1998) Eur. J. Biochem. 256 271–278 [DOI] [PubMed] [Google Scholar]
- 69.Boswell, A. P., Moore, G. R., Williams, R. J., Harris, D. E., Wallace, C. J., Bocieck, S., and Welti, D. (1983) Biochem. J. 213 679–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Droghetti, E., Oellerich, S., Hildebrandt, P., and Smulevich, G. (2006) Biophys. J. 91 3022–3031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kapralov, A. A., Kurnikov, I. V., Vlasova, I. I., Belikova, N. A., Tyurin, V. A., Basova, L. V., Zhao, Q., Tyurina, Y. Y., Jiang, J., Bayir, H., Vladimirov, Y. A., and Kagan, V. E. (2007) Biochemistry 46 14232–14244 [DOI] [PubMed] [Google Scholar]
- 72.Schejter, A., Goldkorn, T., and Sokolovsky, M. (1971) Eur. J. Biochem. 20 414–419 [DOI] [PubMed] [Google Scholar]
- 73.Maity, H., Rumbley, J. N., and Englander, S. W. (2006) Proteins 63 349–355 [DOI] [PubMed] [Google Scholar]
- 74.Berghuis, A. M., Guillemette, J. G., Smith, M., and Brayer, G. D. (1994) J. Mol. Biol. 235 1326–1341 [DOI] [PubMed] [Google Scholar]
- 75.Birnboim, H. C., Lemay, A. M., Lam, D. K., Goldstein, R., and Webb, J. R. (2003) J. Immunol. 171 528–532 [DOI] [PubMed] [Google Scholar]
- 76.Rodríguez-Roldán, V., García-Heredia, J. M., Navarro, J. A., Rosa, M. A., and Hervás, M. (2008) Biochemistry, in press [DOI] [PubMed]
- 77.Lee, I., Salomon, A. R., Yu, K., Doan, J. W., Grossman, L. I., and Huttemann, M. (2006) Biochemistry 45 9121–9128 [DOI] [PubMed] [Google Scholar]
- 78.Kong, S. K., Yim, M. B., Stadtman, E. R., and Chock, P. B. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 3377–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]