

Abstract
In a previous study, we examined thermodynamic parameters for 20 alanine mutants inβ-lactamase inhibitory protein (BLIP) for binding to TEM-1 β-lactamase. Here we have determined the structures of two thermodynamically distinctive complexes of BLIP mutants with TEM-1 β-lactamase. The complex BLIP Y51A-TEM-1 is a tight binding complex with the most negative binding heat capacity change (ΔG = ∼-13 kcal mol-1 and ΔCp = ∼-0.8 kcal mol-1 K-1) among all of the mutants, whereas BLIP W150A-TEM-1 is a weak complex with one of the least negative binding heat capacity changes (ΔG = ∼-8.5 kcal mol-1 and ΔCp = ∼-0.27 kcal mol-1 K-1). We previously determined that BLIP Tyr51 is a canonical and Trp150 an anti-canonical TEM-1-contact residue, where canonical refers to the alanine substitution resulting in a matched change in the hydrophobicity of binding free energy. Structure determination indicates a rearrangement of the interactions between Asp49 of the W150A BLIP mutant and the catalytic pocket of TEM-1. The Asp49 of W150A moves more than 4 Å to form two new hydrogen bonds while losing four original hydrogen bonds. This explains the anti-canonical nature of the Trp150 to alanine substitution, and also reveals a strong long distance coupling between Trp150 and Asp49 of BLIP, because these two residues are more than 25 Å apart. Kinetic measurements indicate that the mutations influence the dissociation rate but not the association rate. Further analysis of the structures indicates that an increased number of interface-trapped water molecules correlate with poor interface packing in a mutant. It appears that the increase of interface-trapped water molecules is inversely correlated with negative binding heat capacity changes.
Protein-protein interactions play essential roles in most biological processes, from the control of enzymatic catalysis to signal transduction. Recent advances in structural biology have revealed the complexity of structure-function relationships in protein-protein interactions (1, 2). Many factors that influence the strength of a binding interaction have been recognized (3). The design of new protein-specific binding partners is beginning to show some results but is not yet generally applicable (4-7). A protein-protein binding interaction typically involves many contact residues and a relatively large interface that is rather planar. There are several aspects of the contact interface that could play critical roles in the binding interaction, such as the flexibility of the contact residues, chemical and physical complementarity, as well as trapped solvent molecules in the interface (1, 8, 9). Alanine-scanning mutagenesis results indicate that only a small fraction of the contact residues contribute significantly to the binding free energy. Substitution of these hotspot contact residues with alanine results in a large loss in binding free energy (more than 2 kcal) (10, 11). Understanding this phenomenon is an active area of research in the structural biology of protein interactions. An in-depth and thorough assessment of binding characteristics of these alanine-substituted mutants will provide an increased understanding of the interactions.
β-Lactamases are enzymes produced by bacteria that provide resistance to β-lactam antibiotics, which are the most widely used antimicrobial therapeutics. β-Lactamases provide for resistance to these antibiotics by selectively cleaving the active peptide bond in the β-lactam ring. There are four major classes of β-lactamases (A-D) (12) with the class A β-lactamases being the most widespread in the clinical setting (12). Among the class A β-lactamases, TEM-1 is the most prevalent among Gram-negative bacteria and has been a subject of concern because it has a broad specificity to hydrolyze a wide array of β-lactam-based antibiotics (13).
BLIP2 is a 165-amino acid protein produced by the soil bacterium Streptomyces clavuligerus that potently inhibits the TEM-1 β-lactamase (14, 15). BLIP has a tandem repeat structure of a 76-amino acid αβ domain. These two tandem domains form a β-saddle with a relatively hydrophobic concave surface. The concave saddle surface of BLIP binds on the loop-helix region of TEM-1 and extends two loops into the substrate binding pocket of TEM-1. The Asp49 residue of BLIP on one of the loops (loop1) occupies the catalytic site of TEM-1 (15). A BLIP-TEM-1 complex structure is shown in Fig. 1A.
FIGURE 1.

The structure of the BLIP-TEM-1 β-lactamase complex indicating the location of several interesting residues and their binding thermodynamics. A, structural representation of BLIP-TEM-1 complex. TEM-1 is represented as a schematic and colored in cyan. BLIP is represented in a white surface model, and the TEM-1 contact residues are colored in yellow. BLIP Trp150 is represented as a CDW model colored in red, and Tyr51 is represented as a CDW model colored in green. Asp49 and Trp112 are represented in bond models colored red. B, plots of different forms of free energy versus temperature for the formation of four different BLIP mutant-TEM-1 β-lactamase complexes. The thin curves represent the binding free energy, ΔG0s, as function of temperature, and are calculated from Equation 1 using the experimentally determined ΔH, ΔS, and ΔCp values. × indicates the specific experimental measurements (16). The hypothetical hydrophobic component “ΔG0hydrs” is calculated using the Equation 2 with the experimentally determined ΔCp and is shown as thick dark curves. The nonhydrophobic component, “ΔG0nonhydrs,” is calculated from Equation 3 in the text and represented as linear dashed lines.
The BLIP-β-lactamase complex has become a protein-protein interaction model system with increasingly more detailed information on structure-function relationships becoming available (15-24). Alanine-scanning mutagenesis of residues on the BLIP binding surface revealed the following: 1) there are two hotspots of binding energy, and 2) one mutation (Y50A) actually increases binding affinity for TEM-1 by 50-fold (17). Our lab previously determined the complete binding thermodynamics of the available alanine-substituted mutants of BLIP that are involved in complex formation with TEM-1 β-lactamase. In that study, the isoenthalpic temperature (temperature at which the binding enthalpy is zero) was used to gauge the role of hydrophobicity in binding. An increase in the isoenthalpic temperature suggests an increase in the hydrophobic nature of the binding. A contact residue whose alanine substitution results in a matched change in isoenthalpic temperature with the contribution of hydrophobicity to binding is called canonical. For example, a charged contact residue is deemed canonical if its alanine substitution, which increases hydrophobicity, also exhibits an increase in the binding isoenthalpic temperature. In contrast, a residue whose alanine substitution results in a nonmatching change in isoenthalpic temperature and hydrophobic contribution to binding is termed anti-canonical. Canonical hotspot contact residues likely contribute directly to the driving force for binding. Anti-canonical hotspot contact residues, however, likely provide binding driving forces indirectly through energetically coupled changes linked to other contact residues (16). The previous thermodynamics experiments also revealed a large variation in binding heat capacity changes in complex formation between the various BLIP mutants and β-lactamase. This leads to the question of what is the information content of the heat capacity changes.
Here a detailed analysis of two complexes was performed to extend the previous studies and address the following two issues. 1) What changes in the contact interface and binding interactions are associated with alanine substitutions? 2) What are the effects of the alanine substitutions on the binding kinetics of complex formation? This information, in conjunction with the previously determined thermodynamic information, provides another level of detail toward understanding the driving forces in protein-protein interactions.
The results provide some insight into the long observed variation among binding heat capacity changes and buried surface area. It is unclear why the relationship between buried surface area and binding heat capacity changes is so unpredictable. The results obtained in this study argue that binding heat capacity changes are related to a combination of changes in the buried surface area and the number of trapped solvent molecules. The results suggest that the origin of the increased number of trapped water molecules is subtle structural mismatches in the interface introduced by an alanine substitution. These mismatches disrupt short range binding interactions that directly impact the dissociation process, which in turn modulates the binding free energy.
EXPERIMENTAL PROCEDURES
Materials—Talon resin was purchased from Clontech. Ion-exchange media and columns (Mono Q 5/50 GL, HighTrap Q, Q-Sepharose Fast Flow, and DEAE-Sepharose Fast Flow) and sizing columns (Superdex 75 10/300, Superdex 75 prep grade, and Superdex 200 10/300 GL) were purchased from Amersham Biosciences. Cephalosporin C was purchased from Sigma (product name cephalosporin C zinc salt, 22237 BioChemika). All other reagents were reagent grade from Sigma.
Crystallization screening kits were purchased from Hampton Research, Emerald Biostructure Inc., and Nextal Biotechnologies (Montreal, Quebec, Canada). Reagents used in crystallization were of the highest purity available from Sigma. Filter concentrators were from Millipore Inc. (Amicon concentrators) and from Sartorius Group (Vivaspin concentrators).
Protein Expression and Purification—All expression DNA clones were constructed previously, and the proteins were expressed and purified using a previously described procedure (16, 17). Briefly, Escherichia coli bacteria (strain RB791) containing the BLIP mutant expression plasmid were grown in LB media containing 17.5 μg/ml chloramphenicol at 37 °C from a single clone and induced with 2 mm lactose and 1% glycerol instead of isopropyl 1-thio-β-d-galactopyranoside when the absorbance at 600 nm was ∼1.2. The culture was further grown for 6 h and harvested by centrifugation at 5000 rpm for 10 min at 6 °C. The bacterial pellets were resuspended in B-PER bacterial protein extraction solution (Pierce) or a prepared equivalent (1% Triton X-100 in 10 mm Tris-HCl, pH 8.0, 150 mm NaCl) at a ratio of 15 ml for 1 g of bacterial pellet. The resuspensions were vigorously shaken for 20 min at room temperature and then centrifuged at high speed (15,000 rpm in a Beckman type 35 rotor) for 30 min. The supernatants were mixed by stirring with Talon cobalt resin overnight. The Talon cobalt resin was allowed to settle by gravity and then collected and washed three times. The bound BLIP mutant proteins were eluted from the Talon cobalt resins using 150 mm imidazole in TBS, and then further purified on a Superdex-75 sizing column. The bacteria containing the TEM-1 β-lactamase expression plasmid DNA were grown at 37 °C and induced at A600 nm ∼0.6 with 0.3 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h at 37 °C. The expressed periplasmic protein TEM-1 was extracted by osmotic shock in which bacteria were soaked with 20% sucrose solution at room temperature and centrifuged, and the resulting pellet was resuspended in 5 mm ice-cold magnesium sulfate. The osmotic shock fluid was fractionated on a DEAE column in 10 mm Tris-HCl buffer at pH 7 with 100 mm NaCl. Flow-through material was diluted 10-fold with distilled water and fractionated on the DEAE column again. TEM-1 β-lactamase was eluted with a 0-2 m NaCl gradient in Tris-HCl buffer at pH 7.0. The protein was further purified using a Superdex-75 sizing column in 50 mm phosphate buffer at pH 7.0 with 150 mm NaCl. The purity of the proteins was confirmed by SDS-PAGE analysis. Protein concentrations were determined using absorbance measurements at 280 nm with 8 m guanidine HCl denatured protein, based on the theoretically calculated extinction coefficients according to the amino acid sequence.
Stopped-flow Fluorescence Spectrometric Measurements of Association—The kinetic association measurements were carried out using an SLM 48000S fluorescence spectrometer equipped with a MilliFlow stopped-flow reactor accessory. Equal concentrations of TEM-1 and BLIP mutants were loaded into the two injection syringes. The excitation wavelength was 286 nm with an 8 nm bandwidth, and the emission wavelength was 330 nm with a 4 nm bandwidth. The instrument has a 10-ms mixing deadtime and can collect data at a 10-ms interval. In this study, we determined the constants only at room temperature (∼25 °C). Typically, 10-20 time course traces were averaged. The association constants were determined by fitting the data with a second order kinetic time course.
Enzymatic Activity-based Kinetic Measurements—For slow complex dissociation, the recovery of activity was followed by competitive displacement. The active wild type TEM-1 β-lactamase was first complexed with a BLIP mutant, and then the active TEM-1 was recovered by adding an excess amount of an inactive TEM-1 mutant (S70A). The BLIP mutant binds to the inactive TEM-1 S70A mutant β-lactamase as it is dissociated from the pre-formed complex thus leaving the dissociated wild type TEM-1 as active monomer. The time course of the recovery of the active TEM-1 enzyme activity is an effective first order dissociation. By measuring the TEM-1 activity at various time points after the addition of the inactive TEM-1 S70A mutant, the time course of the recovered activity allowed determination of the dissociation time. The activity was determined using cephalosporin C (100 μm) as substrate in phosphate-buffered saline at room temperature. The hydrolysis of cephalosporin C was monitored optically at 280 nm absorbance with an extinction coefficient of 7500 m-1 cm-1. The amount of active enzyme was calculated using [TEM-1] = (V(S + Km)/(S × kcat)), where V and S are hydrolysis rate and cephalosporin concentration, respectively. Km was determined to be 700 μm, and kcat was 11 s-1 (17, 25). The time course of the amount of active TEM-1 was fitted with first order kinetics to extrapolate the kinetic parameters.
For the fast dissociation of the complex of BLIP W150A-TEM-1, 1 volume of a concentration of 10 μm of preformed W150A-TEM-1 complex was mixed with 7.3 volumes of the assay solution containing cephalosporin C as substrate. This was done by driving the protein solution in a 3-ml syringe and the substrate solution in a 30-ml syringe in a syringe pump. These two streams of solutions were mixed immediately before entering an optical monitoring cell. The estimated time from mixing to optical cell is less than 1 s. The hydrolysis of cephalosporin C was continuously monitored. The recording of the time courses was started when the syringe pump was stopped. The time courses were fitted with on- and off-rates simultaneously. Given the affinity of this complex of 180 nm and the above scheme in the diluted solution, the free TEM-1 will increase from ∼200 to ∼700 nm. This range of the changes in TEM-1 allows kinetic determination with reasonable accuracy.
Fluorescence Spectrometric Measurements of Dissociation—An apparatus similar to the dilution enzymatic activity assay was constructed, in which a fluorescence cell optical monitoring cell was used in place of the optical monitoring cell to fit into the fluorescence spectrometer. The mixing syringes were replaced with 1-ml and 50-ml size syringes. A solution of 3 μm premixed BLIP W150A-TEM-1 complex was loaded into the 1-ml syringe and phosphate-buffered saline solution into the 50 ml syringe. Both syringes were mounted on a syringe pump to drive the solution at the same constant piston speed. The sample was mixed with the buffer immediately before entering the fluorescence cell at a ratio of 1:37. The fluorescence signals were recorded at 100-ms intervals. The kinetic recording starts when the flow is stopped, after about 3 ml of the mixture flows through. Our experimental setup requires about 1-2 ml of mixture to flow through for the signal to become stable. Typically the noise level was about 10% of the total fluorescence signal under the conditions of the recording for individual traces. Our estimates (based on fluorescence differences of the bound complex and the unbound proteins and the amount of the dilution and the affinity of the complex) show the signal was about 5% of the fluorescence signal. Multiple traces (more than 50) were averaged to increase the signal to noise.
Crystallization Screening and Crystal Quality Improvements—Based on our previous determination of the binding thermodynamics of the interactions between TEM-1 β-lactamase and the BLIP mutants, we selected several thermodynamically distinctive complexes for initial crystallization screens. The selected complexes were Y50A-TEM-1, Y51A-TEM-1, W112A-TEM-1, and W150A-TEM-1. Initial batches of complexes used in the crystallization screens were from resulting complexes of isothermal titration calorimetric measurements reported previously (16). The complexes were repurified on an ion-exchange column followed by a sizing column. The repurified complexes were concentrated to more than 10 mg/ml using filter concentrators with a cutoff size of 10 kDa. Crystallization screening was set up in a 96-well plate format using commercial screening kits (crystal screening kits from Hampton Research Inc., Wizard kits from Emerald Biotechnology, and PEGs and MPD and AmSO4 suite screening kits from Qiagen were used in these studies). The high resolution x-ray diffraction quality crystals were crystallized in conditions of 0.1 m phosphate/citric acid, pH 4.4, 14% PEG 8000, 0.1 m NaCl (for the Y51A-TEM-1 complex), and 0.1 m phosphate/citric acid, pH 4.8, 15% PEG 8000, 0.1 m NaCl (for the W150A-TEM-1 complex), and 0.1 m MES, pH 6.9, 2 m NaCl, 0.1 m KH2PO4, 0.1 m NaH2PO4 (for the W112A crystal). Microseeding was used to yield high quality crystals.
X-ray Diffraction and Data Collection—The high quality crystals were mounted in cryo-loops and soaked in a cryo-protectant of 20% glycerol in mother liquor and then flash-frozen in liquid nitrogen. The x-ray diffraction data were collected at the Gulf Coast Protein Crystallography Consortium synchrotron beamline at the Center for Advanced Microstructures and Devices at Louisiana State University in Baton Rouge, LA.
Data Processing and Refinement—All x-ray diffraction datasets had a resolution that was better than 2.7 Å, with the highest resolution of 2.07 Å. Diffraction data were indexed in P21 symmetry, integrated using DENZO, and scaled using SCALEPACK from the HKL software package (HKL Research Inc. (26)). We converted the merged intensity data into structural factors using the TRUNCATE method in the Scalepack2MTZ program in the CCP4 package, and 5% of data were tagged with free R-flag.
Structural Determination and Refinement—The crystal structures were determined by molecular replacement using both Molrep and AmoRe programs in the CCP4 package with one complex from the 1JTG.pdb file (Protein Data Bank (15)) as the search model. The resulting structures were refined using program Refmac5 in the CCP4 package at the initial stages. These preliminary structures along with the electron density maps were subjected to manual structure modification, such as changing the mutant sites and adding the TEM-1 residue 214 that was missing in the 1JTG coordinates. These modified structures were further subjected to several cycles of restrained refinement in the Refmac5 program. Afterward, simulated annealing refinement was performed using CNS (27). The resulting structures were further refined using TLS and restraint refinement. We added water molecules using the CNS program and additional waters in COOT (28).
Structural Analysis—The structures were analyzed mostly using the programs from the CCP4 package as follows (29). The alignment was carried out for the whole sequences or several segments using the SUPERPOSE program in the CCP4 package. The buried surface area was calculated using AreaiMol program in the CCP4 package with the standard 1.4 Å solvent probe. Hydrogen bonds were calculated using the contact program in the CCP4 package. The shape complementarity statistics (SC value) were calculated using the sc.exe program in the CCP4 package (30). Structure graphic programs used were VMD, CCP4 mg, and Swiss PDB Viewer (31-33).
RESULTS
The selection of the BLIP-β-lactamase complexes for this study was based on an analysis of the binding thermodynamics of BLIP alanine mutants with TEM-1 β-lactamase determined previously (16). One of the selection criteria was to choose complexes with the most discrepant binding heat capacity changes. Among the mutant complexes being studied, the BLIP W150A-TEM-1 complex has one of the least negative binding heat capacity changes (-275 cal/mol/K), whereas the BLIP Y51A-TEM-1 has the most negative binding heat capacity change of -809 cal/mol/K. In addition, the BLIP W150A-TEM-1 complex is the weakest complex (K of ∼2 × 106 m-1), and BLIP Y51A-TEM-1 is one of the tightest complexes (K of ∼1.5 × 109 m-1) (see Fig. 1A for the locations of the residues). We hypothesized that such a large difference could be manifest in some distinctive structural and kinetic features that will provide some insight into the driving forces for binding.
It has long been accepted that hydrophobic interactions are the major driving force for protein-protein binding interactions. It is also recognized that binding heat capacity changes are related to buried surface area changes that are in turn related to hydrophobic interactions. However, detailed quantitative assessments have been frustrating due to the inconsistencies of the assessments because of a high system and methodology dependence of the measurements.
Because of the difficulties associated with quantitation of the driving forces, a rather general and coarse approach was used here in combination with the experimental data to assess the hydrophobic and nonhydrophobic driving forces. This approach allowed an estimation of the contribution from hydrophobic and nonhydrophobic interactions at an imprecise, yet informative level. Because the approach is imprecise, we elect to use the terms “hydrophobic driving force” and “nonhydrophobic driving force” within the context of this study.
The temperature dependence of the binding free energy for each mutant can be described by Equation 1,
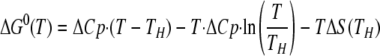 |
(Eq.1) |
where ΔCp is the experimentally determined binding heat capacity change, TH is the isoenthalpic temperature (at which the binding enthalpy is zero), and ΔS(TH) is the binding entropy at the isoenthalpic temperature. This binding free energy can be separated into two terms as shown in Equations 2 and 3,
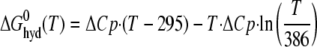 |
(Eq.2) |
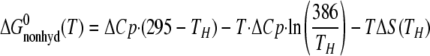 |
(Eq.3) |
where 295 and 386 K are chosen based on the hydrophobic model treatment proposed by Baldwin (34). Using this formula implies that the ΔCp is completely because of hydrophobic interactions, a highly debatable assumption. Nevertheless, the binding free energy can be separated into two terms as follows: one is the hydrophobic interaction free energy with the experimentally determined binding heat capacity changes similar to Baldwin's treatment, and the other linear temperature-dependent (Fig. 1B). The linear temperature-dependent “nonhydrophobic” term can be interpreted as the traditional free binding energy with constant binding enthalpy and entropy over the temperature range of interest. The “hydrophobic” nonlinear temperature-dependent term arises from the compensation between the enthalpy-entropy terms (34). This analysis suggests that the binding heat capacity change provides a large hydrophobic driving force that is counter-balanced by a nonhydrophobic force. The result is a moderate binding affinity. By comparing the corresponding complex structures, we hoped to assess various nonhydrophobic driving forces by inspecting van der Waals contacts, salt bridges, and hydrogen bonds, and hydrophobic driving forces by examining the characteristics of the buried surface areas.
General Observations on the Structures—After several rounds of crystallization improvements the crystals of BLIP Y51A-TEM-1, W112A-TEM-1, and W150A-TEM-1 diffracted to high resolution. Both BLIP Y51A-TEM-1 and W150A-TEM-1 crystals have similar morphology and similar symmetry in the diffraction data and crystal lattice dimensions. The BLIP W112A-TEM-1 crystal has quite different morphology, crystal symmetry, and lattice dimensions. When the structure was solved, only the BLIP W112A mutant was apparent without the TEM-1 protein (data not shown). Many of these crystals could x-ray diffract to a resolution better than 3 Å. The x-ray diffraction data collection and processing statistics are shown in Table 1. The structures of the two complexes were solved by molecular replacement methods. In this study, the emphasis is on the structures of the complexes. The structural refinement statistics are shown in Table 2. The crystal contacts of the two protein complexes and of the published wild type complex share many residues, suggesting the intercomplex interactions are retained in different crystallization conditions. The cell dimensions of these crystals are also quite similar.
TABLE 1.
X-ray diffraction data collection statistics
Rsym = ∑(Ihkl - I<hkl>) / (∑Ihkl).
| W150A/TEM-1 | Y51A/TEM1 | |
|---|---|---|
| Wavelength (Å) | 1,3808 | 1,3808 |
| Angular increment per frame | 1° | 0.5° |
| Total rotation range | 360° | 180° |
| Crystal to detector distance | 75 mm | 125 mm |
| Space group | P21 | P21 |
| Unit cell parameters | ||
| a | 49.09 Å | 48.67 Å |
| b | 129.48 Å | 129.04 Å |
| c | 80.13 Å | 78.90 Å |
| α | 90.00 Å | 90.00 Å |
| β | 91.86° | 91.20° |
| γ | 90.00° | 90.00° |
| Matthews coefficient | 2.69 Å3/Da | 2.65 Å3/Da |
| Solvent content | 53.98% | 53.14% |
| No. molecule/asymmetric unit | 2 | 2 |
| Data reduction | ||
| No. of measured reflections | 50,562 | 54,728 |
| Resolution limits | 32.4-2.20 Å | 31.0-2.07 Å |
| No. of unique reflections (>3σ) | 37,425 | 38,282 |
| Data completeness | 99.7% | 92.60% |
| Redundancy | 7.2 | 3.1 |
| Overall Rsym | 0.099 (0.339) | 0.061 (0.303) |
TABLE 2.
Structural refinement statistics
| W150A/TEM-1 | Y51A/TEM1 | ||
|---|---|---|---|
| No. of reflections (total) | 50,562 | 54,728 | |
| No. of reflections for refinement | 47,954 | 51,713 | |
| No. of reflections for R free | 2566 | 2780 | |
| No. of protein atoms/<B> | 6512/25.4 Å2 | 6516/31.3 Å2 | |
| No. of water | 236 | 172 | |
| <B> for solvent | ∼30 Å2 | ∼28 Å2 | |
| Resolution range | 29.7-2.20 Å | 31.0-2.07 Å | |
| R value for working set | 20.8% | 21.4% | |
| R free (5% reserved) | 23.0 | 24 | |
| Weighted root mean square deviation from ideality | |||
| Bond length | 0.026 Å | 0.012 Å | |
| Bond angle | 1.926° | 1.403° | |
Structural Analysis of the BLIP Y51A-TEM-1 Complex—This crystal has a solvent content of 53.14% (Table 2). The structures of the two complexes that are present in one asymmetric unit have a root mean square deviation value of 0.89 Å for all atoms. This indicates there are some structural differences in these two complexes, and the differences are located mostly in the crystal contacts. A comparison of these complexes and the published wild type complexes indicate root mean square deviation of Cα values between 0.55 and 0.64 Å. The side chain of Gln99 of TEM-1 appears to rearrange to have the amide group pointing toward the protein side rather than the solvent side, which is the case in wild type structure.
The direct intermolecular hydrogen bonds are almost identical for the BLIP Y51A-TEM-1 and wild type complexes (Table 3). The hydrogen bond bridging Gln99 of TEM-1 and His148 of the BLIP Y51A mutant is different from that of the wild type complex (Fig. 2C). The side chain of Gln99 of TEM-1 in the BLIP Y51A-TEM-1 complex assumes a different conformation than that in the wild type complex. In addition, Ser113 of the BLIP Y51A mutant forms a new hydrogen bond to Glu110 of TEM-1 (Ser113 Oγ to Glu110 Oε2). This may reflect the fact that there is a weak hydrogen bond between these two atoms in the wild type complex (2.90 Å and 101°). It was observed that the characteristics of the identified direct intermolecular hydrogen bonds do change but remain within the detection threshold. There are dramatic differences, however, in the water-bridged hydrogen bonds. This can be explained by the fact that the interface in the Y51A-TEM-1 complex has less identified water molecules.
TABLE 3.
Thermodynamic, structural, and kinetic characteristics of BLIP mutant/TEM-1 complexes
| BLIP/TEM-1 | W150A/TEM-1 | Y51A/TEM-1 | |
|---|---|---|---|
| Binding parameters | |||
| K (108m−1)a | 90 ± 10 | 0.02 ± 0.001 | 15.0 ± 1.5 |
| ΔG (kcal/mol)a | −13.8 ± 0.5 | −8.4 ± 0.1 | −12.6 ± 0.5 |
| ΔCp (ITC) (cal/mol/K)a | −667 ± 51 | −275 ± 28 | −809 ± 37 |
| ΔCp (predicted) (cal/mol/K)b | −346 (−379) | −316 (−350) | −337 (−402) |
| THa | 18.8 ± 0.9 °C | 32.4 ± 3.3 °C | 18.9 ± 0.9 °C |
| Intermolecular interactions | |||
| Hydrogen bonds (direct)c | 13 | 12 | 14 |
| (water-bridged)c | 11 (11) | 13 (6) | 6 (2) |
| Salt bridgesd | 3 | 1 | 3 |
| van der Waals contactsd | 313 (324) | 312 (292) | 292 (265) |
| Buried surface areae | 2800 (2715) Å | 2738 (2696) Å | 2761 (2713) Å |
| ΔSASpolar | 46.0% (43.7%) | 47.1% (45.1%) | 46.2% (42.5%) |
| ΔSASapolar | 54.0% (56.3%) | 52.9% (54.9%) | 53.8% (57.5%) |
| Shape complementarity | |||
| SC statisticsf | 0.59 (0.60) | 0.60 (0.60) | 0.62 (0.62) |
| S(T → B) | 0.58 (0.59) | 0.60 (0.60) | 0.59 (0.61) |
| S(B → T) | 0.60 (0.61) | 0.61 (0.61) | 0.64 (0.62) |
| Kinetic rates | |||
| kon (105m−1 s−1)g | 2.0 ± 1.0 | 2.9 ± 1.5 (8.4 ± 4)h | 3.9 ± 1.0 |
| koff (10−4 s−1)i | 1.3 ± 0.7 | 1900 ± 500i (500 ± 300)j | 0.9 ± 0.5 |
These thermodynamic parameters were reported previously (16).
These values are calculated using ΔCp = −0.45 × ΔSASap + 0.26 × ΔSASp (47).
Hydrogen bond distance is ≤3.5 Å, and number in parentheses refers to the second complex in asymmetric units.
van der Waals contacts are ≤4.0 Å, and number in parentheses refers to the second complex in asymmetric units.
Number in parentheses refers to the second complex in asymmetric units.
T is for TEM-1, and B for BLIP mutant. Number in parentheses refers to the second complex in asymmetric units.
Stopped-flow fluorescence spectrometry measurements at ambient temperature (~23 °C).
Activity-based dilution assay fitted with on-rates (in parentheses) and off-rates simultaneously.
Activity-based using inactive TEM-1 S70A mutant displacement assay.
Fluorescence dilution assay fitted with on-rates (in parentheses) and off-rates simultaneously.
FIGURE 2.

Crystallographic analysis of BLIP mutant-TEM-1 complexes. A, selection of four regions of the electron density maps centered on the mutation sites shows the maps match the mutated residues. B, three-dimensional alignment of α-carbon chains of BLIP mutants from three complex structures. Regions that exhibit large conformational changes are circled. C, comparison of the structures around the Gln99 of TEM-1 β-lactamase. The wild type complex is colored red. The Y51A complex is colored blue, showing the Gln99 side chain is positioned differently from that in wild type despite the fact that both complexes have the Gln99 contact with the Trp150 of BLIP, with different hydrogen bond configuration between the Gln99 and His148 of the BLIP. The green represents the W150A mutant complex, showing the Gln99 residue undergoes a rather large conformational change but retains the original hydrogen bond between TEM-1 Gln99 and His148 of BLIP. D, close-up view of the Asp49 region of the BLIPs and the catalytic pocket of TEM-1. It shows the large movement of Asp49 in the W150A complex (green). The hydrogen bonds are completely different between Asp49 of the BLIP W150A mutant and the catalytic pocket of the TEM-1.
The calculated values for the buried surface area of the interaction interface for the two complexes within the same unit cell are from 2761 Å2 (AB complex with 46.2% polar and 53.8% nonpolar) to 2713 Å2 (CD complex with 42.5% polar and 57.5% nonpolar), which are comparable with that of the published wild type BLIP complex with TEM-1 (our calculated values are 2800 Å2 for the AB complex with 46.0% polar and 54.0% nonpolar; and 2713 Å2 for the CD complex with 43.7% polar and 56.3% nonpolar. See Table 3). Thus, the alanine substitution at BLIP Tyr51 does not greatly alter the binding interface between BLIP and TEM-1 β-lactamase.
The BLIP Y51A-TEM-1 complex structure exhibits almost no differences from that of the wild type even at the mutated site. The aromatic rings of the two adjacent tyrosines (Tyr50 and Tyr51) form a perpendicular configuration that optimizes the CH-π interaction. Without this CH-π interaction in the Y51A mutant, the complex still formed and the backbone structures of these residues were almost identical with those in the wild type complex. A possible explanation is that the alanine may allow better intermolecular hydrophobic packing than the tyrosine. This difference may in part explain the almost 200 cal/mol/K change in the binding heat capacity.
Structural Analysis of BLIP W150A-TEM-1 Complex—The W150A-TEM-1 complex structure exhibits some profound differences from the wild type structure at the mutated site (Fig. 2, C and D). Around the interacting region of Trp150 of BLIP, Gln99 from TEM-1 makes contacts with Trp150 of the wild type BLIP. Without this contact, the TEM-1 Gln99 has its β and γ carbons rotated and its amide group lies in a configuration that has been rotated almost 180°, pointing toward BLIP rather than toward the solvent as it does in the wild type complex. The calculated buried surface areas are 2738 Å2 for the AB complex with 47.1% polar and 52.9% nonpolar; and 2696 Å2 for the CD complex with 45.1% polar and 54.9% nonpolar. The loss of Trp150 side chain appears to have dramatic effect on the interaction between Asp49 of BLIP and the catalytic pocket of TEM-1. Asp49 no longer forms hydrogen bonds with Ser130, Ser235, and Arg 243 of the TEM-1 target. This is a surprise considering the distance between Trp150 and Asp49 is more than 25 Å. This shows the long distance effect of the point mutation at Trp150. This observation supports the interpretation that residues such as Trp150 with anti-canonical thermodynamic properties act through energetic coupling between different sites (16). Asp49 of the W150A mutant of BLIP moves more than 4 Å toward residue Asn132 of TEM-1 (from 8.87 Å between the Asn132 Cγ of TEM-1 and Asp49 Cγ of BLIP in the wild type complex to 4.43 Å between Asn132 Cγ of TEM-1 and Asp49 Cγ of the W150A mutant in the W150A-TEM-1 complex). Asp49 of the BLIP W150A mutant forms two hydrogen bonds with Asn132 of TEM-1 in the W150A-TEM-1 complex compared with the four hydrogen bonds distributed among Ser130, Ser235, and Arg243 in the Y51A-TEM-1 and wild type complexes (Table 3 and Fig. 2D). This results in a net loss of two direct intermolecular hydrogen bonds, a fact that is consistent with the binding thermodynamics indicating a much less favorable enthalpy (16). This suggests that the altered Asp49 interactions may result in a loss of enthalpy driving forces.
With regard to the remaining hydrogen bonds, it is surprising that there are more water-bridged intermolecular hydrogen bonds in the BLIP W150A-TEM-1 complex than the Y51A-TEM-1 or the wild type complexes (see Table 3). Furthermore, the extra interface-trapped water molecules contribute to an increased number of water-bridged hydrogen bonds. These water-bridged hydrogen bonds actually result in a lower enthalpic driving force for binding and also disrupt hydrophobic stacking interactions, which is another factor contributing to the anti-canonical nature of the W150A substitution.
Analysis of Trapped Water Molecules—Water molecules were located using the standard crystallographic criteria (B value less than 50, and σ cutoff of 1.5). Fig. 3, A and B, shows the distribution of identified water molecules around the complexes and the locations of these water molecules in the interfaces of the complexes. There are some differences in the interface-trapped water molecules in the two structures within the same asymmetric unit (Table 3). Nevertheless, there are clearly more water molecules trapped in the interface of the BLIP W150A-TEM-1 complex than that of the Y51A-TEM-1 complex. A total of 46 water molecules were identified in the two interfaces of the W150A-TEM-1 complexes, and 33 water molecules in the two interfaces of the Y51A-TEM-1 complex structures. The published wild type BLIP-TEM-1 complex structure contains a total of 69 water molecules in the two interfaces. It is known that the identification and location of water molecules are highly dependent on the crystals (35, 36). The wild type complex crystal was formed in quite different conditions than those used here, and it also exhibited a higher diffraction resolution (15). The BLIP mutant crystals were grown in similar conditions and exhibit similar morphology and symmetry as well as similar diffraction resolution. Therefore, comparison of water molecules between our structures may be more valid. Large variations in location are associated with the peripheral water molecules. When only interface water molecules that have zero solvent-exposed area (with a probe of 1.4 Å) are considered, the wild type complex contains a total of 16 water molecules; the W150A-TEM-1 complex contains 13 water molecules, and the Y51A-TEM-1 complex contains a total of 9 water molecules. When we aligned the Y51A-TEM-1 and W150A-TEM-1 complexes using SUPERPOSE in the CCP4 package and determined the distances between the interface-trapped water molecules from one complex to the other complex, about half (22 of 46 in W150A-TEM-1 and 15 of 33 in Y51A-TEM-1) of the interface-trapped water molecules from one complex are within 1 Å of some interface-trapped water molecules from the other complex, suggesting these water molecules share the same hydration sites.
FIGURE 3.

Analysis of interface-trapped water molecules. A, locations of all the identified water molecules in the crystal structures of Y51A-TEM-1 and of W150A-TEM-1 complexes. The complexes were superposed using SUPERPOSE in the CCP4 package. TEM-1 is represented as orange tubes, and the BLIP mutant is in green schematic representation. Green CPK balls are the identified water molecules from the AB complex of W150A-TEM-1 crystal structure, and green dotted circles represent water molecules from the CD complex. Red CPK balls are the identified water molecules from the AB complex of Y51A-TEM-1 crystal structure, and red dotted circles are the water molecules from the CD complex of Y51A-TEM-1 crystal. B, locations of the identified interfacial water molecules on the surface of the BLIP mutants. The BLIP mutants are represented as molecular surfaces. The interfacial water molecules are represented as red balls (for the AB complex in the asymmetric unit) or as dotted spheres (for the CD complex in the asymmetric unit). The W150A-TEM-1 complex is at left, the wild type complex is at center, and to the right is the Y51A-TEM-1 complex. C, histogram of the identified electron density peaks within the BLIP mutant-TEM-1 interfaces. The electron density peaks with B values less than 50 Å2 are assigned as water.
The identification and assignment of water molecules to electron density peaks are highly variable and subject to many factors. Therefore, we expanded our search and identification of electron density peaks and then assigned them as tentative water molecules to calculate their B values using the CNS program. Fig. 3C shows a histogram of the electron density peaks within the interfaces. The peaks with B values less than 50 are the assigned water molecules, and there are numerous peaks with B values larger than 50. Even if the larger B value peaks are included, the W150A-TEM-1 complex interface contains more electron density peaks than the Y51A-TEM-1 complex interface.
Shape Complementarity Analysis of the Interface—An analysis of shape complementarity of the interface in the complexes of W150A-TEM-1, Y51A-TEM-1, and of wild type BLIP-TEM-1 was performed using the sc.exe program in the CCP4 package (30). The results are tabulated in Table 3. The shape complementary statistics increase from the wild type complex (average SC = 0.591) to the W150A complex (average SC = 0.601) and theY51A complex (average SC = 0.613). Interestingly, the substatistics of (S(BLIP mutant→TEM-1)) show more differences than the substatistics of (S(TEM-1→ BLIP mutant)). In general, the shape complementarity statistics for these complexes are within the range of 0.58 to 0.64, which are considered poor complementarity statistics according to Lawrence and Colman (30).
Distribution of Intermolecular Atom Pairs between BLIP Mutants and TEM-1—To evaluate the packing of the complex interface, we calculated the distribution of intermolecular atom pairs on the TEM-1 surface (see Fig. 4). The distance of an intermolecular atom pair is defined as the distance from a BLIP atom to the nearest TEM-1 atom. This distribution shows that the BLIP Y51A mutant has a 20% higher number of atom pairs at 3.6 Å than the W150A mutant (Fig. 4). This indicates that Y51A BLIP is better packed with TEM-1 than the W150A mutant. This is consistent with the fact that the Y51A mutant is a stronger binder with TEM-1 than is W150A BLIP.
FIGURE 4.

Plot of the number of the selected intermolecular atom pairs located within various distance ranges from the TEM-1 surface versus the distance. Selection criterion is the shortest intermolecular atom pairs for each BLIP atom. The solid thick line with the square symbols indicates the intermolecular atom pair distribution of the Y51A-TEM-1 complex that is 20% denser than W150A-TEM-1 complex (thin line with circle symbols) at ∼3.6 Å.
Kinetics of Association of the Complexes—To better understand if the changes in affinity observed for the mutants are due to either a change in the rate of association or dissociation or both, the binding kinetics of formation of these complexes were determined using a stopped-flow fluorescence spectrometer. Fig. 5 shows the time courses of the change of the intrinsic fluorescence of the interacting proteins. In these experiments, equal concentrations of both interacting proteins were used. At the concentration of 5 μm, the association was treated as irreversible. The second order kinetics between TEM-1 and the BLIP mutants can be described by Equation 4,
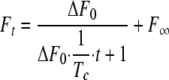 |
(Eq.4) |
where ΔF0 is the amplitude of the changes of intrinsic fluorescence; Ft is the intrinsic fluorescence signal at time t, and ΔF∞ is the intrinsic fluorescence after the association is complete. The binding of these two proteins causes a quench of the intrinsic fluorescence (see the left panels in Fig. 5. λex = 275 nm, λem = 335 nm). The changes in intrinsic fluorescence are proportional to changes in the amount of free unbound proteins because the fluorescence signals are proportional to the protein concentrations to at least 5 μm in our experimental conditions (data not shown). Tc is the time parameter extracted from fitting the above equation to the data. The association rate constant kon is calculated from Equation 5,
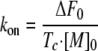 |
(Eq.5) |
where [M]0 is the initial concentration of the unbound proteins. The association rates for all three complexes are quite similar in the tested experimental conditions (see Fig. 5, 50 mm phosphate, pH 7.0, 150 mm NaCl, temperature is ∼23 °C). The on-rate values differ only 1-2-fold (2.9 × 105 m-1 s-1 for W150A-TEM-1, 3.9 × 105 m-1 s-1 for Y51A-TEM-1, and 2.0 × 105 m-1 s-1 for the wild type complex; see Table 3). These results show that the changes in binding thermodynamics are due to changes in the dissociation rate rather than the association rate.
FIGURE 5.

Intrinsic fluorescence signals from the proteins and complexes. Time course traces of the intrinsic fluorescence signals that accompany the association of the BLIP mutants and TEM-1 β-lactamase from stopped-flow spectrometry measurements are shown in the right panels (signals are shown as percent of the average of the equilibrium fluorescence signals). Fluorescence emission spectra are shown in the left panels with the signals normalized to the maximum of the TEM-1 fluorescence peaks. Spectra are labeled with each individual protein name for the corresponding spectra; “complex” is for the spectra of complex solution, and the “sum” labels the arithmetic sum of the two individual protein spectra. At top is the association of the W150A BLIP mutant and TEM-1 fitted with second order kinetics (red line) with a kon of 2.9 × 105 m-1 s-1. The middle graph is from the association of the Y51A BLIP mutant and TEM-1 fitted with second order kinetics (red line) with a kon value of 3.9 × 105 m-1 s-1. At bottom is the association of the W150A BLIP mutant and TEM-1 fitted with second order kinetics (red line) with kon value of 2.0 × 105 m-1 s-1.
Enzymatic Activity-based Dissociation Measurement—To confirm the above kinetic assessment, measurements of the time course of activity recovery were performed to determine the dissociation kinetics. For the weak complex BLIP W150A-TEM-1, a high concentration of complex was diluted by 7.3-fold into substrate solution, and the hydrolysis of the cephalosporin C substrate was monitored. The rationale is that dilution of the complex will favor the equilibrium to the free, unbound active TEM-1 enzyme and the BLIP W150A mutant. Because of the relatively weak affinity of the W150A-TEM-1 complex, the dilution scheme works well. Fig. 6 shows that the β-lactamase activity increases after the dilution in a time-dependent pattern. This time dependence appears to follow the binary binding kinetics mentioned under “Experimental Procedures.” The data were fitted with reversible dissociation Equation 6 as follows (see Fig. 6),
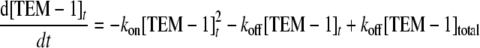 |
(Eq.6) |
where [TEM-1]t is the active free TEM-1 enzyme, [TEM-1]total is the total concentration of TEM-1, and kon and koff are the association and dissociation rate constants of the BLIP W150A-TEM-1 binding. This equation is only valid when both BLIP W150A and TEM-1 have the same concentration. Equations 7 and 8 are analytical solutions of this equation derived by symbolic computation
FIGURE 6.

Measurements of dissociation rates of the BLIP mutant-TEM-1 β-lactamase complexes. A, plot of the time course of cephalosporin C hydrolysis by TEM-1 after a 7.3-fold dilution of 10 μm of preformed W150A-TEM-1 complex (circles). The curve is a fit of the on- and off-rates simultaneously with an on-rate of 8.4 ± 4 × 105 m-1 s-1 and an off-rate of 0.19 ± 0.05 s-1. B, plot of the time course of intrinsic fluorescence after a 37-fold dilution of 3 μm of preformed W150A-TEM-1 complex (cross). The curve is a fit of the on- and off-rates simultaneously with an on-rate of 3.4 ± 2 × 105 m-1 s-1 and an off-rate of 0.05 ± 0.03 s-1. C, plot of the time course of cephalosporin C hydrolysis activity that is reflective of the fractional amount of TEM-1 after dilution and mixing of the preformed Y51A-TEM-1 complex into 10 μm of inactive S70A TEM-1 mutant. The curve is a first order kinetics fit with koff = 0.00009/s. D, plot of the time course of cephalosporin C hydrolysis by TEM-1 after dilution and mixing of the preformed wild type complex into 10 μm of inactive S70A TEM-1 mutant. The curve is a first order kinetics with koff = 0.00013/s.
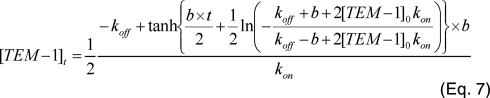
using the program Maple 8 (Waterloo Maple Inc. Waterloo, Ontario, Canada), where b is calculated as follows:
 |
(Eq.8) |
Curve fitting yields a dissociation constant of 0.19 ± 0.1 s-1, an association of 1.2 ± 0.3 × 106 m-1 s-1, [TEM-1]0 = 53 nm, and [TEM-1]total = 9400 nm. Both [TEM-1]0 and [TEM-1]total values match the experimental setup. This association constant is close to that determined by the stopped-flow measurement.
In addition to following complex dissociation with the enzymatic activity-based assay, we also carried out measurements of the fluorescence signals after dilution of the preformed TEM-1/W150A complexes as described under “Experimental Procedures.” To optimize the signal to noise ratio, we selected a dilution factor and concentration that would yield a reasonable signal and averaged over 50 traces to increase the signal to noise ratio. Fig. 6B shows the averaged time course for recovery of intrinsic fluorescence signals after the 37-fold dilution. Fitting the data to Equations 7 and 8 with the initial concentration of free proteins of 9 nm and total concentration of 82 nm resulted in best-fit parameters for koff of 0.05 ± 0.03 s-1 and kon of 3.4 ± 2 × 105 m-1 s-1. The values are consistent with those measured from the stopped-flow and the enzymatic activity-based measurements.
To measure the dissociation constant of the tight BLIP Y51A-TEM-1 complex, the protein pair was diluted into a solution of 20 μm of the TEM-1 mutant S70A. The TEM-1 S70A mutant has a cephalosporin C hydrolysis rate of kcat of 0.02 s-1, a value 500-fold less than that of the wild type TEM-1 (kcat of ∼11 s-1). This hydrolysis rate represents less than 10% of total activity being monitored at the concentration of the mutant used in the assay. At designated time points after the mixing of the complex and S70A TEM-1 mutant, aliquots of the mixture were tested in the enzymatic activity assay. Because of the over-whelming amount of S70A TEM-1 enzyme compared with the wild type TEM-1, the appearance of the active TEM-1 enzyme from the complex is essentially first order kinetics as described by Equation 9,
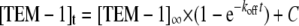 |
(Eq.9) |
where [TEM-1]t is the amount of free TEM-1 enzyme estimated by the enzymatic activity assay; [TEM-1]∞ is the maximum amount of active enzyme in the experiment; koff is the dissociation constant, and C is a fitting constant representing the background hydrolysis rate (including the TEM-1 mutant S70A activity).
Fig. 6C shows the time course of recovered β-lactamase activity from the BLIP Y51A-TEM-1 complex after mixing with the inactive TEM-1 S70A mutant. The dissociation constant for the Y51A-TEM-1 complex was determined from the time course to be 9 × 10-5 s-1. Using the same approach, the dissociation constant for the wild type BLIP-TEM-1 complex was measured at 1.3 × 10-4 s-1 (Fig. 6D). Both association constant and dissociation constants yield similar equilibrium constants as the ITC measurements previously reported (Kd of 1.5 × 10 9 m-1 for the wild type and 4.3 × 10 8 m-1 for Y51A complexes), thus confirming the kinetic measurements are valid (16, 17, 23). Both the association rate and dissociation rate determinations indicate that the three complexes studied exhibit changes mainly in the dissociation step.
DISCUSSION
Structural, Kinetic, and Thermodynamic Consequences of the BLIP Substitutions—One of the main issues addressed here is how single amino acid substitutions alter binding affinity. The structures of the two mutant complexes presented argue the following: 1) structural matching (complementarity), 2) alteration of the contact interactions, and 3) water molecules trapped in the interface are affected by the substitutions examined. These three issues are closely related. In the structure of the BLIP Y51A-TEM-1 complex, the interface appears to be reasonably complementary between the BLIP mutant and TEM-1. The calculated shape complementarity statistics indicate a better shape fit for Y51A-TEM-1 than for both the W150A-TEM-1 and wild type complexes. In addition, the intermolecular atom pair distribution on the TEM-1 contact surface shows that the BLIP Y51A mutant packs more densely than the W150A mutant (Fig. 4 and Table 3). Analysis of the extensive information (both structural and thermodynamic) of these two complexes reveals correlations between the structural details and thermodynamics, which provides insight into the interactions. The results also demonstrate the complexity of protein-protein interactions, which require large amounts of detailed information to describe.
Anti-canonical Nature of the BLIP Trp150 Residue—Previously, we identified Trp150 as an anti-canonical residue. An anti-canonical residue is one whose alanine substitution results in a change in the hydrophobic contribution to binding (indicated by changes in the binding isoenthalpic temperature) that does not match with the hydrophobicity change resulting from the mutation (16). An alanine substitution of the tryptophan at position 150 of BLIP should decrease the hydrophobicity around position 150 of BLIP. However, the W150A substitution increases the TEM-1 binding isoenthalpic temperature, indicating an increase in the hydrophobic nature of binding. The x-ray structure of the W150A-TEM-1 complex explains this thermodynamic result. The alanine substitution of Trp150 of BLIP introduces a number of geometric shape defects in the interface of the two proteins, which results in rearranged interactions between Asp49 of the BLIP mutant and the TEM-1 β-lactamase catalytic pocket. The rearrangement results in the net loss of two hydrogen bonds (Fig. 2D) and the loss of about 1% of the nonpolar interface (Table 3) compared with the Y51A-TEM-1 complex. This net loss of two hydrogen bonds, which are hydrophilic interactions, explains why BLIP W150A-TEM-1 binding is more driven by hydrophobic forces (a higher isoenthalpic temperature (16)), thus explaining the anti-canonical nature of Trp150. This net loss of two hydrogen bonds may also result in the increased rate of dissociation of the two proteins (Figs. 5 and 6). Because Asp49 is located within a BLIP binding hotspot and is more than 25 Å away from residue Trp150, which is locating within the other binding hotspot, the above observation shows there is a strong energetic coupling between these two hotspots. The binding thermodynamics of the W150A-TEM-1 interaction (TH = 32.4 °C, ΔCp = -275 cal/mol/K, and ΔG = -8.5 kcal/mol) indicates the W150A mutant loses both hydrophobic and “hydrophilic” driving forces as shown in Fig. 1B. The higher isoenthalpic temperature (TH = 32.4 °C) shows that the loss of hydrophilic driving forces is disproportionally larger, which can be attributed to the loss of two hydrogen bonds. The structural information augments the extensive thermodynamic information, which not only provides insights on the nature of the driving forces but also predicts an energetic coupling with some distal sites based on the anti-canonical nature (16).
Interface-trapped Water Molecules—The number of the interface-trapped water molecules appears to follow an inverse trend relative to the experimentally determined binding heat capacity changes in that more negative binding heat capacity changes are associated with fewer interface-trapped water molecules (Fig. 3).
It is known that the identification and localization of water molecules by protein x-ray crystallography is affected by many factors, including the crystallization conditions and resolution of the x-ray diffraction (35, 36). One reason for this is that interface-trapped water molecules are highly mobile. The structures solved in this study exhibit variation in the number of interface-trapped water molecules in the two complexes of the same asymmetry unit. The structures presented here are from similar crystallization conditions and x-ray diffraction resolution, and therefore comparison of the water molecules between these two complex structures is quite informative. When the electron density peaks search was expanded, the number of the identified peaks did increase but exhibited higher B values (Fig. 3B). Even with the higher B value peaks, the W150A-TEM-1 interface contains more electron density peaks than that of the Y51A-TEM-1 complex. This shows that the difference in the numbers of the interface-trapped water molecules is unlikely to be due to experimental artifacts.
Correlation with the Difference in ΔCp for These Two Mutant Complexes—A comparative analysis of these two mutant complexes suggests the sources of the difference of the binding heat capacity changes of the complexes. It is generally accepted that the binding ΔCp is mainly due to the changes of solvent-exposed area of the interacting proteins before and after binding, which in turn changes the hydration states. Because the interprotein interactions in protein-protein binding are similar to the intraprotein interactions in the folding of a protein, it was proposed that the empirical equation for predicting the heat capacity changes of protein unfolding be applied to protein-protein association (37, 38). This equation calculates the heat capacity changes based on the changes in polar and nonpolar solvent-accessible surface areas between the initial and final states, with polar residues contributing to a positive value and nonpolar to a negative value. This equation is mainly based on the assumption that the major contribution to ΔCp in a protein-protein association is the interacting interface, which releases a large number of hydration water molecules when the interacting interface is covered up during binding. This equation predicts the binding heat capacity changes reasonably well for several antibody-antigen binding complexes (39-41) and small protein binding interactions (42, 43). However, this equation tends to underestimate the magnitude of the large negative binding heat capacity changes of many other protein-protein associations, especially for the binding interactions of large proteins. For instance, barnase-barstar binding has a ΔCp of about -300 cal/K/mol, whereas the predicted value is close to zero (44). The binding of point mutants of human growth hormone to human growth hormone receptor have binding heat capacity changes that range from -600 cal/K/mol to -800 cal/K/mol (45), and the binding of multisite mutation mutants of human growth hormone and the receptor have an even wider range, -600 to -1000 cal/K/mol (46). These interactions are predicted to have binding heat capacity changes around -300 cal/K/mol by the empirical equation. The binding of BLIP mutants to TEM-1 exhibit binding heat capacity changes that range from -275 to -809 cal/K/mol, whereas the predicted values are around -300 cal/K/mol (16). Literature surveys have shown that many protein-protein binding systems do not match the proposed empirical equation prediction, although some systems do (47, 48). The structural information presented here illustrates that similar interacting interfaces may not exhibit similar patterns of hydration. One of the reasons is the residual hydration in an interface. This residual hydration, or interface-trapped water molecules, could be a significant factor in determining the ΔCp. This is because the trapped water molecules are most likely in the hydration water states when the proteins are unbound. These hydration water molecules are in a thermodynamic state different from that of bulk water molecules. The water molecules that change from the hydration state to the bulk solution state will change the thermodynamic parameters, such as heat capacity, of the system. The larger the number of water molecules that change state, the more dramatic change in the heat capacity (in this case, the more negative the binding heat capacity change will be). Therefore, having more hydration water molecules trapped in the interface leads to fewer water molecules changing their states and results in less negative binding heat capacity changes. However, our results do not exclude other potential contributions to the ΔCp. One such potential contribution is from changes in hydration states outside of the binding interface.
Relationships between Trapped Water and Binding Free Energy—Based on our results that the interface of the weaker complex traps more water molecules than the stronger complex, we argue that increases in interface-trapped water molecules tend to weaken interactions. One reason for this is that water is required to fill in gaps in the interface that occur because the geometric complementarity of the contact surfaces is not precise. The difference in number of waters between complexes is consistent with the fact that the two structures exhibit differences in their calculated shape complementarity statistics (Table 3). By this view, defects in shape geometric complementarity result in trapping more water molecules, which then correlates with less negative binding heat capacity changes. Therefore, less negative binding heat capacity changes indicate larger defects in shape complementarity and more disruption of direct intermolecular interactions such as hydrogen bonds. This explanation agrees well with the observed results for the weaker complex of BLIP W150A-TEM-1. It may be a general rule that an increase in the interface-trapped water molecules tends to diminish the binding strength. A survey study shows that the weak interfaces such as crystal contacts have 50% more interface-trapped water molecules per unit area than the stronger, specific protein-protein binding interfaces (36). In the strong complex of barnase-barstar, the interface traps a large number of water molecules. This is because the binding is dominated by electrostatic interactions, which tend to associate with the highly polarizable water molecules, some of which do show bridging interactions between barnase and barstar (49). Mutations that affect the electrostatic interactions also affect the interface-trapped water molecules. It is very difficult to separate the contributions of direct electrostatic interactions and water-bridged interactions. It is interesting to note that the complex of barnase with the D35A barstar mutant trapped an extra water molecule within the interface cavity created by the alanine substitution, and this extra water molecule destabilized the complex by about 0.5 kcal (50). In the complex of the anti-lysozyme antibody D1.3 and lysozyme, mutation of VL92 tryptophan creates an interface cavity that traps a number of water molecules (51). It was shown that there is a strong correlation between the loss of hydrophobic interactions and the loss of the strength of binding. This suggests that the interface-trapped water molecules actually destabilize the complex. This argues that the interface trapped water molecules could attenuate strong intermolecular hydrophobic interactions by replacement with significantly weaker water-bridge interactions. For the non-hotspot residues in the complex of BLIP-TEM-1, double mutations can eliminate the water-bridge interactions and show little effect on the binding strength (20). These observations provide further indication that water-bridge interactions make only limited contributions to the binding.
Given the large differences in ΔCp values among the point alanine substitution mutant complexes, the changes in the hydration states of the interface may not be sufficient in explaining the differences, even to a first order of approximation. Based on Sturtevant's proposal, there are many other possible contributions to ΔCp such as the hydrophobic effect (hydration-structured water around nonpolar groups), hydrogen bonds, electrostatic interactions, conformational entropy, and intramolecular vibrations (52). It has also been proposed that additional linked equilibria could contribute to ΔCp (53, 54), and hydration states outside of the interface could also contribute (55). The results presented here point to the interface-trapped water molecules as an important factor but do not rule other contributions. Future experiments with more complexes and different approaches are needed to confirm and assess other potential contributions to ΔCp.
Forces Driving Binding, Specificity and Cross-reactivity—Both of the mutant complexes as well as the wild type complex contain at least 13 direct intermolecular hydrogen bonds, a high number compared with many other protein-protein interfaces (56). These observations argue that the hydrogen bonds are a major source of the binding driving forces. The high number of hydrogen bonds may be necessary to overcome the moderate to poor shape complementarity (calculated shape complementarity statistics of about 0.6 compared with the good shape complementarity statistics of 0.76, see Lawrence and Colman (30)). Based on the assumption that high levels of shape complementarity lead to high selectivity, the relatively poor shape complementarity of BLIP and β-lactamase may lead to a broad selectivity and allow BLIP to bind to many class A β-lactamases. Consistent with this reasoning is the fact that BLIP can bind to many β-lactamases (18).
Tight Energetic Coupling between the Two Hot Spots and Implications for Inhibitor Design—The hotspot contact residues of BLIP are located in two regions of the binding surface: one is in the area of loop 1 (Asp49, Phe36, His41, and Tyr53), and the other in the middle of the concave interaction surface (Lys74, Trp112, Phe142, His148, Trp150, and Arg160). The crystal structure of BLIP W150A-TEM-1 indicates there is a strong energetic coupling between Trp150 and Asp49. The coupling may extend from loop 1 to other hotspot residues around Trp150. The hotspot residues around Trp150 (Lys74, Trp112, His148, Trp150, and Arg160) form two hydrogen bonds (BLIP Arg160 NH1 to TEM-1 Asn100 O, and BLIP His148 Nε2 to TEM-1 Gln99 Nε2). In addition, there are several TEM-1 surface residues that form a hydrogen bond network with BLIP in this region (TEM-1 Glu104 Oε1 to BLIP Tyr143 N; TEM-1 Tyr105 N to BLIP Glu73 Oε1; TEM-1 Tyr105 N to BLIP Glu73 Oε2; TEM-1 Tyr105 OH to BLIP Gly141 O; TEM-1 Ser106 N to BLIP Glu73 Oε1; TEM-1 Glu110 Oε1 to BLIP Ser71 Oγ; TEM-1 Glu110 Oε2 to BLIP Ser71 Oγ). These hydrogen bond-forming surface residues of TEM-1 could be an attractive target for inhibitor design. One possibility is to design an anchoring molecular moiety that links with previously identified inhibitor peptides (57-60). Increased understanding of the determinants of binding energy in the BLIP-β-lactamase interface may facilitate the design of other new inhibitors against this class of β-lactamases.
Acknowledgments
We thank Drs. Henry Bellamy and David Neau for their assistance in data collection and processing.
The atomic coordinates and structure factors (codes 3c7u and 3c7v) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by a Scientist Development Grant 0435186N from the American Heart Association and startup funding from University of Houston and the Institute for Molecular Design (to D. C.). This work was also supported in part by National Institutes of Health Grant AI32956 (to T. P.). Data used in this publication were collected at the Gulf Coast Protein Crystallography Beamline at the Center for Advanced Microstructures and Devices. This beamline is supported by the National Science Foundation Grant DBI-9871464 with co-funding from the National Institutes of Health (NIGMS). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: BLIP, β-lactamase inhibitory protein; PEG, polyethylene glycol; MES, 4-morpholineethanesulfonic acid.
References
- 1.Janin, J., Rodier, F., Chakrabarti, P., and Bahadur, R. P. (2007) Acta Crystallogr. Sect. D Biol. Crystallogr. 63 1-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janin, J. (2008) Science 319 165-166 [DOI] [PubMed] [Google Scholar]
- 3.Nooren, I. M., and Thornton, J. M. (2003) EMBO J. 22 3486-3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fletcher, S., and Hamilton, A. D. (2007) Curr. Top. Med. Chem. 7 922-927 [DOI] [PubMed] [Google Scholar]
- 5.Grueninger, D., Treiber, N., Ziegler, M. O., Koetter, J. W., Schulze, M. S., and Schulz, G. E. (2008) Science 319 206-209 [DOI] [PubMed] [Google Scholar]
- 6.Thanos, C. D., DeLano, W. L., and Wells, J. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 15422-15427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitty, A., and Kumaravel, G. (2006) Nat. Chem. Biol. 2 112-118 [DOI] [PubMed] [Google Scholar]
- 8.Chothia, C., and Janin, J. (1975) Nature 256 705-708 [DOI] [PubMed] [Google Scholar]
- 9.Janin, J. (1995) Biochimie (Paris) 77 497-505 [DOI] [PubMed] [Google Scholar]
- 10.Clackson, T., and Wells, J. A. (1995) Science 267 383-386 [DOI] [PubMed] [Google Scholar]
- 11.Wells, J. A. (1991) Methods Enzymol. 202 390-411 [DOI] [PubMed] [Google Scholar]
- 12.Bush, K., Jacoby, G. A., and Medeiros, A. A. (1995) Antimicrob. Agents Chemother. 39 1211-1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majiduddin, F. K., Materon, I. C., and Palzkill, T. G. (2002) Int. J. Med. Microbiol. 292 127-137 [DOI] [PubMed] [Google Scholar]
- 14.Strynadka, N. C., Jensen, S. E., Johns, K., Blanchard, H., Page, M., Matagne, A., Frere, J. M., and James, M. N. (1994) Nature 368 657-660 [DOI] [PubMed] [Google Scholar]
- 15.Strynadka, N. C., Jensen, S. E., Alzari, P. M., and James, M. N. (1996) Nat. Struct. Biol. 3 290-297 [DOI] [PubMed] [Google Scholar]
- 16.Wang, J., Zhang, Z., Palzkill, T., and Chow, D. C. (2007) J. Biol. Chem. 282 17676-17684 [DOI] [PubMed] [Google Scholar]
- 17.Zhang, Z., and Palzkill, T. (2003) J. Biol. Chem. 278 45706-45712 [DOI] [PubMed] [Google Scholar]
- 18.Zhang, Z., and Palzkill, T. (2004) J. Biol. Chem. 279 42860-42866 [DOI] [PubMed] [Google Scholar]
- 19.Reichmann, D., Cohen, M., Abramovich, R., Dym, O., Lim, D., Strynadka, N. C., and Schreiber, G. (2007) J. Mol. Biol. 365 663-679 [DOI] [PubMed] [Google Scholar]
- 20.Reichmann, D., Phillip, Y., Carmi, A., and Schreiber, G. (2008) Biochemistry 47 1051-1060 [DOI] [PubMed] [Google Scholar]
- 21.Reichmann, D., Rahat, O., Albeck, S., Meged, R., Dym, O., and Schreiber, G. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 57-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reynolds, K. A., Thomson, J. M., Corbett, K. D., Bethel, C. R., Berger, J. M., Kirsch, J. F., Bonomo, R. A., and Handel, T. M. (2006) J. Biol. Chem. 281 26745-26753 [DOI] [PubMed] [Google Scholar]
- 23.Albeck, S., and Schreiber, G. (1999) Biochemistry 38 11-21 [DOI] [PubMed] [Google Scholar]
- 24.Selzer, T., Albeck, S., and Schreiber, G. (2000) Nat. Struct. Biol. 7 537-541 [DOI] [PubMed] [Google Scholar]
- 25.Cantu, C., III, Huang, W., and Palzkill, T. (1997) J. Biol. Chem. 272 29144-29150 [DOI] [PubMed] [Google Scholar]
- 26.Otwinowski, Z., and W. Minor. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 27.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 28.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 29.Collaborative Computational Project Number 4 (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 30.Lawrence, M. C., and Colman, P. M. (1993) J. Mol. Biol. 234 946-950 [DOI] [PubMed] [Google Scholar]
- 31.Guex, N., and Peitsch, M. C. (1997) Electrophoresis 18 2714-2723 [DOI] [PubMed] [Google Scholar]
- 32.Humphrey, W., Dalke, A., and Schulten, K. (1996) J. Mol. Graphics 14 33-38 [DOI] [PubMed] [Google Scholar]
- 33.Potterton, E., Briggs, P., Turkenburg, M., and Dodson, E. (2003) Acta Crystallogr. Sect. D Biol. Crystallogr. 59 1131-1137 [DOI] [PubMed] [Google Scholar]
- 34.Baldwin, R. L. (1986) Proc. Natl. Acad. Sci. U. S. A. 83 8069-8072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dauter, Z. (2003) Methods Enzymol. 368 288-337 [DOI] [PubMed] [Google Scholar]
- 36.Rodier, F., Bahadur, R. P., Chakrabarti, P., and Janin, J. (2005) Proteins 60 36-45 [DOI] [PubMed] [Google Scholar]
- 37.Murphy, K. P., Xie, D., Garcia, K. C., Amzel, L. M., and Freire, E. (1993) Proteins 15 113-120 [DOI] [PubMed] [Google Scholar]
- 38.Murphy, K. P., and Freire, E. (1992) Adv. Protein Chem. 43 313-361 [DOI] [PubMed] [Google Scholar]
- 39.Hibbits, K. A., Gill, D. S., and Willson, R. C. (1994) Biochemistry 33 3584-3590 [DOI] [PubMed] [Google Scholar]
- 40.Schwarz, F. P., Tello, D., Goldbaum, F. A., Mariuzza, R. A., and Poljak, R. J. (1995) Eur. J. Biochem. 228 388-394 [PubMed] [Google Scholar]
- 41.Tsumoto, K., Ueda, Y., Maenaka, K., Watanabe, K., Ogasahara, K., Yutani, K., and Kumagai, I. (1994) J. Biol. Chem. 269 28777-28782 [PubMed] [Google Scholar]
- 42.Gomez, J., and Freire, E. (1995) J. Mol. Biol. 252 337-350 [DOI] [PubMed] [Google Scholar]
- 43.Baker, B. M., and Murphy, K. P. (1997) J. Mol. Biol. 268 557-569 [DOI] [PubMed] [Google Scholar]
- 44.Frisch, C., Schreiber, G., Johnson, C. M., and Fersht, A. R. (1997) J. Mol. Biol. 267 696-706 [DOI] [PubMed] [Google Scholar]
- 45.Pearce, K. H., Jr., Ultsch, M. H., Kelley, R. F., de Vos, A. M., and Wells, J. A. (1996) Biochemistry 35 10300-10307 [DOI] [PubMed] [Google Scholar]
- 46.Kouadio, J. L., Horn, J. R., Pal, G., and Kossiakoff, A. A. (2005) J. Biol. Chem. 280 25524-25532 [DOI] [PubMed] [Google Scholar]
- 47.Perozzo, R., Folkers, G., and Scapozza, L. (2004) J. Recept. Signal. Transduct. Res. 24 1-52 [DOI] [PubMed] [Google Scholar]
- 48.Stites, W. E. (1997) Chem. Rev. 97 1233-1250 [DOI] [PubMed] [Google Scholar]
- 49.Buckle, A. M., Schreiber, G., and Fersht, A. R. (1994) Biochemistry 33 8878-8889 [DOI] [PubMed] [Google Scholar]
- 50.Ikura, T., Urakubo, Y., and Ito, N. (2004) Chemical Physics 307 111-119 [Google Scholar]
- 51.Sundberg, E. J., Urrutia, M., Braden, B. C., Isern, J., Tsuchiya, D., Fields, B. A., Malchiodi, E. L., Tormo, J., Schwarz, F. P., and Mariuzza, R. A. (2000) Biochemistry 39 15375-15387 [DOI] [PubMed] [Google Scholar]
- 52.Sturtevant, J. M. (1977) Proc. Natl. Acad. Sci. U. S. A. 74 2236-2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horn, J. R., Brandts, J. F., and Murphy, K. P. (2002) Biochemistry 41 7501-7507 [DOI] [PubMed] [Google Scholar]
- 54.Horn, J. R., Russell, D., Lewis, E. A., and Murphy, K. P. (2001) Biochemistry 40 1774-1778 [DOI] [PubMed] [Google Scholar]
- 55.Ladbury, J. E., and Williams, M. A. (2004) Curr. Opin. Struct. Biol. 14 562-569 [DOI] [PubMed] [Google Scholar]
- 56.Xu, D., Tsai, C. J., and Nussinov, R. (1997) Protein Eng. 10 999-1012 [DOI] [PubMed] [Google Scholar]
- 57.Huang, W., Beharry, Z., Zhang, Z., and Palzkill, T. (2003) Protein Eng. 16 853-860 [DOI] [PubMed] [Google Scholar]
- 58.Rudgers, G. W., and Palzkill, T. (2001) Protein Eng. 14 487-492 [DOI] [PubMed] [Google Scholar]
- 59.Rudgers, G. W., Huang, W., and Palzkill, T. (2001) Antimicrob. Agents Chemother. 45 3279-3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang, W., Zhang, Z., and Palzkill, T. (2000) J. Biol. Chem. 275 14964-14968 [DOI] [PubMed] [Google Scholar]