

Abstract
N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) lesions of the locus coeruleus (LC), the major brain noradrenergic nucleus, exacerbate the damage to nigrostriatal dopamine (DA) terminals caused by the psychostimulant methamphetamine (METH). However, because noradrenergic terminals contain other neuromodulators and the noradrenaline (NA) transporter, which may act as a neuroprotective buffer, it was unclear whether this enhancement of METH neurotoxicity was caused by the loss of noradrenergic innervation or the loss of NA itself. We addressed the specific role of NA by comparing the effects of METH in mice with noradrenergic lesions (DSP-4) and those with intact noradrenergic terminals but specifically lacking NA (genetic or acute pharmacological blockade of the NA biosynthetic enzyme dopamine β-hydroxylase; DBH). We found that genetic deletion of DBH (DBH −/− mice) and acute treatment of wild-type mice with a DBH inhibitor (fusaric acid) recapitulated the effects of DSP-4 lesions on METH responses. All three methods of NA depletion enhanced striatal DA release, extracellular oxidative stress (as measured by in vivo microdialysis of DA and 2,3-dihydroxybenzoic acid), and behavioural stereotypies following repeated METH administration. These effects accompanied a worsening of the striatal DA neuron terminal damage and ultrastructural changes to medium spiny neurons. We conclude that NA itself is neuroprotective and plays a fundamental role in the sensitivity of striatal DA terminals to the neurochemical, behavioural, and neurotoxic effects of METH.
Keywords: Methamphetamine, Noradrenaline, Dopamine, Parkinsonism, Drugs of Abuse, Locus coeruleus
Introduction
Methamphetamine (METH) is a psychostimulant drug that is widely abused and causes long-term neurotoxicity (Seiden et al., 1976; Kogan et al., 1976; Hotchkiss and Gibb, 1980; Ricaurte et al., 1980). Neurotoxicity occurs in rodents (Schmidt et al., 1985; Sonsalla et al., 1985; Sonsalla and Heikkila 1988; Sonsalla et al., 1989; O'Callaghan and Miller, 1994; Albers and Sonsalla, 1995; Fornai et al., 2004), as well as non-human primates (Seiden et al. 1976; Villemagne et al. 1998), and humans (Wilson et al., 1996), leading to partially reversible dopamine (DA) depletion (Volkow et al., 2001a), which is greater in the caudate than the putamen nucleus (Wilson et al., 1996; McCann et al., 1998; Volkow et al., 2001a; Volkow et al., 2001b; Fitzmaurice et al., 2006).
The noradrenaline (NA) system is generally neuroprotective and influences the vulnerability of nigrostriatal DA neurons as shown by various neurotoxic models like 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP, Mavridis et al., 1991; Marien et al., 1993; Bing et al., 1994; Fornai et al., 1995; Fornai et al., 1997a; Gesi et al., 2000) and 6-hydroxydopamine (Srinivasan and Schmidt, 2003, 2004). Lesions of NA axons arising from the locus coeruleus (LC) exacerbate the damage to DA neurons produced by METH in mice (Fornai et al., 1995; Fornai et al., 1999). However, because these experiments were performed within the context of damaged NA terminals, it is unclear whether the loss of NA terminals or NA itself is deleterious. It has been suggested that the plasma membrane NA transporter (NET), which is expressed by NA neurons and can take up METH, can act as a “buffer” by sequestering toxins such as METH (Han and Gu, 2006) and MPTP away from DA neurons (Rommelfanger et al., 2004). In addition, damage to NA neurons likely causes changes in the release of potential neuroprotective noradrenergic co-transmitters such as neuropeptide Y and galanin (Yoshihara et al., 1996). Thus, a specific role for endogenous NA itself remains questionable.
In addition to the influence on METH-induced neurotoxicity, a prominent role for NA in the rewarding and addictive properties of DA-releasing drugs has also been described (Weinshenker and Schroeder, 2007). For example, we found that mice specifically lacking NA are hypersensitive to the behavioural effects of cocaine, but responses to METH were not tested (Weinshenker et al., 2002; Schank et al., 2006).
In the present study we compared the neurochemical, behavioural, and neurotoxic effects induced by different doses of METH in intact wild-type mice, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) treated mice with damaged noradrenergic terminals, dopamine β-hydroxylase knockout (DBH −/−) mice that have structurally intact noradrenergic terminals but specifically lack NA (Thomas et al., 1995; 1998), and in mice treated with the DBH inhibitor fusaric acid. Specifically, we examined METH-induced DA release, METH-induced behavioural effects, METH-induced oxidation and neurotoxicity as well as alterations of ultrastructural morphology. The results of the present study enhance our understanding of the neuroprotective interaction between NA and the nigrostriatal DA system, as well as the role of endogenous NA in the neurochemical and behavioural effects of addictive DA-releasing agents.
Materials and Methods
Animals
Male C57Bl mice (8 months old, N = 50) were purchased from Charles River (Calco, Italy). Male DBH −/− mice (8−9 months old, N= 20), maintained on a mixed 129/SvEv and C57Bl6/J background, were developed and generated at Emory University (Atlanta, GA, USA) as previously described (Thomas et al., 1995; 1998). Because DBH is required for NA synthesis, DBH −/− mice completely lack NA. Heterozygous (DBH +/−) mice have normal brain NA levels and are indistinguishable from wild-type (DBH +/+) littermates for all previously tested phenotypes, including MPTP toxicity (e.g. Thomas and Palmiter, 1997; Thomas et al., 1998; Szot et al., 1999; Bourdelat-Parks et al., 2005; our unpublished data) and were therefore used as controls for most experiments. As METH toxicity is sensitive to several environmental parameters, mice were housed in stable and controlled conditions, consisting of constant room temperature (21°C), number of mice per cage (N= 5) and size of the cage (15 × 30 × 15 cm). Mice were handled in accordance with the Policy on Humane care and Use of Laboratory Animals of the United States Public Health Service. All the experiments were approved by the Local Ethical Committee for Animal Studies.
Experimental groups and drug administration
The experimental protocol was designed to compare the effects produced by the absence of NA per se with the damage of NA-containing axons. Therefore, saline or different doses of METH were administered to untreated control mice 3 days following the systemic injection of the neurotoxin DSP-4-hydrochloride (50 mg/kg; Sigma Chemical Co., St. Louis, MO, U.S.A.), which selectively destroys LC-arising NA axons; 15 min after each administration of the DBH inhibitor fusaric acid (40 mg/kg × 2, 2 h apart; Sigma), or to DBH −/− mice. In a pilot series of experiments we measured NA levels at 1, 4, and 8 h following 4 different doses of fusaric acid. Based on these experiments, the dose of fusaric acid of 40 mg/kg × 2, 2 h apart was selected as the most effective in depleting brain NA in a variety of brain areas including the striatum (Table I).
Table 1.
Time course of changes in NA levels (ng/mg protein) within different brain areas following various dosing paradigms of fusaric acid.
| STRIATUM | FRONTAL CORTEX | HIPPOCAMPUS | CEREBELLUM | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 h | 4 h | 8 h | 1 h | 4 h | 8 h | 1 h | 4 h | 8 h | 1 h | 4 h | 8 h | |
| Control | 0.90± 0.20 | 0.89± 0.04 | 1.03± 0.20 | 3.87± 0.19 | 3.56± 0.10 | 3.98± 0.62 | 4.08± 0.27 | 3.97± 0.24 | 4.03± 0.30 | 3.66± 0.32 | 3.43± 0.21 | 3.81± 0.31 |
| FA 20mg/kg×1 | 0.71± 0.01 | 0.80± 0.04 | 1.10± 0.30 | 3.40± 0.21 | 3.22± 0.31 | 3.85± 0.40 | 3.62± 0.31 | 3.50± 0.21 | 3.46± 0.29 | 3.21± 0.25 | 3.06± 0.22 | 3.49± 0.29 |
| FA 20mg/kg×2 | 0.68± 0.10 | 0.50± 0.01* | 0.58± 0.11* | 2.10± 0.33* | 1.80± 0.22* | 1.84± 0.31* | 2.13± 0.21* | 1.75± 0.19* | 1.91± 0.20* | 2.08± 0.14* | 1.96± 0.15* | 2.20± 0.17* |
| FA 40mg/kg×1 | 0.11± 0.01 a | ND | 0.60± 0.30* | 1.56± 0.21* | 1.06± 0.22* | 1.45± 0.27* | 0.64± 0.19§ | 0.53± 0.12§ | 0.78± 0.21§ | 1.80± 0.15§ | 1.02± 0.12§ | 2.07± 0.18§ |
| FA 40mg/kg×2 | ND | ND | ND | 0.70± 0.10§ | 0.49± 0.20§ | 0.53± 0.15§ | 0.30± 0.09§ | 0.18± 0.07§ | 0.22± 0.18§ | 0.59± 0.11§ | 0.29± 0.08§ | 0.37± 0.10§ |
Noradrenaline levels were measured in male C57/B1 mice at various time intervals after administration of either saline or different dosing of the DBH inhibitor, fusaric acid (20 mg/kg ×1 and ×2 or 40 mg/kg ×1 or ×2, 2 h apart) to induce a loss of NA. Results were obtained from 5 mice per group and are expressed as mean ±SEM values, in ng/mg of protein.
Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis:
p<0.05 compared with controls.
p<0.05 compared with control and lower dosing of FA.
Immediately before and during METH administration (5 mg/ kg × 3, 2 h apart) 5 mice from each group were used for the in vivo measurement of striatal extra-cellular levels of DA and ROS by brain dialysis. These mice were then sacrificed 1 week later to measure striatal tissue DA content, TH immunohistochemistry and striatal ultrastructure by electron microscopy. Separate groups of mice (N=5 per group) were administered saline or METH (5 mg/kg × 1 or 5 mg/kg × 2) and were sacrificed after 1 week for the same assays. METH hydrochloryde (Sigma), dissolved in saline solution, was administered intraperitoneally (i.p.). The doses of METH refer to the free base and were selected based on previous studies to obtain a mild (low, subtoxic dose) or severe (high, toxic dose) neurotoxic effect on DA nigrostriatal system (Fornai et al., 1999; 2004; Battaglia et al., 2002). DSP-4 hydrochloryde and fusaric acid were dissolved in distilled water and administered i.p.. Seven days after treatment, mice were sacrificed and brain samples were processed either for biochemical or morphological analysis. Briefly, mice were killed by decapitation and brains were quickly removed from the skull. For each mouse, monoamine assays were carried out on brain samples collected from the right hemiencephalon, whereas light microscopy and immunoblotting were performed on the left hemiencephalon (see below).
Behavioral observations
Behavioral observations consisted of measurement of the locomotor activity in open field and evaluation of stereotypies, such as licking and biting as defined by previous work (Seiden et al. 1993). The open field box consisted of an 80 cm × 80 cm × 15 cm glassy holder, with the floor divided into 64 squares (10 cm × 10 cm). For each mouse, locomotor activity was measured by counting open field activity and rearing episodes. These were measured, daily, by two independent, blind observers. In particular, open field activity was evaluated by the number of squares crossed by the mice with all four legs during constant time intervals, each lasting 2 min. Rearing behaviour (vertical movements) was evaluated using the same box with transparent walls, with a normal, white floor. Rearing episodes were counted during periods lasting 2 min and each episode was defined by the rearing of the mouse standing on the hind limbs. Total observation period began 210 min before METH or saline administration and continued for 420 min after first METH administrations. Specific behaviour was simultaneously observed in order to describe the occurrence of different stereotypies and locomotor activity during the same time interval. Data are shown as the means ± SEM of the single values collected for each mouse belonging to the same group during a time interval of 1 h.
Monoamine assay
Mice were sacrificed and their brains quickly removed to dissect the different brain areas (striatum, and for the pilot experiments also the cerebellum, the hippocampus and the frontal cortex) on a plate wheat of ice-cold saline. Briefly, the ice-cold dissected tissue was homogenized with a sonicator. 50 μL was collected and processed to measure the protein concentrations according to the Lowry's procedure, while the remaining 550 μL was centrifuged, and 20 μL of the clear supernatant was immediately injected into HPLC. When striatal dialysate samples were collected, 20 μL of the perfusate was directly injected into an HPLC. The HPLC system consisted of an autosampler 507, a programmable solvent module 126, a C18 reverse-phase column (Beckmann Instruments, Fullerton, CA, USA) and a Coulochem III with an analytical cell 5011A (ESA Inc. Chelmsford, MA, USA). NA, DA, serotonin and metabolites were analyzed in a ion-pairing, reverse phase mode. The analytical cell was composed of two electrodes the first working at +300 mV, and the second at −300 mV. The signal was recorded at both electrodes. Before and during the assay of the samples, standard solutions were run in the system to build two regression curves (one for the oxidizing and the other for the reducing electrode). On this basis, the striatal amount of DA was determined by an absolute quantitative assay per protein unit (striatal homogenates) or extracellular volume unit (striatal microdialysates).
Striatal microdialysis in freely moving mice
Mice were implanted with microdialysis intracerebral guides (CMA/7 Guide Cannula, CMA/Microdialysis, Stockholm, Sweden). The site of implantation was left or right striatum (coordinates in mm: AP = +0.6 from bregma; ML = ±1.7 lateral to the midline; DV = −2.5−4.5 from the surface of the skull, according to the atlas of Paxinos and Franklin 2004). Concentric vertical microdialysis probes 2 mm long and 0.24 mm in outer diameter having a cuprophane membrane with a molecular cut-off of 6000 Da (CMA/7 Microdialysis Probe, CMA/Microdialysis) were used. The probes were perfused continuously with artificial cerebrospinal fluid (ACSF), at a flow rate of 1.5 μL/min, using a microinjection pump (Bioanalytical System Inc., West Lafayette, IN, USA). The ACSF contained 150 mmol/L NaCl, 3 mmol/L KCl, 1.7 mmol/L CaCl2, and 0.9 mmol/L MgCl2. This solution was not buffered, and the pH was typically 6.5. After three sample fractions, used to determine the basal levels of striatal DA and ROS, mice received three injections of METH (5 mg/kg, i.p., 2 h apart).
Measurement of reactive oxygen species (ROS)
Formation of ROS was examined by monitoring the amount of the product 2,3-dihydroxybenzoic acid (2,3-DHBA), which is derived from the reaction of salicylate (5 mmol/L, added to ACSF) with hydroxyl radicals occurring in the extracellular space. Thus, the amount of 2,3-DHBA provides an indirect measure of the extracellular ROS levels (Giovanni et al. 1995). Analysis of 2,3- DHBA was performed by HPLC with electrochemical detection (Coulochem II electrochemical detectorESA, Inc., Chelmsford, MA, USA, as previously published, Battaglia et al. 2002). The amount of 2,3-DHBA was expressed as percentage of baseline values.
Morphological analysis and immunocytochemistry
The left brain hemisphere from each mouse was either frozen by immersion in cold isopentane and stored at −80°C or immersed for 24 h in a fixative solution consisting of a buffered formaline solution, containing 4% formaldehyde in phosphate buffer 0.1 mol/L. Fixed brains were included in paraffin as described in Fornai et al. (1999).
For immunocytochemical experiments, frozen brains were cut on a cryostat (20 μm-thick sections), and paraffin-embedded brains were cut on a microtome (5 μm-thick sections). In order to characterize monoaminergic innervation in specific brain areas, we used a primary antibody directed against TH (Chemicon International, Temecula, CA, USA) at working dilution of either 1:1000 (frozen slices) or 1:500 (paraffin-embedded slices), or against glial fibrillary acidic protein (GFAP, 1:400, Chemicon). The secondary biotynilated antibodies (for TH staining, Vector Laboratories, Burlingame, CA) were used at a dilution of 1:200, followed by incubation with an ABC kit and diaminobenzydine (Vector Laboratories), whereas the fluorescein-conjugated secondary antibody (for GFAP staining, Vector Laboratories) was used at a dilution of 1:100.
SDS-Page Immunoblotting
The tissues were homogenized in buffer containing 50 mmol/L Tris-HCl (pH=7.5), 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L PMSF, 0.5% NP-40, 0.25% SDS and 10 μg/ml of protease inhibitors (leupeptin, pepstatin, aprotinin; Sigma). Homogenates were centrifuged at 5,000g for 5 min and the supernatant was subsequently centrifuged at 30,000g for 30 min (Rothman et al., 2003). An aliquot of supernatant was used to determine the protein concentration by a protein assay kit (Sigma). Samples containing 40 μg of total protein were dissolved and electrophoresed on 12% sodium dodecyl sulfate-polyacrylamide gel. Following electrophoresis, the proteins were transferred to PVDF membrane (Millipore, Bedford, MA, USA). The membrane was immersed in blocking solution (5% not fat dried milk in 20 mmol/L Tris, 137 mmol/L NaCl at pH 7.6 containing 0.05% Tween-20) for 3 h at 4°C. Subsequently, the membrane was incubated with an anti-TH primary antibody (1:1000; Sigma) at 4°C overnight. The blot was probed with horseradish peroxidase-labeled secondary antibody (1:2000; Amersham Pharmacia Biotech, Buckinghamshire, UK) and TH bands were visualized with enhanced chemiluminescence reagents (Amersham). Immunoreactive bands were analyzed semi-quantitatively using densitometry (Lin et al., 1999). Films of western blots were scanned by using Adobe Photoshop CS2 version 9.0 (Adobe Systems, Mountain View, CA), and the optical density (OD) of the bands was measured by using NIH IMAGE 1.61. The relative values of each immunoreactive band were calculated by subtracting the background of the OD value from the measured OD of the bands. Groups used for statistical analyses were always determined within the same western blot. The analysis was performed in triplicate for confirmation of the results.
Electron microscopy
For electron microscopy, mice were perfused and the brain were immersed in fixing solution (2% pformaldehyde/0.1% glutaraldehyde in 0.1 mol/L phosphate-buffered saline, pH 7.4) overnight at 4°C. Brains were then removed, and the striatum was dissected and post fixed in 1% OsO4 in buffered solution, dehydrated in ethanol, and embedded in Epon-araldide. Ultrathin sections were stained with uranyl acetate and lead citrate and were examined with a Jeol (Tokyo, Japan) Jem 100SX transmission electron microscope.
Statistical analysis
For the locomotor activity (open field activity and rearing), each mouse was tested for 630 min during drug administration, for periods lasting 2 min every 30 min. Results obtained by each observer for each mouse were averaged for each count. The cumulative data were shown as the mean ± SEM from groups of 5−7 mice. For the biochemical assay, results from each group are expressed as the mean ± SEM of values obtained from each group of mice (N=5−7). All data were compared between groups using one way ANOVA with Sheffè's post-hoc analysis. The null hypothesis was rejected when p<0.05. To count the number of striatal cells with whorls, we analysed electron microscopy samples. In particular, from each mouse (N=5) per group, we randomly chose two tissue blocks dissected from dorso-lateral striatum. From each block, we obtained 10 grids, each one containing several non-serial (for the striatum, size interval = 50 μm) slices in order to observe a total of 10 neurons. The number of the striatal cells containing whorls, out of the total number of neurons under observation, were compared using ANOVA with Sheffè's post hoc analysis. The null hypothesis was rejected when p<0.05.
Results
Fusaric acid induces a specific pattern of NA depletion
Tissue NA levels were measured in the striatum, hippocampus, frontal cortex and cerebellum at various time intervals (1, 4, 8 h) after fusaric acid administration (20 mg/kg ×1 and ×2 or 40 mg/kg ×1 and ×2, 2 h apart, i.p.) (Table 1). A significant NA loss was detected in all areas that reached the maximum effect 4 h after the drug administration. For the highest dose of fusaric acid (40 mg/kg ×2) the depletion of NA was greater than 80% and was still evident after 8 h.
NA depletion enhances METH-induced striatal DA release, locomotor activity, and stereotypies
Baseline extracellular striatal DA levels were reduced by all three methods of NA depletion, and reached significance for DSP-4 and in DBH −/− mice (Fig. 1). Administration of METH (5 mg/kg × 3) produced a significant increase in extracellular striatal DA levels in control mice, as measured by in vivo microdialysis, which was magnified by damage to NA axons elicited 3 days earlier by the neurotoxin DSP-4 (Fig. 1a). This effect of DSP-4 was, in general, mimicked by selective NA depletion by pharmacological (fusaric acid; Fig. 1b) or genetic (DBH −/− mice; Fig. 1c) inhibition of DBH. We did observe one discrepancy between the effects produced by NA damage and the NA depletion (Fig. 1). While all paradigms reduced baseline DA levels, extracellular DA levels were increased following all three METH injections in DSP-4 treated mice compared to controls (Fig. 1a). In contrast, extracellular DA levels were reduced in NA depleted mice following the first METH injection, then increased following the second and third METH injections (Fig. 1b, 1c). The stimulation of DA release was accompanied by an increase in open field activity, which reached the maximum in control (wild-type and DBH +/−) mice soon after the first injection of METH (Fig. 2). This hyperlocomotion showed a progressive decrease starting at about 3 h following the first METH injection, remaining lower than baseline during the rest of the observation period. The onset of decreased locomotor activity was concomitant with the increase of stereotyped behaviours in most mice, and stereotypies were present in all mice following the second METH injection (Fig. 3). Pre-treatment with fusaric acid and the DBH −/− mutation both reduced baseline locomotor activity (30 min prior to METH administration; Fig. 2), as previously described (e.g. Weinshenker et al., 2002), but had no effect on baseline stereotypies (Fig. 3). DSP-4 and fusaric acid enhanced the initial METH-induced locomotor activity (Fig. 2a, 2b), while the locomotor activity of DBH −/− mice started to decrease following the first METH injection, then remained steady following the subsequent METH injections (Fig. 2c). All three NA depletion strategies (DSP-4, fusaric acid, DBH −/− mice) significantly exacerbated the magnitude of METH-induced stereotypies, and onset of stereotypies was faster in DBH −/− mice compared to DBH +/− mice (Fig. 3c). These results are consistent with previous observations of stereotypies in amphetamine-treated DBH −/− mice (Weinshenker et al., 2002). Following the third METH injection, two DBH −/− mice experienced typical limbic seizures, which lasted for ∼ 30 min. DBH −/− mice are known to be hypersensitive to many types of seizures elicited by various chemoconvulsants, including kainic acid-induced limbic seizures (Szot et al., 1999; Weinshenker et al., 2001). Interestingly, we also observe occasional limbic seizures in mice pre-treated with fusaric acid (data not shown)
Figure 1. NA depletion enhances METH-induced striatal DA release.
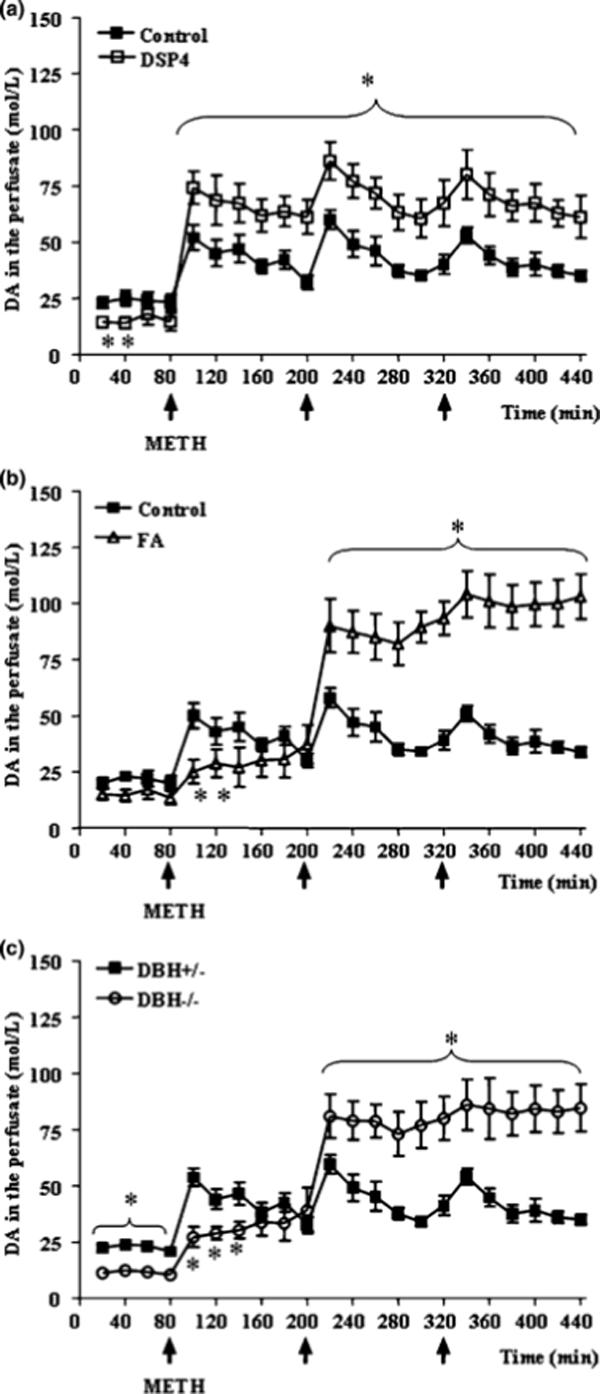
Shown are striatal DA levels, as measured by microdialysis, in mice at baseline and following METH administration (5 mg/kg ×3, 2 h apart; arrows) in (a) mice pretreated with DSP-4 (b) mice pretreated with fusaric acid, and (c) in DBH −/− mice. Results were obtained from 5 animals per group and are expressed as mean ±SEM (bars) values. Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis.
*p<0.05 compared with controls.
Figure 2. NA depletion modifies METH-induced locomotor activity.
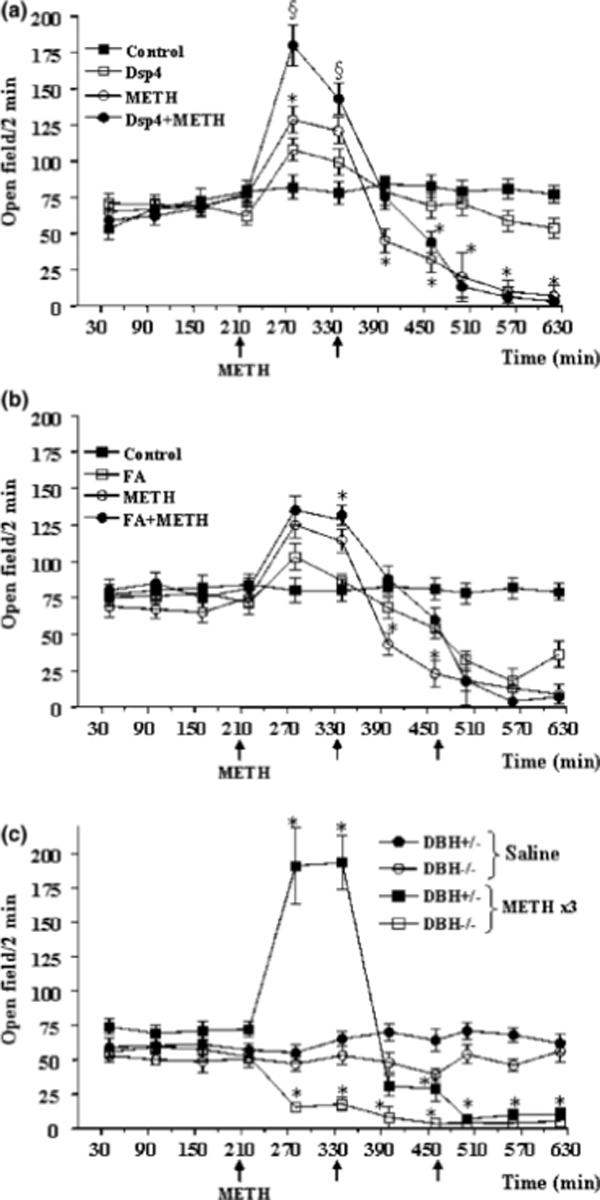
Shown is locomotor activity, as measured by line crossings and rearing episodes in an open field, at baseline or following METH administration (5 mg/kg ×3, 2 h apart; arrows) in (a) mice pretreated with DSP-4, (b) mice pretreated with fusaric acid, and (c) DBH −/− mice. Results were obtained from 5 animals per group and are expressed as mean ±SEM (bars) values. Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis.
*p<0.05 compared with controls, §p<0.05 compared with METH.
Figure 3. NA depletion enhances METH-induced stereotypes.
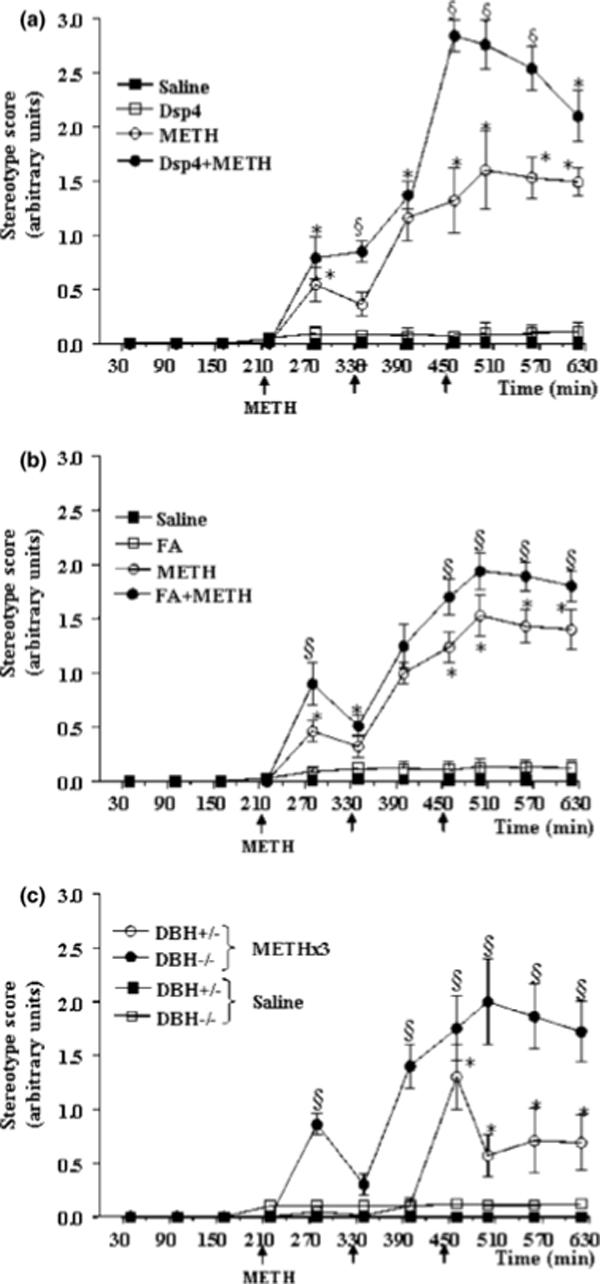
Shown are the onset and frequency of stereotyped behaviours following METH administration (5 mg/kg ×3, 2 h apart; arrows) in (a) mice pretreated with DSP-4, (b) mice pretreated with fusaric acid, and (c) DBH −/− mice. Note that the appearance of stereotypies were concomitant with the decrease in locomotor activity (see Fig. 2).
Results were obtained from 5 animals per group and are expressed as mean ±SEM (bars) values. Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis.
*p<0.05 compared with controls, §p<0.05 compared with METH.
NA depletion increases METH-induced extracellular ROS and exacerbates METH toxicity
METH treatment acutely increased the production of striatal extracellular ROS as measured by 2,3 DHBA in dialysate, and this effect was even more pronounced in mice pre-treated with DSP-4, fusaric acid, and in DBH −/− mice (Fig. 4). Interestingly, the effect of NA depletion on the pattern of extracellular ROS levels was similar to that of extracellular DA levels, with opposite effects of DSP-4 vs. fusaric acid and DBH −/− mice following the first but not second or third METH injection. METH dose-dependently caused damage to DA terminals in the striatum, as measured by reductions in tissue DA content (Fig. 5) and TH immunostaining (Figs. 6a, 7a, and 8a) 1 week later. METH also increased striatal gliosis (immunofluorescence for GFAP) (Fig. 6b, 7b, and 8b). All measures of toxicity were exacerbated by DSP-4 (Fig. 6a-d), fusaric acid (Fig. 7), and in DBH −/−mice (Fig. 8). No change in the number of nigral neurons was observed (Fig. 6e-f). After METH treatment, cytoplasmic and nuclear whorls and double membrane autophagic-like structures were observed in striatal neurons (Figs. 9a, 9b), as we have previously described (Fornai et al., 2004). Strikingly, genetic or pharmacological NA depletion produced these ultrastructural changes even in the absence of METH (Fig. 9c), and slightly enhanced these morphological features following METH treatment (Figs. 9d, 9e).
Figure 4. NA depletion increases METH-induced extracellular radical oxygen species (ROS).
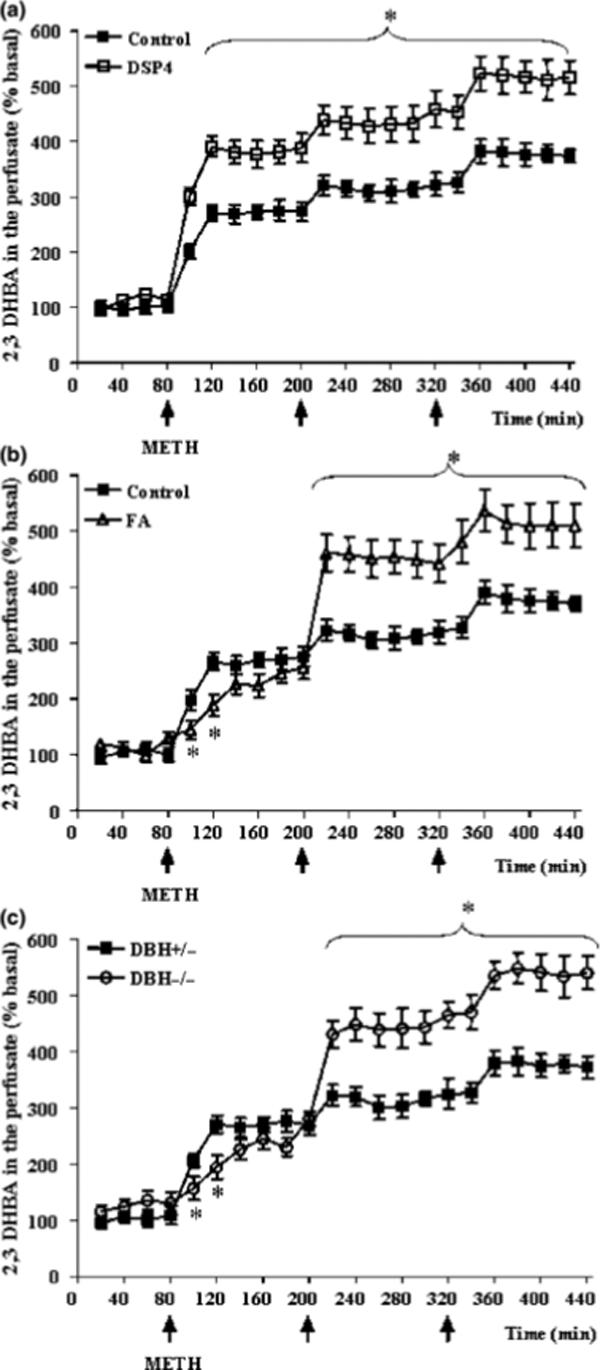
Shown is the formation of ROS, as assessed by striatal dialysate levels of 2,3-DHBA, following METH administration (5 mg/kg ×3, 2 h apart; arrows) in (a) mice pretreated with DSP-4, (b) mice pretreated with fusaric acid, and (c) DBH −/− mice. Results were obtained from 5 animals per group and are expressed as mean ±SEM (bars) values. Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis.
*p<0.05 compared with controls.
Figure 5. NA depletion enhances METH-induced striatal DA loss.
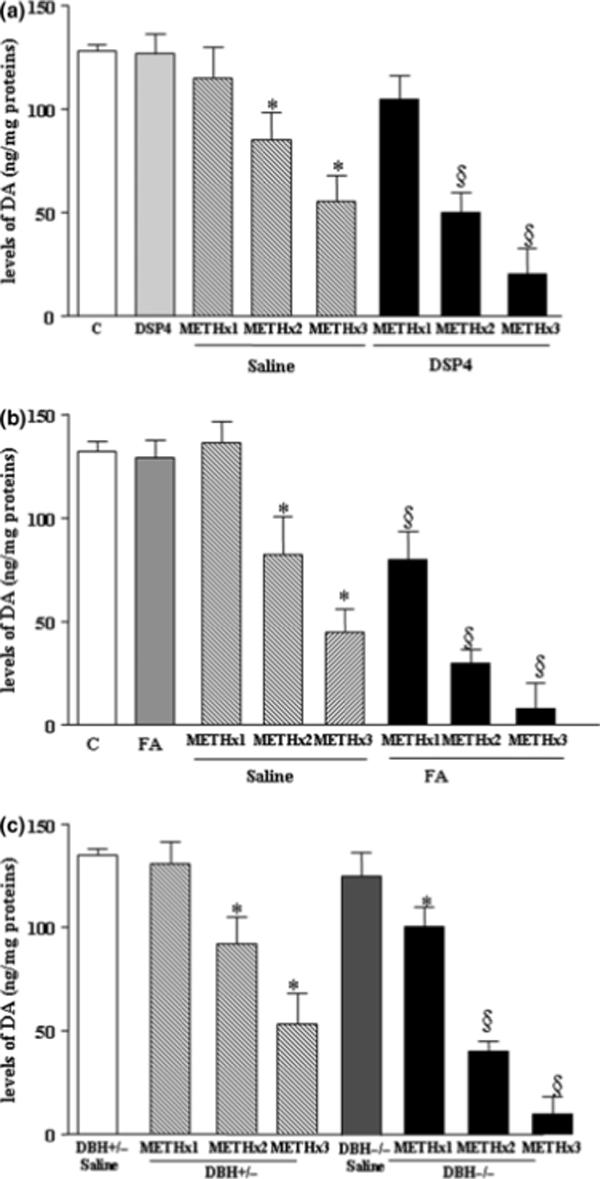
Shown are striatal tissue DA levels 7 days following saline or METH administration (5 mg/kg ×1, 5 mg/kg ×2, and 5 mg/kg ×3, 2 h apart) in (a) mice pretreated with DSP-4, (b) mice pretreated with fusaric acid, and (c) DBH −/− mice. Results were obtained from 5 animals per group and are expressed as the mean ±SEM. Differences among groups were evaluated using ANOVA.
*p<0.05 compared with controls, §p<0.05 compared with METH.
Figure 6. Effects of DSP-4 lesions on METH-induced toxicity.
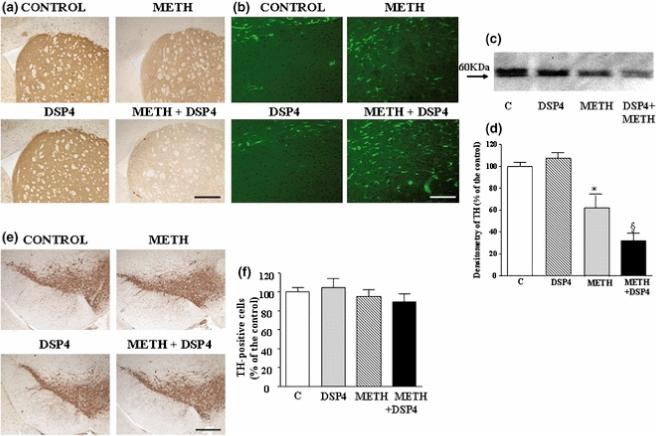
Mice were treated with saline, METH (5 mg/kg ×3, 2 h apart, i.p), DSP-4 (50 mg/kg, i.p.), or DSP-4 + METH (METH was administered 3 days following DSP-4), and striata were analyzed 7 days later. Shown is (a) representative immunohistochemical analysis of striatal tyrosine hydroxylase (TH), (b) representative immunohistochemical analysis of the glial marker GFAP in striatum, (c) representative Western blot for striatal TH, (d) semiquantitative densitometry of TH Western blot, and (e,f) representative analysis of TH immunoreactivity in the substantia nigra.
Densitometry results were obtained from 3 experiments and are expressed as mean ±SEM (bars) values. Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis.
*p<0.05 compared with controls, §p<0.05 compared with METH.
Scale bars= 460 μm (a); 150 μm (b); 400 μm (e).
Figure 7. Effects of fusaric acid treatment on METH-induced toxicity.
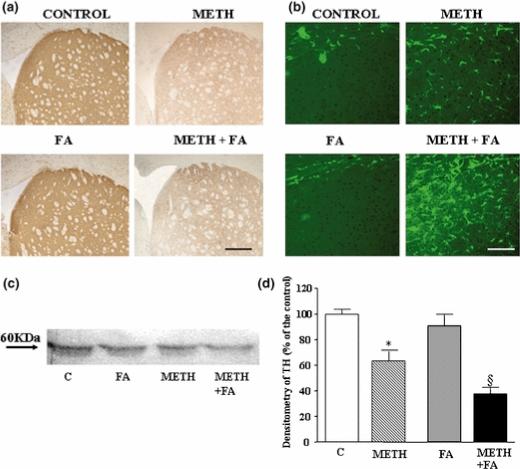
Mice were treated with saline, METH (5 mg/kg ×3, 2 h apart, i.p), fusaric acid (40 mg/kg ×2, 2 h apart, i.p), or DSP-4 + fusaric acid, and striata were analyzed 7 days later. Shown is (a) representative immunohistochemical analysis of striatal tyrosine hydroxylase (TH), (b) representative immunohistochemical analysis of the glial marker GFAP in striatum, (c) representative Western blot for striatal TH, and (d) semiquantitative densitometry of TH Western blot.Densitometry results were obtained from 3 experiments and are expressed as mean ±SEM (bars) values. Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis.
Scale bars= 400 μm (a); 125 μm (b).
Figure 8. METH-induced toxicity in DBH −/− mice.
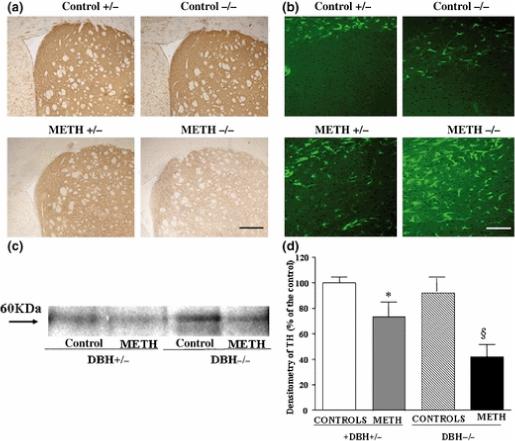
DBH +/− and DBH −/− mice were treated with saline or METH (5 mg/kg ×3, 2 h apart, i.p), and striata were analyzed 7 days later. Shown is (a) representative immunohistochemical analysis of striatal tyrosine hydroxylase (TH), (b) representative immunohistochemical analysis of the glial marker GFAP in striatum, (c) representative Western blot for striatal TH, and (d) semiquantitative densitometry of TH Western blot. Results were and are expressed as mean ±SEM (bars) values. Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis.
Scale bars= 400 μm (a); 115 μm (b)
Figure 9. Effects of NA depletion on baseline and METH-induced whorls in striatal medium spiny neurons.
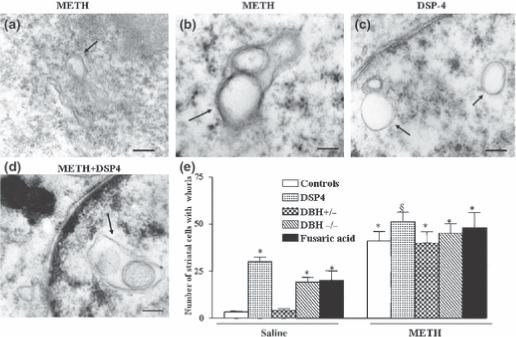
Mice were treated with saline or METH (5 mg/kg ×3, 2 h apart, i.p.), and striatal ultrastructure was analyzed for the presence of membranous multilayer whorls in the cytoplasm (arrows) 7 days after METH treatment (5 mg/kg ×3, 2 h apart; a, b).
Similar morphological changes were produced following pharmacological depletion of NA induced by DSP-4 in the absence of METH (c), this was slightly increased when METH was administered to different kinds of NA depleted mice (d). Bar graphs show the number of whorls-containing neurons for each experimental group (e).
Scale bars=0.56 μm (a); 0.14 μm (b); 0.42 μm (c); 0.2 μm (d)
Results were obtained from 5 animals per group and are expressed as mean ±SEM (bars) values. Differences among groups were evaluated using ANOVA with Sheffè's post hoc analysis.
*p<0.05 compared with controls, §p<0.05 compared with METH.
Discussion
A specific loss of NA enhances the effects of METH
We have previously shown that damage to noradrenergic neurons using DSP-4 enhanced the neurotoxic effects of METH (Fornai et al., 1995; 1997b; 1999). Noradrenergic terminals possess METH uptake sites (the NA transporter), which could sequester METH away from DA neurons. Loss of these uptake sites following DSP-4 lesions may result in the exposure of DA axons to greater concentrations of METH. In addition, noradrenergic terminals contain co-transmitters such as NPY and galanin, and loss of these neuromodulators could also contribute to the enhanced METH-induced toxicity observed after DSP-4 lesion. Thus, it was unclear whether the effects of DSP-4 lesions on METH toxicity were due to a loss METH uptake sites and co-transmitters, or NA itself. A “buffering” role for NA terminals was previously postulated for other DA neurotoxins such as MPP+, which could potentially be sequestered away from DA neurons by NA axons, thereby decreasing its availability to DA neurons (Herkenham et al., 1991; Di Chiara et al., 1992; Fornai et al., 1997a). However, data do not support this hypothesis. We showed that peak striatal METH concentration is not changed by LC lesions (Fornai et al., 1999), and mice with a genetic or pharmacological disruption of the NA transporter are resistant, rather than hypersensitive, to MPTP toxicity (Rommelfanger et al., 2004). We found that the effects of DSP-4 on METH-induced striatal DA release, extracellular ROS production, DA terminal damage, gliosis, and formation of autophagic vacuoles and multilamellar whorls in intrinsic striatal neurons were mimicked by selective genetic or pharmacological blockade of NA synthesis. These results demonstrate that the absence of endogenous NA per se increases the toxicity of METH to the DA system.
The effects of NA depletion on METH-induced DA release
The enhancement of DA release by NA depletion following multiple doses of METH was somewhat unexpected given that NA facilitates DA neuron firing, and DA release induced by other drugs is typically attenuated when NA signalling is blocked (reviewed by Weinshenker and Schroeder, 2007). For example, we have previously shown that both basal and amphetamine-evoked DA release is attenuated in the striatum of DBH −/− mice (Schank et al., 2006). This discrepancy is likely due to the mechanism of METH-induced DA release and the number of METH injections. Despite the fact that amphetamine is a DA releasing agent, its ability to increase extracellular DA in vivo is largely impulse-dependent, and requires noradrenergic innervation (e.g. Darracq et al., 1998; Ventura et al., 2004; Schank et al., 2006). It is possible that, as opposed to amphetamine, METH-induced DA release in vivo has a significant impulse-independent component (e.g. Nishijima et al., 1996). We confirmed that baseline DA release was reduced following all three methods of NA depletion, and DA release was reduced following a single dose of METH in DBH −/− and fusaric acid-treated mice. It was only after the second dose of METH that DA release was markedly enhanced in the absence of NA activity. Because DBH inhibition increases DA production in noradrenergic terminals, it is tempting to speculate that DA overflow coming from noradrenergic neurons can account for this observation. However, this mechanism can be ruled out by our results with the DSP-4 lesioned mice, which do not produce extra DA in noradrenergic neurons but still show an enhancement of METH-induced DA release. We propose the following model to explain the enhancement of DA release produced by multiple METH injections following NA depletion. NA exerts a neuroprotective effect on DA neurons. In NA-depleted mice, the first dose of METH disrupts the structural integrity of DA terminals to a larger extent than in mice with intact noradrenergic innervation, thus exposing intracellular pools of DA to the extracellular environment upon subsequent METH injections. In support of this hypothesis, we found that 7 days following a single METH injection, striatal tissue DA levels were reduced in NA-depleted mice but not control mice (Fig. 5). It is still unclear why DA release is enhanced following the first METH injection in DSP-4 lesioned mice but not DBH −/− or fusaric acid-treated mice. It is possible that the levels of LC co-transmitters that normally inhibit DA release, such as galanin, are reduced by the DSP-4 lesion, whereas these inhibitory neuromodulators remain intact in mice with a specific NA depletion (Tsuda et al., 1998; Weiss et al., 1998; Weiss et al., 2005; our unpublished data).
The effects of NA depletion on METH-induced striatal damage
We observed not only damage to striatal DA terminals following METH administration, but also alterations to intrinsic striatal medium spiny neurons as well. As originally hypothesized by Jakel and Maragos (2000), high extracellular levels of DA within the striatal milieu may affect the medium spiny neurons, either via DA receptors or through oxidative species. All three methods we used to deplete NA enhanced the DA release and the production of extracellular ROS produced by METH administration, and also worsened gliosis and the abundance of striatal whorls. We recently found that the increase in extracellular striatal DA levels is directly responsible, through the activation of D1-like DA receptors, for the ultrastructural alterations in medium spiny neurons generated by METH, in the form of autophagic vacuoles and multilamellar whorls (Lazzeri et al., 2007). Furthermore, we and others have shown that in the absence of NA, there is a compensatory increase in the sensitivity of striatal DA receptors (Donaldson et al., 1976; Lategan et al., 1990; Harro et al., 2000; Schank et al., 2006). Although the stimulation of D1 receptors alone can reproduce the ultrastructural changes triggered by DA (Lazzeri et al., 2007), this effect is modest compared with METH administration. Thus, we hypothesize that a synergistic effect of extracellular ROS and DA receptor stimulation on METH-induced damage to striatal medium spiny neurons takes place, and that NA loss exacerbates both of these mechanisms.
Relationships between METH-neurotoxicity and neurodegeneration
Multiple degenerative disorders of the central nervous system, such as Parkinson's disease (PD) and Alzheimer's disease, are characterized by the loss of NA neurons in the LC (Gesi et al., 2000; Zarow et al., 2003; Marien et al., 2004; Rommelfanger and Weinshenker, 2007). Experimental damage to LC neurons exacerbates the toxicity of a variety of DA neurotoxins, including MPTP (Mavridis et al., 1991; Marien et al., 1993; Fornai et al., 1997), METH (Fornai et al. 1995), 3,4-methylenedioxyamphetamine (MDMA) (Ferrucci et al., 2002), and 6-OHDA (Srinivasan and Schmidt, 2003, 2004), indicating a protective role of this nucleus on the survival of nigrostriatal DA neurons. A similar effect was recently substantiated by pathological (Zarow et al., 2003) and biochemical (Tong et al., 2006) findings in humans. Our results indicate that the protective effect of LC neurons against METH toxicity is at least partially, if not predominantly, mediated by NA itself, although a contribution of other substances contained in NA neurons cannot be ruled out. Our findings have significant implications for both METH addiction and the survival of the nigrostriatal DA system in PD. An important role for the NA system in psychostimulant abuse is now emerging (reviewed by Weinshenker and Schroeder, 2007), while the protective role of the NA system in PD has strong support at pre-clinical and clinical levels. It should be considered that the pattern of nigrostriatal DA denervation that occurs in PD differs from what is reported in long-term abstinent, previously chronic METH abusers. For example, the pattern of striatal DA loss is more pronounced in the putamen than the caudate nucleus of PD patients, while the opposite occurs in METH abusers. The influence of METH on the caudate nucleus may partially underlie the psychiatric symptoms that often occur in these patients (Volkow et al., 2001b). Moreover, although still pronounced long after discontinuation of METH use, there is considerable recovery from the nigrotriatal denervation in these patients (Volkow et al., 2001a), while PD is characterized by irreversible DA neuronal death and disease progression. Nonetheless, NA system involvement in PD and METH abuse may extend to other features of disease, including the long-term side effects of the DA substitution therapy. For example, stereotyped abnormal involuntary movement, which occurs as a consequence of chronic DA replacement therapy in PD with L-3,4-dihydroxyphenylalanine (L-DOPA), occurs earlier and with greater severity when NA neurons are damaged (Fulceri et al., 2007). Interestingly, such a behavioral effect is accompanied by ultrastructural alterations within striatal neurons that are equivalent of those occurring in NA-deficient mice following METH administration (Fulceri et al., 2007). It is important to note that although the neuroprotective effects of NA in PD and METH toxicity appear to be crucial, its precise mechanism of action remains elusive. NA could promote the survival of DA neurons by activating neurotrophins such as NGF and BDNF (Semkowa and Krieglstein, 1999). Alternatively, NA has been shown to possess potent antioxidant properties and protects DA neurons from oxidative stress in vitro (Troadec et al., 2001). Finally, it should be considered that noradrenergic innervation provides both excitatory and inhibitory drive onto DA neurons. Thus, a direct receptor-mediated influence on midbrain DA neurons by NA and subsequent changes in DA release and striatal signaling should be considered.
References
- Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J. Pharmacol. Exp. Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- Battaglia G, Fornai F, Busceti CL, Aloisi G, Cerrito F, De Blasi A, Melchiorri D, Nicoletti F. Selective blockade of mGlu5 metabotropic glutamate receptors is protective against methamphetamine neurotoxicity. J. Neurosci. 2002;22:2135–2141. doi: 10.1523/JNEUROSCI.22-06-02135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing G, Zhang Y, Watanabe Y, McEwen BS, Stone EA. Locus coeruleus lesions potentiate neurotoxic effects of MPTP in dopaminergic neurons of the substantia nigra. Brain. Res. 1994;668:261–265. doi: 10.1016/0006-8993(94)90534-7. [DOI] [PubMed] [Google Scholar]
- Bourdelat-Parks BN, Anderson GM, Donaldson ZR, Weiss JM, Bonsall RW, Emery MS, Liles LC, Weinshenker D. Effects of dopamine beta-hydroxylase genotype and disulfiram inhibition on catecholamine homeostasis in mice. Psychopharmacology (Berl) 2005;183:72–80. doi: 10.1007/s00213-005-0139-8. [DOI] [PubMed] [Google Scholar]
- Darracq L, Blanc G, Glowinski J, Tassin JP. Importance of the noradrenaline-dopamine coupling in the locomotor activating effects of D-amphetamine. J. Neurosci. 1998;18:2729–2739. doi: 10.1523/JNEUROSCI.18-07-02729.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Tanda GL, Frau R, Carboni E. Heterologous monoamine reuptake: lack of transmitter specificity of neuron-specific carriers. Neurochem. Int. 1992;20:231S–235S. [PubMed] [Google Scholar]
- Donaldson IM, Dolphin A, Jenner P, Marsden CD, Pycock C. The involvement of noradrenaline in motor activity as shown by rotational behaviour after unilateral lesions of the locus coeruleus. Brain. 1976;99:427–446. doi: 10.1093/brain/99.3.427. [DOI] [PubMed] [Google Scholar]
- Ferrucci M, Gesi M, Lenzi P, Soldani P, Ruffoli R, Pellegrini A, Ruggieri S, Paparelli A, Fornai F. Noradrenergic Loss enhances MDMA toxicity and induces ubiquitin-positive striatal whorls. Neurol. Sci. 2002;23:S75–S76. doi: 10.1007/s100720200077. [DOI] [PubMed] [Google Scholar]
- Fornai F, Alessandrì MG, Torracca MT, Bassi L, Corsini GU. Effects of noradrenergic lesions on MPTP/MPP+ kinetics and MPTP-induced nigrostriatal dopamine depletions. J. Pharmacol. Exp. Ther. 1997a;283:100–107. [PubMed] [Google Scholar]
- Fornai F, Bassi L, Bonaccorsi I, Giorgi F, Corsini GU. Noradrenaline loss selectivity exacerbates nigrostriatal toxicity in different species of rodents. Funct. Neurol. 1997b;12:193–198. [PubMed] [Google Scholar]
- Fornai F, Bassi L, Torracca MT, Scalori V, Corsini GU. Norepinephrine loss exacerbates methamphetamine-induced striatal dopamine depletion in mice. Eur. J. Pharmacol. 1995;283:99–102. doi: 10.1016/0014-2999(95)00313-a. [DOI] [PubMed] [Google Scholar]
- Fornai F, Giorgi FS, Alessandrì MG, Giusiani M, Corsini GU. Effects of pretreatment with N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) on methamphetamine pharmacokinetics and striatal dopamine losses. J. Neurochem. 1999;72:777–784. doi: 10.1046/j.1471-4159.1999.0720777.x. [DOI] [PubMed] [Google Scholar]
- Fornai F, Lenzi P, Gesi M, Soldani P, Ferrucci M, Lazzeri G, Capobianco L, Battaglia G, De Blasi A, Nicoletti F, Paparelli A. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J. Neurochem. 2004;88:114–123. doi: 10.1046/j.1471-4159.2003.02137.x. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice PS, Tong J, Yazdanpanah M, Liu PP, Kalasinsky KS, Kish SJ. Levels of 4-hydroxynonenal and malondialdehyde are increased in brain of human chronic users of methamphetamine. J. Pharmacol. Exp. Ther. 2006;319:703–709. doi: 10.1124/jpet.106.109173. [DOI] [PubMed] [Google Scholar]
- Fulceri F, Biagioni F, Ferrucci M, Lazzeri G, Bartalucci A, Galli V, Ruggieri S, Paparelli A, Fornai F. Abnormal involuntary movements (AIMs) following pulsatile dopaminergic stimulation: severe deterioration and morphological correlates following the loss of locus coeruleus neurons. Brain Res. 2007;1135:219–229. doi: 10.1016/j.brainres.2006.12.030. [DOI] [PubMed] [Google Scholar]
- Gesi M, Soldani P, Giorgi FS, Santinami A, Bonaccorsi I, Fornai F. The role of the locus coeruleus in the development of Parkinson's disease. Neurosci. Biobehav. Rev. 2000;24:655–668. doi: 10.1016/s0149-7634(00)00028-2. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Liang LP, Hastings TG, Zigmond MJ. Estimating hydroxyl radical content in rat brain using systemic andintraventricular salicylate: impact of methamphetamine. J. Neurochem. 1995;64:1819–1825. doi: 10.1046/j.1471-4159.1995.64041819.x. [DOI] [PubMed] [Google Scholar]
- Grenhoff J, Svensson TH. Prazosin modulates the firing pattern of dopamine neurons in rat ventral tegmental area. Eur. J. Pharmacol. 1993;233:79–84. doi: 10.1016/0014-2999(93)90351-h. [DOI] [PubMed] [Google Scholar]
- Grenhoff J, Nisell M, Ferre S, Aston-Jones G, Svensson TH. Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J. Neural. Transm. Gen. Sect. 1993;93:11–25. doi: 10.1007/BF01244934. [DOI] [PubMed] [Google Scholar]
- Han DD, Gu HH. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC. Pharmacol. 2006;3:6. doi: 10.1186/1471-2210-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harro J, Merikula A, Lepiku M, Modiri AR, Rinken A, Oreland L. Lesioning of locus coeruleus projections by DSP-4 neurotoxin treatment: effect on amphetamine-induced hyperlocomotion and dopamine D2 receptor binding in rats. Pharmacol. Toxicol. 2000;86:197–202. doi: 10.1034/j.1600-0773.2000.d01-35.x. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Little MD, Bankiewicz K, Yang SC, Markey SP, Johannessen JN. Selective retention of MPP+ within the monoaminergic systems of the primate brain following MPTP administration: an in vivo autoradiographic study. Neuroscience. 1991;40:133–158. doi: 10.1016/0306-4522(91)90180-v. [DOI] [PubMed] [Google Scholar]
- Hotchkiss A, Gibb JW. Blockade of methamphetamine-induced depression of tyrosine hydroxylase by GABA transaminase inhibitors. Eur. J. Pharmacol. 1980;66:201–205. doi: 10.1016/0014-2999(80)90143-0. [DOI] [PubMed] [Google Scholar]
- Jakel RJ, Maragos WF. Neuronal cell death in Huntington's disease: a potential role for dopamine. Trends Neurosci. 2000;23:239–245. doi: 10.1016/s0166-2236(00)01568-x. [DOI] [PubMed] [Google Scholar]
- Kogan FJ, Nichols WK, Gibb JW. Influence of methamphetamine on nigral and striatal tyrosine hydroxylase activity and on striatal dopamine levels. Eur. J. Pharmacol. 1976;36:363–371. doi: 10.1016/0014-2999(76)90090-x. [DOI] [PubMed] [Google Scholar]
- Lategan AJ, Marien MR, Colpaert FC. Effects of locus coeruleus lesions on the release of endogenous dopamine in the rat nucleus accumbens and caudate nucleus as determined by intracerebral microdialysis. Brain Res. 1990;523:134–138. doi: 10.1016/0006-8993(90)91646-x. [DOI] [PubMed] [Google Scholar]
- Lategan AJ, Marien MR, Colpaert FC. Suppression of nigrostriatal and mesolimbic dopamine release in vivo following noradrenaline depletion by DSP-4: a microdialysis study. Life Sci. 1992;50:995–999. doi: 10.1016/0024-3205(92)90093-5. [DOI] [PubMed] [Google Scholar]
- Lazzeri G, Lenzi P, Busceti CL, Ferrucci M, Falleni A, Bruno V, Paparelli A, Fornai F. Mechanisms involved in the formation of dopamine-induced intracellular bodies within striatal neurons. J. Neurochem. 2007;101:1414–1427. doi: 10.1111/j.1471-4159.2006.04429.x. [DOI] [PubMed] [Google Scholar]
- Lin L, Georgievska B, Mattsson A, Isacson O. Cognitive changes and modified processing of amyloid precursor protein in the cortical and hippocampal system after cholinergic synapse loss and muscarinic receptor activation. Proc. Natl. Acad. Sci. USA. 1999;96:12108–12113. doi: 10.1073/pnas.96.21.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marien M, Briley M, Colpaert F. Noradrenaline depletion exacerbates MPTP-induced striatal dopamine loss in mice. Eur. J. Pharmacol. 1993;236:487–489. doi: 10.1016/0014-2999(93)90489-5. [DOI] [PubMed] [Google Scholar]
- Marien MR, Colpaert FC, Rosenquist AC. Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res. Brain Res. Rev. 2004;45:38–78. doi: 10.1016/j.brainresrev.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Mavridis M, Degryse AD, Lategan AJ, Marien MR, Colpaert FC. Effects of locus coeruleus lesions on parkinsonian signs, striatal dopamine and substantia nigra cell loss after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in monkeys: a possible role for the locus coeruleus in the progression of Parkinson's disease. Neuroscience. 1991;41:507–523. doi: 10.1016/0306-4522(91)90345-o. [DOI] [PubMed] [Google Scholar]
- McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J. Neurosci. 1998;18:8417–8422. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima K, Kashiwa A, Hashimoto A, Iwama H, Umino A, Nishikawa T. Differential effects of phencyclidine and methamphetamine on dopamine metabolism in rat frontal cortex and striatum as revealed by in vivo dialysis. Synapse. 1996;22:304–312. doi: 10.1002/(SICI)1098-2396(199604)22:4<304::AID-SYN2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J. Pharmacol. Exp. Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- Paladini CA, Williams JT. Noradrenergic inhibition of midbrain dopamine neurons. J. Neurosci. 2004;24:4568–7455. doi: 10.1523/JNEUROSCI.5735-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Academic Press; San Diego: 2004. [Google Scholar]
- Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Weinshenker D. Norepinephrine: The redheaded stepchild of Parkinson's disease. Biochem. Pharmacol. 2007;74:177–190. doi: 10.1016/j.bcp.2007.01.036. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Weinshenker D, Miller GW. Reduced MPTP toxicity in noradrenaline transporter knockout mice. J. Neurochem. 2004;91:1116–1124. doi: 10.1111/j.1471-4159.2004.02785.x. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Jayanthi S, Wang X, Dersch CM, Cadet JL, Prisinzano T, Rice KC, Baumann MH. High-dose fenfluramine administration decreases serotonin transporter binding, but not serotonin transporter protein levels, in rat forebrain. Synapse. 2003;50:233–239. doi: 10.1002/syn.10266. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J. Pharmacol. Exp. Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- Semkova I, Krieglstein J. Neuroprotection mediated via neurotrophic factors and induction of neurotrophic factors. Brain Res. Brain Res. Rev. 1999;30:176–188. doi: 10.1016/s0165-0173(99)00013-2. [DOI] [PubMed] [Google Scholar]
- Seiden LS, Fischman MW, Schuster CR. Long-term methamphetamine induced changes in brain catecholamines in tolerant rhesus monkeys. Drug Alcohol Depend. 1976;1:215–219. doi: 10.1016/0376-8716(76)90030-2. [DOI] [PubMed] [Google Scholar]
- Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu. Rev. Pharmacol. Toxicol. 1993;33:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- Schank JR, Ventura R, Puglisi-Allegra S, Alcaro A, Cole CD, Liles LC, Seeman P, Weinshenker D. Dopamine beta-hydroxylase knockout mice have alterations in dopamine signalling and are hypersensitive to cocaine. Neuropsychopharmacology. 2006;31:2221–2230. doi: 10.1038/sj.npp.1301000. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Gibb JW, Hanson GR. Roles of D1 and D2 dopamine receptor subtypes in mediating the methamphetamine-induced changes in monoamine systems. J. Pharmacol. Exp. Ther. 1986;238:932–937. [PubMed] [Google Scholar]
- Sonsalla PK, Heikkila RE. Neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and methamphetamine in several strains of mice. Prog. Neuropsychopharmacol. Biol. Psychiatry. 1988;12:345–354. doi: 10.1016/0278-5846(88)90054-1. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- Srinivasan J, Schmidt WJ. Potentiation of parkinsonian symptoms by depletion of locus coeruleus noradrenaline in 6-hydroxydopamine-induced partial degeneration of substantia nigra in rats. Eur J Neurosci. 2003;17:2586–2592. doi: 10.1046/j.1460-9568.2003.02684.x. [DOI] [PubMed] [Google Scholar]
- Srinivasan J, Schmidt WJ. Behavioral and neurochemical effects of noradrenergic depletions with N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine in 6-hydroxydopamine-induced rat model of Parkinson's disease. Behav. Brain Res. 2004;151:191–199. doi: 10.1016/j.bbr.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Szot P, Weinshenker D, White SS, Robbins CA, Rust NC, Schwartzkroin PA, Palmiter RD. Norepinephrine-deficient mice have increased susceptibility to seizure-inducing stimuli. J. Neurosci. 1999;19:10985–10992. doi: 10.1523/JNEUROSCI.19-24-10985.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature. 1995;374:643–646. doi: 10.1038/374643a0. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J. Neurochem. 1998;7:2468–2476. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Palmiter RD. Impaired maternal behavior in mice lacking norepinephrine and epinephrine. Cell. 1997;91:583–592. doi: 10.1016/s0092-8674(00)80446-8. [DOI] [PubMed] [Google Scholar]
- Tong J, Hornykiewicz O, Kish SJ. Inverse relationship between brain noradrenaline level and dopamine loss in Parkinson disease: a possible neuroprotective role for noradrenaline. Arch. Neurol. 2006;63:1724–17288. doi: 10.1001/archneur.63.12.1724. [DOI] [PubMed] [Google Scholar]
- Troadec JD, Marien M, Darios F, Hartmann A, Ruberg M, Colpaert F, Michel PP. Noradrenaline provides long-term protection to dopaminergic neurons by reducing oxidative stress. J. Neurochem. 2001;79:200–210. doi: 10.1046/j.1471-4159.2001.00556.x. [DOI] [PubMed] [Google Scholar]
- Tsuda K, Tsuda S, Nishio I, Masuyama Y, Goldstein M. Effects of galanin on dopamine release in the central nervous system of normotensive and spontaneously hypertensive rats. Am. J. Hypertens. 1998;11:1475–1479. doi: 10.1016/s0895-7061(98)00168-x. [DOI] [PubMed] [Google Scholar]
- Ventura R, Alcaro A, Mandolesi L, Puglisi-Allegra S. In vivo evidence that genetic background controls impulse-dependent dopamine release induced by amphetamine in the nucleus accumbens. J. Neurochem. 2004;89:494–502. doi: 10.1111/j.1471-4159.2004.02342.x. [DOI] [PubMed] [Google Scholar]
- Villemagne V, Yuan J, Wong DF, Dannals RF, Hatzidimitriou G, Mathews WB, Ravert HT, Musachio J, McCann UD, Ricaurte GA. Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable to those recreationally abused by humans: evidence from [11C]WIN-35,428 positron emission tomography studies and direct in vitro determinations. J. Neurosci. 1998;18:419–427. doi: 10.1523/JNEUROSCI.18-01-00419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, Gatley SJ, Miller E, Hitzemann R, Ding YS, Logan J. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J. Neurosci. 2001a;21:9414–9418. doi: 10.1523/JNEUROSCI.21-23-09414.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, Sedler MJ, Gatley SJ, Hitzemann R, Ding YS, Logan J, Wong C, Miller EN. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am. J. Psychiatr. 2001b;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, Miller NS, Blizinsky K, Laughlin ML, Palmiter RD. Mice with chronic norepinephrine deficiency resemble amphetamine-sensitized animals. Proc. Natl. Acad. Sci. USA. 2002;99:13873–13877. doi: 10.1073/pnas.212519999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D, Schroeder JP. There and back again: a tale of norepinephrine and drug addiction. Neuropsychopharmacology. 2007;32:1433–1451. doi: 10.1038/sj.npp.1301263. [DOI] [PubMed] [Google Scholar]
- Weinshenker D, Szot P, Miller NS, Palmiter RD. Alpha(1) and beta(2) adrenoreceptor agonists inhibit pentylenetetrazole-induced seizures in mice lacking norepinephrine. J. Pharmacol. Exp. Ther. 2001;298:1042–1048. [PubMed] [Google Scholar]
- Weiss JM, Bonsall RW, Demetrikopoulos MK, Emery MS, West CH. Galanin: a significant role in depression? Ann. N Y Acad. Sci. 1998;863:364–382. doi: 10.1111/j.1749-6632.1998.tb10707.x. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Boss-Williams KA, Moore JP, Demetrikopoulos MK, Ritchie JC, West CH. Testing the hypothesis that locus coeruleus hyperactivity produces depression-related changes via galanin. Neuropeptides. 2005;39:281–287. doi: 10.1016/j.npep.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- Yoshihara T, Honma S, Mitome M, Honma K. Methamphetamine stimulates the release of neuropeptide Y and noradrenaline from the paraventricular nucleus in rats. Brain Res. 1996;707:119–121. doi: 10.1016/0006-8993(95)01343-1. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]