

Abstract
The synthesis and characterization of diiron(II) complexes supported by 9-triptycenecarboxylate ligands (-O2CTrp) is described. The interlocking nature of the triptycenecarboxylates facilitates formation of quadruply bridged diiron(II) complexes of the type [Fe2(μ-O2CTrp)4(L)2] (L = THF, pyridine or imidazole derivative) with a paddlewheel geometry. A systematic lengthening of the Fe-Fe distance occurs with the increase in steric bulk of the neutral donor L, resulting in values of up to 3 Å without disassembly of the paddlewheel structure. Reactions with an excess of water do not lead to decomposition of the diiron(II) core, indicating that these quadruply bridged complexes are of exceptional stability. The red-colored complexes [Fe2(μ-O2CTrp)4(4-AcPy)2] (10) and [Fe2(μ-O2CTrp)4(4-CNPy)2] (11) exhibit solvent-dependent thermochromism in coordinating solvents that was studied by variable temperature UV-vis spectroscopy. Reaction of [Fe2(μ-O2CTrp)4(THF)2] with N,N,N’,N’-tetramethylethylenediamine (TMEDA), tetra-n-butyl ammonium thiocyanate, or excess 2-methylimidazole resulted in the formation of mononuclear complexes [Fe(O2CTrp)2(TMEDA)] (13), (n-Bu4N)2[Fe(O2CTrp)2(SCN)2] (14), and [Fe(O2CTrp)2(2-MeIm)2] (15) having an O4/N2 coordination sphere composition.
Keywords: X-ray structures, thermochromism, O- and N-donor ligands, mononuclear Fe(II) complexes, Mössbauer spectroscopy
Introduction
Dinuclear metal complexes bridged by four carboxylate ligands, such as occurs in the classic paddlewheel “copper acetate” core [1-3], comprise an extensive class of compounds. More than 1600 structures of this type have been reported [4]. Tetracarboxylate-bridged diiron(II) complexes are relatively rare species, however, with only 25 crystallographically characterized structures reported in the literature to date [5-8]. This small number is not surprising, considering that Fe(II) carboxylate complexes are rather kinetically labile and have a strong tendency to form species of higher nuclearity [9,10].
Tetracarboxylate-bridged diiron(II) complexes with sterically encumbered carboxylate ligands have been prepared in order to model enzyme active sites, examples of which are displayed in Chart 1. We and others have investigated the Fe(II) coordination chemistry of these carboxylates with the aim of synthesizing model complexes for the active sites of such carboxylate-bridged diiron metalloproteins [11-14] as soluble methane monooxygenase (sMMOH) [15,16], ribonucleotide reductase (RNR-R2) [17-19], stearoyl-acyl carrier protein (ACP) Δ9-desaturase (Δ9D) [20,21], and toluene monooxygenase (ToMOH) [22]. The active sites of these enzymes share the following structural features: two iron atoms coordinated by four carboxylates from either glutamate and/or aspartate and two N-donor ligands from histidines. However, they differ in the carboxylate coordination mode, metal ion coordination number, and the presence of additional ligands, such as water or hydroxide ion.
Chart 1.

The steric requirements of these sterically demanding carboxylate ligands determine the geometry of the resulting diiron(II) compounds and significantly influence their reactivity. Discrete diiron(II) tetracarboxylate compounds, which have the same ligand stoichiometry as the enzymes, can be obtained from an Fe(II) source and an m-terphenyl-derived carboxylate -O2CArR (R = Tol, 4-FPh, Mes) or the dixylyl-substituted benzoates -O2Cdxl in an efficient self-assembly process [23]. Despite the similarity of the carboxylate ligand structures, the resulting diiron(II) complexes differ in their solid-state geometries, forming either doubly (windmill) or quadruply (paddlewheel) bridged species. Moreover, the physical properties and reactivities of these complexes vary. The compounds with -O2CArR (R = Tol, 4-FPh) undergo dynamic carboxylate shifts between the windmill and paddlewheel forms in solution (Scheme 1) [24], whereas those with -O2CArMes are trapped in the doubly bridged windmill conformation [25]. The former ligand also facilitates formation of triply bridged diiron(II) complexes with bulkier neutral ligands [24]. The Fe-Fe distances range between 2.7 and 4.3 Å for these doubly, triply, and quadruply bridged species. The selective addition of stoichiometric amounts of water affects the stereochemistry of dinuclear complexes and also yields doubly bridged complexes with two additional bridging water molecules [26-28]. The carboxylate ligand -O2Cdxl facilitates formation of paddlewheel diiron(II) complexes, which can undergo carboxylate shifts in reactions with dioxygen to form stable diiron(III) peroxo intermediates, revealing a flexibility of the paddlewheel core [8]. The sterically less hindered biphenylcarboxylate ligand (-O2Cbiph), which is asymmetric, preferentially forms tetranuclear iron(II) complexes. These oligomers, however, can disassemble when N-donors are added to form either linear trinuclear iron(II) or paddlewheel diiron(II) species [29]. Scheme 1 summarizes the structural conformations and transformations that these iron-carboxylate compounds can undergo.
Scheme 1.

In the present paper we describe the synthesis and characterization of quadruply bridged diiron(II) complexes with the general formula [Fe2(μ-O2CTrp)4(L)2], bearing four bridging triptycenecarboxylates (-O2CTrp) and two terminal neutral donor ligands (L). These complexes are noteworthy for their stability with respect to disassembly or rearrangement of the diiron(II) paddlewheel core. Introduction of pyridine ligands with electron-withdrawing groups, such as 4-cyano- or 4-acetylpyridine, results in compounds with red-shifted absorption bands that facilitate the study of solvent-dependent thermochromism in coordinating solvents. We also describe how the paddlewheel complexes can be disassembled into mononuclear species by addition of sterically demanding or chelating neutral or anionic donor ligands.
Experimental Section
General Procedures and Methods
Tetrahydrofuran (THF), diethyl ether (Et2O), pentane, toluene, and dichloromethane (CH2Cl2) were saturated with nitrogen and purified by passage through activated alumina columns under an argon atmosphere. Anhydrous 1,2-dimethoxyethane (DME) and chlorobenzene were purchased from Aldrich. The triptycenecarboxylate HO2CTrp was prepared by a modification of literature procedures [30,31] and the corresponding sodium salt was synthesized by allowing the acid to react with a stoichiometric amount of NaOH in MeOH. Fe(OTf)2·2MeCN was prepared by a reported method [32]. All other reagents were obtained from commercial sources and used as received unless otherwise noted. Air-sensitive manipulations were performed by using Schlenk techniques or under a nitrogen atmosphere in an MBraun glovebox.
Physical Measurements
1H-NMR spectra were recorded on a Varian 300 spectrometer in the Massachusetts Institute of Technology Department of Chemistry Instrument Facility (MIT DCIF). Chemical shifts were referenced to the residual solvent peaks. All spectra were recorded at ambient probe temperature. FT-IR spectra were measured on a Thermo Nicolet Avatar 360 spectrometer with OMNIC software. UV-vis spectra were obtained on a Hewlett-Packard 8453 diode-array spectrophotometer under anaerobic conditions. The temperature was controlled with an Oxford ITC 601 cryostat during variable temperature UV-vis studies. Melting points were acquired on an electrothermal Mel-Temp melting point apparatus. When a compound was prepared by two methods, the composition of the material synthesized by the alternative method was confirmed by a unit cell determination of crystalline material and by IR spectroscopy. Mössbauer spectra were obtained on an MS1 spectrometer (WEB Research Co.) with a 57Co source in a Rh matrix maintained at room temperature. Solid samples of 1, 8, 9, and 13 were prepared by suspending ca. 25 μmol of pulverized crystalline material in Apiezon N grease and loading the suspension into a sample holder made from Delrin®. All data were collected at 4.2 K and the isomer shift (δ) values are reported with respect to natural iron foil that was used for velocity calibration at room temperature. The spectra were fit to Gaussian lines by using the WMOSS plot and fit program [33].
[Fe2(μ-O2CTrp)4(THF)2] (1a)
Method A
A portion of NaO2CTrp (845 mg, 2.64 mmol) in THF (20 mL) was added to a solution of [Fe(H2O)6](BF4)2 (446 mg, 1.32 mmol) in THF (30 mL) and the reaction mixture was stirred for 1 d in the presence of molecular sieves (4 Å). The suspension was filtered and the solvent removed to yield a dark pink microcrystalline solid. The crude material was recrystallized from CH2Cl2/THF/pentane to yield colorless blocks of 1a and pink-brown crystals of the side-product 1b, both suitable for X-ray crystallographic analysis. Yield: 517 mg (54%).
Method B
A solution of NaO2CTrp (50 mg, 156 μmol) in THF (5 mL) was added dropwise to a rapidly stirred THF solution (5 mL) of Fe(OTf)2·2MeCN (33 mg, 76 μmol). The resulting pale yellow solution was stirred overnight. The solvent was removed and the product extracted with CH2Cl2 (2 × 5 mL) to yield 26 mg of crude material, which was recrystallized by diffusing pentane into a solution of CH2Cl2 that had been layered with THF to yield colorless crystals (8 mg, 14%) suitable for X-ray crystallography. FT-IR (cm-1, KBr): 3059 (w), 3036 (w), 3015 (w), 2957 (w), 2891 (w), 1612 (s), 1459 (s), 1446 (s), 1408 (s), 1314 (m), 1292 (m), 1178 (m), 1031 (m), 943 (m), 928 (m), 873 (m), 786 (m), 763 (m), 748 (s), 722 (m), 687 (m), 648 (m), 626 (s), 609 (m). Anal. Calcd. for 1a·0.75CH2Cl2 (C92.75H69.5Cl1.5O10Fe2): C, 73.83 ; H, 4.61. Found: C, 73.48; H, 4.41. Mp: 240 °C dec.
[Fe2(μ-O2CTrp)4(py)2] (2)
A solution of 1 (55 mg, 38 μmol) in chlorobenzene (8 mL) was treated with pyridine (py) (10 μL, 116 μmol) under vigorous stirring. Pale yellow rods of 2 formed after 5 d by introduction of Et2O into this solution. Block-shaped crystals of 2 suitable for X-ray crystallography were obtained by vapor diffusion of Et2O into a solution of 2 in DME. Yield: 40 mg (72%). FT-IR (cm-1, KBr): 3059 (w), 2955 (w), 2856 (w), 1618 (s), 1486 (w), 1458 (m), 1447 (s), 1408 (s), 1292 (m), 1218 (w), 1179 (w), 1111 (w), 1083 (w), 1068 (w), 1018 (w), 947 (w), 873 (w), 846 (w), 785 (m), 746 (s), 722 (m), 699 (m), 688 (w), 658 (w), 648 (w), 625 (s), 479 (w), 457 (m), 417 (w). Mp: 235 °C dec.
[Fe2(μ-O2CTrp)4(1-MeIm)2] (3)
1-Methylimidazole (1-MeIm) (14 mg, 220 μmol) was added to a solution of 1 (100 mg, 69 μmol) in toluene (4 mL) and stirred for 1.5 h. The colorless microcrystalline precipitate was isolated by filtration and washed with Et2O. Colorless block crystals suitable for X-ray crystallography were obtained by vapor diffusion of Et2O into a solution of 3 in DME. Yield: 67 mg (67%). FT-IR (cm-1, KBr): 3129 (w), 3058 (w), 2956 (w), 1620 (s), 1535 (m), 1458 (m), 1446 (s), 1407 (s), 1287 (m), 1234 (w), 1178 (w), 1142 (w), 1110 (m), 1089 (m), 1032 (w), 947 (w), 873 (w), 786 (w), 749 (s), 721 (m), 688 (w), 658 (w), 648 (w), 625 (s), 610 (w), 481 (w), 464 (w). Mp: 235 °C dec.
[Fe2(μ-O2CTrp)4(2-MeIm)2] (4)
This compound was prepared from 1 (100 mg, 69 μmol) and 2-methylimidazole (2-MeIm) (14 mg, 220 μmol) by a procedure analogous to that used for synthesizing 3. The colorless microcrystalline precipitate of 4 was recrystallized in hot DME followed by vapor diffusion of Et2O to yield colorless crystals suitable for X-ray crystallography. Yield: 86 mg, (86%). FT-IR (cm-1, KBr): 3383, 3190, 3059, 2957, 1598, 1485, 1458, 1445, 1402, 1287, 1177, 1155, 1128, 1109, 1033, 1016, 874, 806, 786, 750, 722, 688, 646, 625, 610, 482. Mp: 240 °C dec.
[Fe2(μ-O2CTrp)4(2-i-PrIm)2] (5)
This compound was prepared from 1 (200 mg, 138 μmol) and 2-isopropylimidazole (2-i-PrIm) (34 mg, 440 μmol) by a procedure analogous to that used to obtain 3. The colorless microcrystalline precipitate of 5 was recrystallized in hot DME followed by vapor diffusion of Et2O to yield colorless block crystals suitable for X-ray crystallography. Yield: 120 mg (46%). FT-IR (cm-1, KBr): 3403 (w), 3247 (w), 3058 (w), 2955 (w), 2926 (w), 2875 (w), 2818 (w), 1623 (s), 1562 (m), 1458 (s), 1446 (s), 1405 (s), 1291 (m), 1178 (w), 1132 (w), 1092 (m), 1062 (w), 1032 (w), 872 (w), 848 (w), 785 (m), 750 (s), 721 (m), 687 (w), 647 (m), 625 (s), 610 (m), 481 (m), 457 (m). Anal. Calcd. for 5·1.5C4H10O2, C102H87N4O11Fe2: C, 73.96; H, 5.29; N, 3.38. Found: C, 73.81; H, 5.05; N, 3.56. Mp: 225 °C dec.
[Fe2(μ-O2CTrp)4(2-PhIm)2] (6)
This compound was prepared from 1 (100 mg, 69 μmol) and 2-phenylimidazole (2-PhIm) (22 mg, 220 μmol) by a procedure analogous to that used for synthesizing 3. The colorless microcrystalline precipitate of 6 was recrystallized in hot DME followed by vapor diffusion of Et2O to yield colorless crystals suitable for X-ray crystallography. Yield: 75 mg (68%). FT-IR (cm-1, KBr): 3136 (w), 3059 (w), 3007 (w), 2954 (w), 2827 (w), 1623 (s), 1458 (s), 1446 (s), 1404 (s), 1290 (m), 1176 (w), 1107 (m), 1086 (m), 1031 (m), 873 (w), 783 (m), 763 (m), 749 (m), 723 (w), 648 (w), 627 (s), 475 (w), 442 (w). Mp: 255 °C dec.
[Fe2(μ-O2CTrp)4(1-R-2-R’-Im)2] Compounds 7 (R = Et, R’ = i-Pr), 8 (R = Pr, R’ = i-Pr), and 9 (R = Pr, R’ = Ph)
Information on the synthesis and characterization of these compounds is supplied in the Supporting Information.
[Fe2(μ-O2CTrp)4(4-AcPy)2] (10)
Bright red-orange crystals of 10, suitable for X-ray crystallography, were isolated by vapor diffusion of pentane into a reaction mixture of 1 (60.0 mg, 42.1 μmol) and 4-acetylpyridine (4-AcPy) (10.4 mg, 91.2 μmol) in CH2Cl2 (2.5 mL). Yield: 24 mg (37%). FT-IR (cm-1, KBr): 3058 (w), 3015 (w), 2953 (w), 2869 (w), 1701 (m, ν C=O(acetyl)), 1631 (s), 1614 (s), 1557 (m), 1458 (s), 1446 (s), 1407 (s), 1361 (m), 1291 (m), 1264 (m), 1175 (w), 1061 (w), 1033 (w), 1018 (w), 928 (w), 872 (w), 822 (w), 784 (w), 751 (s), 721 (m), 648 (w), 626 (s), 595 (w). UV (CH2Cl2) γmax, nm (εM: 450 (1050). Mp: 320 °C dec.
[Fe2(μ-O2CTrp)4(4-CNPy)2] (11)
Method A
Intensely red-orange colored crystals of 11, suitable for X-ray crystallography, were isolated by vapor diffusion of pentane into a reaction mixture of 1 (80.0 mg, 55.4 μmol) and 4-cyanopyridine (4-CNPy) (11.5 mg, 111 μmol) in CH2Cl2 (6 mL). Yield: 49 mg (58%).
Method B
To a rapidly stirred THF solution (5 mL) of Fe(OTf)2·2MeCN (33 mg, 78 μmol) and NaO2CTrp (50 mg, 156 μmol) was added dropwise a solution of 4-CNPy (8.1 mg, 78 μmol) in THF (5 mL). The resulting suspension was stirred overnight. The solvent was removed and the product was extracted with CH2Cl2 (2 × 3 mL), filtered, and recrystallized from CH2Cl2/pentane to yield red-orange colored crystals of 11. Yield: 34 mg (57%). FT-IR (cm-1, KBr): 3057 (w), 3016 (w), 2954 (w), 2870 (w), 2234 (s, νC≡N), 2070 (w), 1611 (s), 1487 (w), 1458 (m), 1446 (m), 1407 (w), 1291 (m), 1261 (w), 1215 (w), 1178 (w), 1156 (w), 1065 (w), 1031 (w), 1017 (w), 944 (w), 926 (w), 872 (w), 829 (w), 786 (m), 763 (m), 749 (s), 721 (m), 688 (w), 648 (w), 626 (s), 609 (w), 600 (w). UV (CH2Cl2) γmax, nm (εM): 475 (1230). Anal. Calcd. for 11·0.75CH2Cl2, C96.75H61.5N4O8Cl1.5Fe2: C, 73.88; H, 3.94; N, 3.56. Found: C, 74.15; H, 4.47; N, 3.36. Mp: 315 °C dec. The presence of residual CH2Cl2 was confirmed by H-NMR spectroscopy of 11 in CDCl3.
[Fe2(μ-O2CTrp)4(4-PPy)2] (12)
Yellow crystals of 12, suitable for X-ray crystallography, were isolated by vapor diffusion of Et2O into a reaction mixture of 1 (40 mg, 28 μmol) and 4-pyrrolidinopyridine (4-PPy) (8.2 mg, 56 μmol) in CH2Cl2 (2.5 mL). Yield: 24 mg (54%). FT-IR (cm-1, KBr): 3058 (w), 2955 (w), 2858 (w), 1719 (w), 1609 (s), 1532 (m), 1482 (w), 1458 (m), 1446 (m), 1408 (s), 1349 (w), 1316 (w), 1290 (w), 1226 (m), 1178 (w), 1156 (w), 1105 (w), 1033 (w), 1015 (m), 948 (w), 926 (w), 873 (w), 809 (w), 786 (w), 749 (m), 722 (m), 688 (w), 658 (w), 647 (w), 626 (m), 609 (w), 526 (w).
[Fe(O2CTrp)2(TMEDA)] (13)
Method A
A CH2Cl2 solution (2 mL) of 1 (60 mg, 42 μmol) was allowed to react with N,N,N’,N’-tetramethylethylenediamine (TMEDA) (7.5 mg, 84 μmol) for 10 min under stirring. Vapor diffusion of pentane into the pale yellow colored solution afforded colorless blocks of 13. Yield: 36 mg (57%).
Method B
To a rapidly stirred THF solution (5 mL) of Fe(OTf)2·2MeCN (34 mg, 78 μmol) and NaO2CTrp (50 mg, 156 μmol) was added dropwise a solution of TMEDA (9.5 mg, 82 μmol) in THF (5 mL) and the resulting suspension was stirred overnight. The solvent was removed and the product was extracted into CH2Cl2 (3 × 3 mL) and recrystallized from CH2Cl2/pentanes to yield colorless crystals of 13. Yield: 34 mg (57%). FT-IR (cm-1, KBr): 3060 (w), 3003 (w), 2951 (w), 2848 (w), 2797 (w), 1566 (s), 1459 (s), 1445 (s), 1417 (s), 1400 (s), 1286 (m), 1263 (m), 1176 (m), 1029 (m), 950 (m), 875 (m), 797 (m), 764 (s), 732 (m), 688 (m), 645 (m), 625 (s), 608 (w). Anal. Calcd. for 13·CH2Cl2, C49H44N2O4Cl2Fe: C, 69.11; H, 5.21; N, 3.29. Found: C, 68.50; H, 5.18; N, 3.34. Mp: 250 °C dec.
(n-Bu4N)2[Fe(O2CTrp)2(NCS)2] (14)
Colorless block crystals of 14, suitable for X-ray crystallography, were obtained by vapor diffusion of Et2O into a filtered, pale yellow reaction mixture of 1 (50 mg, 35 μmol) and tetrabutylammonium thiocyanate (n-Bu4NSCN) (38.2 mg, 142 μmol) in CH2Cl2 (3 mL) that had been stirred for five min. Yield: 48 mg (55%). FT-IR (cm-1, KBr): 3065 (w), 3014 (w), 2957 (s), 2932 (m), 2873 (m), 2078 (vs, νC≡N), 1584 (s), 1573 (m), 1479 (m), 1458 (m), 1446 (m), 1408 (m), 1379 (m), 1287 (w), 1177 (w), 1152 (w), 1030 (w), 874 (w), 800 (m), 768 (m), 750 (m), 725 (w), 688 (w), 645 (w), 626 (m). Anal. Calcd. for 14, C76H98N4FeO4S2: C, 72.93; H, 7.89; N, 4.48. Found: C, 72.74; H, 7.73; N, 4.48. Mp: 210 °C dec.
[Fe(O2CTrp)2(2-MeIm)2] (15)
Colorless block crystals of 15 (ca. 32 mg), suitable for X-ray crystallography, were obtained from Et2O vapor diffusion into a reaction mixture of 1 (40 mg, 28 μmol) containing 6.0 equiv of (2-MeIm) (13.6 mg, 166 μmol) in a solution of DME (3 mL).
Reactions with Water
In a typical experiment, a CH2Cl2 solution of 1 (20 mg, 14 μmol) was prepared and two equiv of the N-donor were added, followed by the addition of a deoxygenated solution of H2O in THF. The reaction mixture was stirred vigorously, filtered, and subjected either to pentane or Et2O vapor diffusion. X-ray quality crystals were taken directly from the reaction vessel for analysis.
A. Reaction of 7 with H2O
The general protocol described above was followed. Here, 13 equiv of water (1.08 mL, 0.18 mmol, 0.17 M) were added. After stirring for 5 min, the filtered reaction mixture was subjected to pentane vapor diffusion. A small amount of colorless crystals of 7, suitable for X-ray crystallography, were recovered.
B. Reaction of 12 with H2O
The general protocol above was followed. To a solution of 1 (20 mg, 14 μmol) in CH2Cl2 (1.5 mL) was added 4-PPy (4.1 mg, 28 μmol) and the resulting yellow solution was allowed to react for 5 min. Then, either 70 or 350 equiv of water in THF solution (0.49 mL, 0.98 mmol, 2.0 M or 2.44 mL, 4.65 mmol, 2.0 M) were added, and the resulting solutions were stirred for either 5 min (70 equiv) or 2.5 h (350 equiv), before being subjected to vapor diffusion of Et2O. Pale yellow blocks with unit cell parameters identical to those of 12 were recovered in both cases.
X-ray Crystallographic Studies
Single crystals were coated with paratone-N oil, mounted at room temperature on the tips of glass fibers or nylon loops (Oxford magnetic mounting system), and cooled under a stream of cold N2 maintained by a KRYO-FLEX low-temperature apparatus. Intensity data were collected on a Bruker APEX CCD diffractometer with graphite-monochromated Mo Kα radiation (γ = 0.71073 Å) controlled by a Pentium-based PC running the SMART software package [34]. A total of 2800 frames were acquired for each measurement. The structures were solved by direct or Patterson methods and refined on F2 by using the SHELXTL software [35,36]. Empirical absorption corrections were applied with SADABS [37] and the structures were checked for higher symmetry with PLATON [38]. All non-hydrogen atoms were refined anisotropically. In general, hydrogen atoms were assigned idealized positions and given thermal parameters equivalent to either 1.5 (methyl hydrogen atoms) or 1.2 (all other hydrogen atoms) times the thermal parameter of the atom to which they were attached. Treatment of solvent disorder and other aspects of the crystal structure refinements are reported in the Supporting Information.
Crystal data, data collection parameters, and structure refinement details for all compounds are provided in Table S1. Bond distances and angles of 2 - 15 are displayed in Table S2, and ORTEP diagrams of the molecular structures of 2 - 12 are shown in Figures S1 - S11 of the Supporting Information.
Results and Discussion
Ligand Synthesis and Metalation; Preparation and Structural Characterization of [Fe2(μ-O2CTrp)4(THF)2] (1a)
The sodium salt of the triptycenecarboxylic acid HO2CTrp was prepared in three steps by modification of literature procedures. A summary of the synthetic steps is provided in Scheme 2 [30,31]. 9-Bromotriptycene was obtained by a Diels-Alder reaction between 9-bromoanthracene and the benzyne intermediates, generated from the unstable diazonium salt prepared from anthranilic acid. Treatment of this product with a lithiating agent led to the formation of 9-triptycyllithium, which was carbonated and subsequently protonated to form the triptycenecarboxylic acid HO2CTrp. Initially, this crude product was purified by recrystallization in 1,4-dioxane/petroleum ether and then treated with NaOH to obtain the metal salt NaO2CTrp. This salt was employed in anion metathesis reactions with [Fe(H2O)6](BF4)2 or Fe(OTf)2·2MeCN in anhydrous THF to synthesize the starting material 1 and the resultant iron(II) complexes. The carboxylate salt did not show any impurities when analyzed by 1H-NMR spectroscopy. Impurities derived from the 9-bromo-10-triptycenecarboxylic acid, however, were observed in crystal structures of the resulting iron complexes. This brominated ligand impurity could be eliminated by purification of HO2CTrp on silica gel using ethyl acetate/hexanes (1:1) as eluent. Some of the crystal structures reported herein were determined for material synthesized with NaO2CTrp containing fractions of the brominated side product (up to 6%), which cocrystallized as observed in the corresponding X-ray structures of the diiron compounds. Because the bromine atom is not located near the iron coordination site, it had no effect on the coordination geometry.
Scheme 2.

Reaction of [Fe(H2O)6](BF4)2 with two equivalents of NaO2CTrp in anhydrous THF afforded the crude starting material 1. Molecular sieves were present in this reaction mixture in order to keep it anhydrous. Recrystallization of the dark pink powder led to colorless crystals of the paddlewheel complex 1a and larger, pink-brown block crystals of a side-product 1b. We estimate by visual inspection the ratio of these two species to be approximately 7:1. Compounds 1a and 1b, which have an identical iron-to-carboxylate ratio, form in 54% yield based on 1a and were employed in further reactions with N-donor ligands. For simplicity, we hereafter define this mixture of 1a and 1b as starting material 1 and base all yields on 1a. An alternative route (B) for the synthesis of pure 1a was established, in which Fe(OTf)2·2MeCN was used a in metathesis reaction with the sodium salt of the carboxylate. This route yielded pure 1a in smaller yields, however, so the starting material prepared by the former method (A) was employed in further reactions.
The colorless complex 1a exhibits the well-known paddlewheel structure with two iron atoms bridged by four carboxylates. The crystal structure of 1a is displayed in Figure 1, selected bond angles and distances are depicted in Table 1, and the crystallographic information is given in Table S1 (Supporting Information). The two Fe(II) atoms are separated by 2.7307(8) Å and bridged by four carboxylate ligands, which are related by a pseudo-C4 axis along the Fe-Fe vector. The structure of this paddlewheel complex is analogous to those of the diiron(II) compounds [Fe2(μ-O2CR)4(THF)2], where R = Ar4-FPh and dxl (Chart 1), which have very similar Fe-Fe distances of 2.7277(7) Å and 2.735 Å, respectively [8,24]. In all cases, the coordination sphere around the iron atoms is square pyramidal with the THF donor in the axial position.
Figure 1.
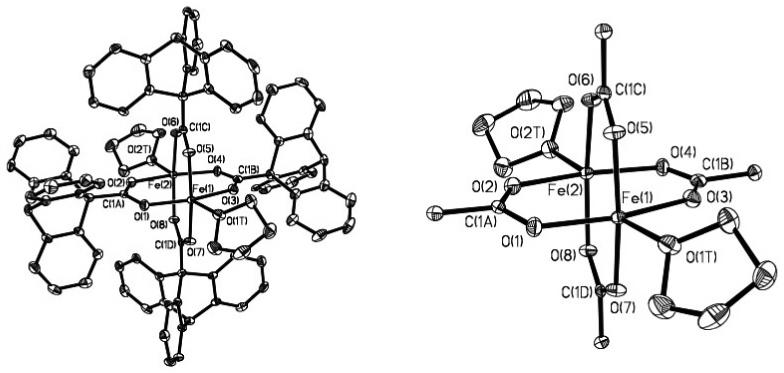
ORTEP diagrams of [Fe2(μ-O2CTrp)4(THF)2] (1a) showing 50% probability thermal ellipsoids for all non-hydrogen atoms. Left: The complete molecule. Right: The molecule omitting triptycene units.
Table 1.
Selected Bond Lengths (Å) and Angles (deg) for 1a
| Interatomic Distances | Interbond Angles | ||
|---|---|---|---|
| Fe(1)-Fe(2) | 2.7307(8) | Fe(1)-O(1)-C(1A) | 119.0(3) |
| Fe(1)-O(1T) | 2.083(3) | Fe(1)-O(3)-C(1B) | 132.8(3) |
| Fe(1)-O(2T) | 2.069(3) | Fe(1)-O(5)-C(1C) | 126.8(3) |
| Fe(1)-O(1) | 2.043(3) | Fe(1)-O(7)-C(1D) | 122.8(2) |
| Fe(1)-O(3) | 2.110(3) | Fe(2)-O(2)-C(1A) | 131.2(3) |
| Fe(1)-O(5) | 2.060(3) | Fe(2)-O(4)-C(1B) | 117.3(3) |
| Fe(1)-O(7) | 2.039(3) | Fe(2)-O(6)-C(1C) | 122.9(3) |
| Fe(2)-O(2) | 2.090(3) | Fe(2)-O(8)-C(1D) | 127.5(2) |
| Fe(2)-O(4) | 2.057(3) | ||
| Fe(2)-O(6) | 2.054(3) | ||
| Fe(2)-O(8) | 2.069(3) |
Atoms are labeled as indicated in Figure 1.
During the synthesis of the paddlewheel complex [Fe2(μ-O2CTrp)4(THF)2] from [Fe(H2O)6](BF4)2 and two equivalents of NaO2CTrp, pale pink-brown blocks of 1b formed as a side-product. X-ray crystallographic analysis exhibited a neutral, trinuclear Fe(II) complex. A triply bridging fluoride ion is located in the center of a nearly equilateral triangle formed by the three iron atoms and is raised only slightly above this plane (ca. 0.14 Å). Two pairs of iron atoms are linked by two bridging carboxylates, whereas, the other iron pair is only singly bridged by another carboxylate. The coordination spheres at these two iron atoms are completed by a sixth carboxylate ligand that is protonated and is therefore only bound in a monodentate fashion. Unfortunately, the quality of the data set was not to sufficient to provide a reliable assignment for the occupancy of this carboxylic acid ligand nor the remaining ligands for this coordination site and, therefore, we do not provide detailed crystallographic information here. The proposed chemical structure of 1b is displayed as Fig. S12 in the Supporting Information.
This type of [Fe3(μ-F)] core has been observed before, but only in pentanuclear complexes in which two equivalent [Fe3(μ-F)] triangles share a common vertex [39]. As in the synthesis of these complexes, the fluoride ion in 1b apparently originates from the BF4- anion of the starting material. It is well-known that fluoride ions are released from BF4- in the presence of strong bases; in this case hydroxide ions are present in the reaction mixture, derived from water molecules deprotonated by the triptycenecarboxylate [40].
Further evidence for 1b only containing ferrous iron is provided by Mössbauer spectroscopy of the starting material 1. Its Mössbauer spectrum, acquired at 4.2 K at zero field is displayed in Figure S13 in the Supporting Information and consists of a somewhat broader quadrupole doublet (δ = 1.33(2) mm/s, ΔEQ = 2.64(2) mm/s and Γ = 0.54 mm/s). These values are characteristic for high spin Fe(II) in an oxygen-rich coordination environment and are in agreement with the proposed structure for 1b [28]. The Mössbauer spectra of 8 and 9 that were acquired under the same conditions, exhibit nearly identical parameters (δ = 1.16(2) mm/s, ΔEQ = 2.77(2) mm/s and Γ = 0.33 mm/s for 8 and δ = 1.16(2) mm/s, ΔEQ = 2.69(2) mm/s and Γ = 0.39 mm/s for 9) which are characteristic for high-spin iron sites in an N/O environment [28]. The corresponding spectra are shown in the Supporting Information in Figures S14 and S15, respectively.
Synthesis and Structural Characterization of Quadruply Bridged Diiron(II) Complexes [Fe2(μ-O2CTrp)4(L)2], 2 - 12
Compounds 2 - 12 having the general formula [Fe2(μ-O2CTrp)4(L)2] were prepared from 1 by displacement of the weakly bound THF molecules upon addition of two equiv of N-donor ligand (L) in modest to excellent yields (37% - 86%). Scheme 2 summarizes the reactions and the corresponding products. The synthetic procedures for these paddlewheel complexes differ depending on the solubilities of the products. Compounds 3 - 6 are quite insoluble and a strategy to prepare them was to precipitate the microcrystalline product from a reaction mixture in toluene, which could then be recrystallized from boiling 1,2-dimethoxyethane followed by Et2O vapor diffusion to obtain X-ray quality crystals. In addition, we alkylated 2-PhIm and 2-i-PrIm in the N1-position (R’ = propyl, ethyl) to enhance the solubility of the corresponding diiron complexes. This reaction involved deprotonation of the imidazole followed by nucleophilic substitution with an alkyl iodide. The resulting compounds 7 - 9 as well as those with pyridine ligands, 2, 10 - 12, had improved solubility, and X-ray quality crystals could be isolated by vapor diffusion of either Et2O or pentane into the corresponding reaction mixtures. Paddlewheel diiron(II) compounds that are soluble in CH2Cl2 can be prepared in a one-pot reaction between Fe(OTf)2·2MeCN, the sodium salt of the triptycenecarboxylate, and the N-donor in a 1:2:1 ratio. This alternative method was utilized for the synthesis of 11, as well as for that of 1a, vide supra.
The crystal structures of 2 - 12 are shown in Figures S1 - S11 and selected bond distances and angles are displayed in Table S2. Crystal data and data collection parameters are presented in Table S1. All the paddlewheel complexes have structures with parameters comparable to those of compound 1a, the only difference being the neutral donor ligand L. In all compounds except 11, two five-coordinate iron centers are related by a crystallographic inversion center, requiring the iron atoms and four oxygen atoms from two of the bridging carboxylates to be positioned in a common plane. One N-donor ligand is bound axially to each of the iron atoms, which have square-pyramidal coordination geometry. Compound 11 crystallizes in a tetragonal space group with one molecule in the asymmetric unit and has analogous metrical parameters to those of the other paddlewheel complexes.
The Fe-Fe distances in 1a, 2 - 12 range from 2.7307(8) to 3.007(2) Å. The smallest distance occurs in the paddlewheel complex with L = THF followed by those with pyridine donor ligands, which have distances between 2.7718(14) and 2.8694(15) Å. In the series of paddlewheel complexes with imidazole-derived ligands the Fe-Fe distances increase significantly, from 2.8603(8) to 3.007(2) Å, with increasing bulkiness of the substituent R on the C2-position of the heterocycle (R = H < Me < i-Pr < Ph). Compared to this 0.15 Å increase in Fe-Fe distance for the imidazole-substituted paddlewheel complexes, those in the series of pyridine-substituted compounds vary less, by ca. 0.10 Å. This difference can be explained by the fact that the residues R in the imidazole ligands sterically interact with the neighboring triptycenecarboxylates, causing them to be pushed away from the Fe-center and the average Fe-O-C angle to increase. The para-substituents on the pyridine derivatives are directed away from the steric bulk of the triptycenecarboxylates and have no significant steric influence on the diiron distance.
A summary of Fe-Fe, Fe-L, and average Fe-O distances, and average Fe-O-C angles of 1a, 2 - 12 is presented in Table 2. It reveals the aforementioned Fe-O-C angle variation (125.0° - 128.3°) that reflects the increasing bulkiness of the N-donor, but no major change in Fe-Oavg bond lengths. This difference is to be expected because angle deformations require less energy than lengthening or shortening of bonds [41]. Figure 2 illustrates the linear relationship between Fe-Fe distances and Fe-O-C angles in 1a, 2 - 12, revealing a systematic trend. These results stand in contrast to those obtained for a series of diiron(II) paddlewheel complexes with m-terphenylcarboxylate ligands of the type [Fe2(μ-O2CAr4-FPh)4(L)2], where L = THF, 1-MeIm, py and t-BuPy [24]. Here, the Fe-Fe distances for all the compounds with N-donor ligands are essentially the same (2.8247 - 2.8249 Å) and the Fe-O-C angles span a narrow range (123.86° - 124.89°); only the THF complex has smaller distances and angles. These dinuclear complexes disassemble to form a mononuclear species when the steric bulk on the N-donor increases (L = 1-benzylimidazole, 1-methylbenzimidazole) [42]. These observations reveal that the triptycenecarboxylate imparts a significantly greater stability to the dinuclear core than other sterically hindered carboxylates.
Table 2.
Comparison of Fe-Fe and Fe-L Distances (Å) and the Average Fe-O-C Angles (deg) in Complexes of the Type [Fe2(μ-O2CTrp)4(L)2]
 | ||||
|---|---|---|---|---|
| Ligand L | Fe-Fe | Fe-O-Cavg | Fe-L | Fe-Oavg |
| THF (1a) | 2.7307(8) | 125.0 | 2.076(3)a | 2.065 |
| 4-CNPy (11) | 2.772(2) | 125.5 | 2.086(3) | 2.054 |
| 4-PPy (12) | 2.822(2) | 126.3 | 2.084(4) | 2.077 |
| Py (2) | 2.8461(7) | 126.5 | 2.119(2) | 2.077 |
| 4-AcPy (10) | 2.8694(15) | 126.6 | 2.127(4) | 2.068 |
| 1-MeIm (3) | 2.8603(8) | 126.8 | 2.080(3) | 2.077 |
| 2-MeIm(4) | 2.9036(9) | 126.8 | 2.085(3) | 2.074 |
| 1-Pr-2-i-PrIm(8) | 2.906(1) | 126.9 | 2.088(2) | 2.087 |
| 2-i-PrIm (5) | 2.912(1) | 127.2 | 2.086(3) | 2.080 |
| 1-Et-2-i-PrIm (7) | 2.921(1) | 127.1 | 2.094(2) | 2.077 |
| 1-Pr-2-PhIm(9) | 2.964(1) | 128.3 | 2.110(4) | 2.079 |
| 2-PhIm(6) | 3.007(2) | 128.3 | 2.132(3) | 2.090 |
Average value.
Figure 2.

Correlation between Fe-Fe Distances (Å) and the Average Fe-O-C Angles in Complexes of the Type [Fe2(μ-O2CTrp)4(L)2] 2 - 12.
The Fe-Fe distances of 2.9036(9) Å to 3.007(2) Å observed for 4 - 9 are about 0.14 Å longer than the largest one previously reported, for the quadruply-bridged diiron(II) complex [Fe2(μ-O2Cdxl)4(1-MeIm)2] (2.864 Å) [8]. The Fe-Fe distance range that can be accommodated in these paddlewheel complexes is significant, 0.27 Å, compared to other dimetallic complexes with different carboxylates in which the distances between the metals usually fall into a narrow range.
Stability of the Diiron Paddlewheel Core
As pointed out above, despite an increase in steric bulk of the neutral donor L, the paddlewheel core stays intact and undergoes neither a carboxylate shift nor disassembly into mononuclear species. In order to investigate further the stability of these diiron paddlewheels, we allowed the complexes to react with water and checked for decomposition of the core or carboxylate shifts. The influence of water on the conformational stability was previously discovered for m-terphenylcarboxylate diiron(II) complexes [26-28,43]. Here, addition of less than ca. 17 equiv of water to the diiron compound resulted in coordination of one water molecule to each iron center, converting it from a quadruply to a doubly bridging diiron carboxylate complex, as portrayed in Scheme 4.
Scheme 4.
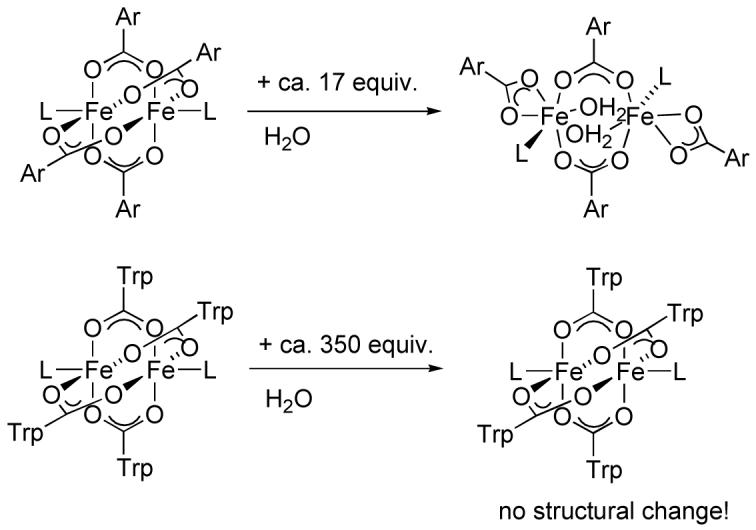
We chose compounds 7 and 12 for water reactivity studies because of their good solubility in CH2Cl2. Solutions of water in anhydrous THF were added to CH2Cl2 solutions of the diiron complexes in an excess of 13 to 350 equiv. The reaction mixtures were stirred for either five min or 2.5 h before being set up for crystallization by vapor diffusion. In all cases, even where a large excess of water had been added and the reaction time was extended to 2.5 h, the resulting crystalline products maintained their structural integrity. This behavior is remarkable, considering that a hexaaquairon(II) cation forms in the case of m-terphenylcarboxylate diiron(II) compounds with the addition of only 35 equiv of water to the dinuclear starting material [27]. We therefore conclude that the triptycene-bridged diiron(II) core has unprecedented conformational stability, which we attribute to interligand steric interactions between the triptycene units. These are locked together in a tongue-in-groove fashion, whereby one phenyl group is embedded between two phenyl rings of the neighboring triptycenecarboxylate, preventing them from undergoing carboxylate shifts. Two space-filling diagrams of compound 1a are displayed in Figure 3 to illustrate this interaction.
Figure 3.
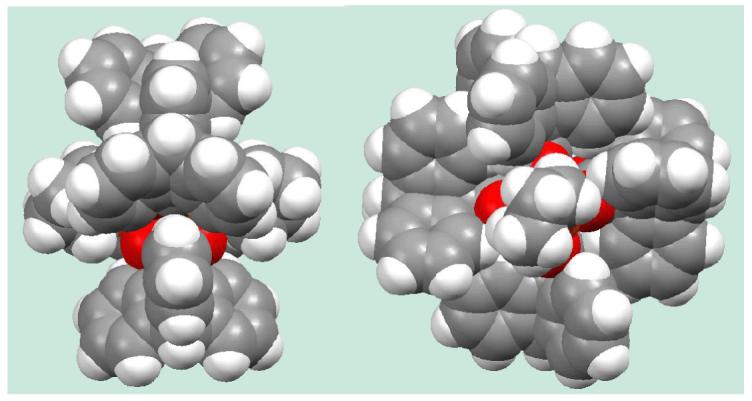
Space-filling diagram of [Fe2(μ-O2CTrp)4(THF)2] (1a). Left: View showing the Fe-Fe vector. Right: View along the Fe-Fe vector.
Electronic Spectroscopy and Equilibria in Coordinating Solvents
High-spin iron(II) complexes with a carboxylate-rich environment are typically colorless, making it difficult to apply UV-vis spectroscopic methods to investigate their chemical reactivity. For this reason, pyridine ligands with electron-withdrawing substituents, such as 4-cyano-(4-CNPy) and 4-acetylpyridine (4-AcPy), were introduced into triptycenecarboxylate-rich diiron(II) complexes, which resulted in the intensely colored complexes [Fe2(μ-O2CTrp)4(4-AcPy)2] (10) and [Fe2(μ-O2CTrp)4 (4-CNPy)2] (11). In 10 and 11, absorptions occur in the visible region at γmax = 450 nm (εM = 1050 M-1 cm-1) and γmax = 475 nm (ε= 1230 M-1 cm-1) in CH2Cl2 solutions, respectively. This approach was applied recently in our group for m-terphenyl-derived carboxylate diiron(II) compounds. Investigation by resonance Raman and electronic absorption spectroscopy confirmed that the colors originate from a charge-transfer transition from the ferrous iron to a π* orbital of the pyridine ligand (MLCT) [28,43]. Methylene chloride solutions of these diiron(II) complexes absorb at somewhat longer wavelengths than the present diiron(II) compounds with triptycenecarboxylate ligands. For example, the complex [Fe2(μ-O2CArTol)4(4-CNPy)2] has an absorption maximum at γmax = 510 nm (ε= 2200 M-1 cm-1), but that of a compound having the more electron-withdrawing carboxylate ligands, [Fe2(μ-O2CAr4-FPh)4(4-CNPy)2] (εM = 2300 M-1 cm-1), is blue-shifted to γmax = 480 nm. This observation is consistent with the assignment of the electronic transition as a MLCT. The triptycenecarboxylate-bridged diiron(II) complex 11 absorbs at a wavelength similar to that of the diiron complex with the more electron-withdrawing terphenylcarboxylate ligands, which leads to the conclusion that the electron-donating capability of the triptycenecarboxylate is about the same as that of -O2 CAr4-FPh. The absorbance maximum of 10, which has the neutral donor 4-AcPy, is blue-shifted by comparison to that of 11. This trend is consistent with the hypsochromic shift in absorbance maximum when L is changed from 4-CNPy to 4-AcPy in the complex [Fe2(μ-O2 CArTol)4(L)2].
When compounds 10 and 11 are dissolved in a coordinating solvent, such as THF, the color disappears, only to reappear when the solutions are cooled to low temperatures. This solvent-dependent thermochromism was investigated for 11 by variable-temperature UV-vis-spectroscopy, monitoring the absorbance change over a temperature range from 193 to 293 K. The equilibrium between 11 and the colorless species can be described by eq 1.
| (1) |
A mathematical analysis that fits the data to a model represented by eq 1, which presumes constant extinction coefficients over the complete temperature range, has been described recently for a similar system [28]. The change in absorbance spectra and a least-squares fit are displayed in Figure 4. The thermodynamic parameters derived from this experiment are ΔH = 18.5(2) kJ mol-1 and ΔS = -16.3(10) J mol-1 K-1, which are very similar to those determined from an analogous study of the windmill complex [Fe2(μ-O2CArTol)2 (O2CArTol)2(4-CNPy)2], ΔH = 19.0(6) kJ mol-1, ΔS = -16.2(16) J mol-1 K-1 [28].
Figure 4.

Top: Temperature-dependent UV-vis spectra of [Fe2(μ-O2CTrp)4(4-CNPy)2] (11) in THF solution. Bottom: The absorbance change at 475 nm fit to eq 1; the solid line represents a least-squares fit to the data points and the estimated errors for the derived thermodynamic parameters are given in the text.
Considering that the geometric structure of the latter is quite different from that of paddlewheel complex 11, this result may seem somewhat surprising. However, we previously determined that these terphenylcarboxylate complexes undergo dynamic carboxylate-shifts from doubly to quadruply bridged structures in a process that does not require much energy. The thermodynamic parameters for exchange of 4-CNPy by THF solvent are therefore mainly determined by the relative strengths of the THF-Fe and (4-CNPy)-Fe bonds and by solvation energies. Preliminary kinetic measurements of a reaction between 11 with excess pyridine suggest that the diiron(II) paddlewheel core stays intact in solution during ligand substitution of 4-CNPy by pyridine. More extensive kinetic studies are planned.
Dimetallic Core Disassembly to Form Mononuclear Fe(II) Complexes. Synthesis and Structural Characterization of 13 - 15
The crystal structure, bond lengths, and interbond angles for 13 - 15 are displayed in Figure 5 and Table 3. Compound 13 can be prepared from 1 by direct ligand substitution using TMEDA or by self-assembly from Fe(OTf)2·2MeCN, NaO2CTrp, and TMEDA in a 1:2:1 ratio in anhydrous THF. Colorless crystals of [Fe(O2CTrp)2(TMEDA)] (13), suitable for X-ray crystallography, were isolated in good yields (57%) by either method. Two carboxylates and one bidentate TMEDA molecule comprise the ligand sphere around the six-coordinate iron(II) atom. The carboxylate ligands each have a long and a short Fe-O bond (ΔFe-O ≈ 0.18 Å), with the shorter bond being trans to the amine donors. The Mössbauer spectrum of 13, displayed in Figure S16 in the Supporting Information, was acquired at zero field and 4.2 K. It shows a sharp quadrupole doublet with a linewidth of Γ = 0.31 mm/s, from which an isomer shift of δ = 1.13(2) mm/s and a quadrupole splitting parameter ΔEQ = 3.01(2) mm/s could be derived. These values are characteristic for a high-spin (S = 2) Fe(II) center [28,42]. Reaction of 1 with four equivalents of tetrabutylammonium thiocyanate led to instant disassembly of the dinuclear starting material to afford colorless blocks of (n-Bu4N)2[Fe(O2CTrp)2(SCN)2] (14) in good yield (55%). The compound has a crystallographically required C2-axis bisecting the molecule. Two symmetry-related bidentate carboxylates and two isothiocyanate ligands support a distorted octahedral coordination environment around the Fe(II) atom. The carboxylates bind in an asymmetric fashion, which is reflected by the two distinct Fe-O distances with ΔFe-O ≈0.21 Å. The longer Fe-O bond is situated anti to the isothiocyanate ligand that binds through its nitrogen (N1) atom at a distance of 2.045(4) Å. The thiocyanate anion is nearly linear, with an N(1)-C(1)-S(1) angle of 178.2(4)°. The angle of coordination, Fe(1)-N(1)-C(1), is 163.1(4)°.
Figure 5.

ORTEP Drawing of [Fe(O2CTrp)2(TMEDA)] (13), [Fe(O2CTrp) (SCN)2]2- (14) and [Fe(O2CTrp)2(2-MeIm)2] (15) (illustrating 50% probability thermal ellipsoids for all non-hydrogen atoms).
Table 3.
Selected Bond Lengths (Å) and Angles (deg) for 13 - 15
| Bond lengths | Bond angles | ||
|---|---|---|---|
| 13 | |||
| Fe(1)-O(1) | 2.097(2) | O(1)-Fe(1)-O(2) | 59.86(7) |
| Fe(1)-O(2) | 2.281(2) | O(1)-Fe(1)-O(3) | 150.71(8) |
| Fe(1)-O(3) | 2.087(2) | O(1)-Fe(1)-O(4) | 97.29(7) |
| Fe(1)-O(4) | 2.257(2) | O(3)-Fe(1)-O(4) | 60.57(7) |
| Fe(1)-N(1) | 2.145(2) | O(2)-Fe(1)-O(4) | 85.71(7) |
| Fe(1)-N(2) | 2.202(2) | O(1)-Fe(1)-N(1) | 109.59(9) |
| O(1)-Fe(1)-N(2) | 100.88(9) | ||
| O(2)-Fe(1)-N(1) | 105.27(9) | ||
| O(2)-Fe(1)-N(2) | 160.50(9) | ||
| O(3)-Fe(1)-N(1) | 93.20(9) | ||
| O(3)-Fe(1)-N(2) | 99.88(9) | ||
| O(4)-Fe(1)-N(1) | 153.00(8) | ||
| O(4)-Fe(1)-N(2) | 94.50(8) | ||
| 14 | |||
| Fe(1)-O(1) | 2.099(3) | C(1)-N(1)-Fe(1) | 163.1(4) |
| Fe(1)-O(2) | 2.312(3) | N(1)-C(1)-S(1) | 178.2(4) |
| Fe(1)-N(1) | 2.045(4) | O(1)-Fe(1)-O(2) | 58.7(1) |
| S(1)-C(1) | 1.643(6) | O(1)-Fe(1)-O(2A) | 95.3(1) |
| C(1)-N(1) | 1.139(6) | O(1)-Fe(1)-O(1A) | 146.5(2) |
| 2.202(2) | O(2)-Fe(1)-O(2A) | 84.2(2) | |
| N(1)-Fe(1)-O(1) | 101.2(1) | ||
| N(1)-Fe(1)-O(1A) | 98.91(12) | ||
| N(1)-Fe(1)-O(2) | 89.14(13) | ||
| N(1)-Fe(1)-N(1A) | 105.8(2) | ||
| 15 | |||
| Fe(1)-O(1) | 2.117(2) | O(1)-Fe(1)-O(2) | 58.82(6) |
| Fe(1)-O(2) | 2.315(2) | O(1)-Fe(1)-O(3) | 93.52(7) |
| Fe(1)-O(3) | 2.222(2) | O(1)-Fe(1)-O(4) | 146.82(7) |
| Fe(1)-O(4) | 2.1482) | O(3)-Fe(1)-O(4) | 60.12(7) |
| Fe(1)-N(1) | 2.095(2) | O(2)-Fe(1)-O(4) | 97.30(7) |
| Fe(1)-N(1A) | 2.099(2) | O(3)-Fe(1)-O(2) | 86.93(7) |
| O(1)-Fe(1)-N(1) | 101.30(9) | ||
| O(1)-Fe(1)-N(1A) | 104.10(8) | ||
| O(2)-Fe(1)-N(1) | 90.34(8) | ||
| O(2)-Fe(1)-N(1A) | 162.79(8) | ||
| O(3)-Fe(1)-N(1) | 160.80(8) | ||
| O(3)-Fe(1)-N(1A) | 92.50(8) | ||
| O(4)-Fe(1)-N(1) | 101.50(8) | ||
| O(4)-Fe(1)-N(1A) | 97.30(8) | ||
Atoms are labeled as indicated in Figure 5.
Compound 15 was isolated from a mixture in which 1 was inadvertently allowed to react with an excess (ca. 6.0 equivalents) of 2-MeIm. Only a few colorless blocks of this compound, which were suitable for X-ray crystallography, formed, together with an amorphous precipitate, and no attempt was made to optimize the synthesis. The Fe(II) atom is coordinated by two bidentate carboxylates and two imidazole ligands. One of the carboxylate ligands binds asymmetrically with ΔFe-O≈ 0.198 Å, whereas the other one coordinates in a more symmetric fashion. The distances between the iron and the imidazole N-atoms are nearly identical.
Addition of a stoichiometric amount of N-donor to 1 results in minimal perturbation of its diiron paddlewheel core to yield 2 - 12. Reactions of 1 with the bidentate N-donor ligand TMEDA, an excessive amount of monodentate ligand 2-MeIm, or the thiocyanate anion, converted the dinuclear core to mononuclear complexes. Monoiron(II) complexes having two carboxylate and two N-donor ligands are relatively rare, and a CSD search [3] revealed that they all contain sterically hindered m-terphenylcarboxylate-ligands [42,44]. The complexes [Fe(O2CArTol)2(TMEDA)] and [Fe(O2CArMes)2(TMEDA)] have a stoichiometry analogous to that of 13, but differ in the carboxylate-binding mode. In particular, only one carboxylate is bidentate and the second is monodentate, which results in five-coordinate species. A complex similar to 15, [Fe(O2CArMes)2(1-MeIm)2], has an unusual, nearly tetrahedral coordination environment. The m-terphenylcarboxylate compounds feature a low coordinate geometry because the steric bulk hinders bidentate coordination. In contrast, for 13 - 15, which contain triptycenecarboxylate ligands, no bulky residues point toward the iron atoms, allowing the carboxylates to bind in a bidentate manner while preventing higher oligomerization.
Compound 14 was prepared in an attempt to synthesize an anionic tetracarboxylate-bridged diiron(II) complex with terminal thiocyanate ligands. No complex of this type has been reported in the literature. When thiocyanate was added to the dinuclear complex 1 in a 2:1 ratio, however, the structure converted to mononuclear 14 with two isothiocyanate ligands per iron atom. Although many structures have two thiocyanates coordinated to Fe(II) [45,46], no compound with the carboxylate-coordination environment of 14 has been previously described.
Conclusions
A general strategy is described for synthesizing 9-triptycenecarboxylate-bridged diiron(II) complexes. With increasing steric demand of the neutral donor axial ligand (L) in these [Fe2(μ-O2CTrp)4(L)2] compounds, a systematic increase in Fe-Fe distance occurs and, for the first time, a diiron(II) paddlewheel complex with an Fe-Fe distance greater than 3 Å was obtained. The reactivity of these complexes with excess water was tested and revealed neither disassembly of the dinuclear core nor a carboxylate shift, leading to the conclusion that the triptycene tetracarboxylate framework has unusual kinetic stability. The thermochromism of the colored compound 11 was used in variable UV-vis temperature studies to determine thermodynamic parameters for ligand substitution. Reactions of 1 with chelating or anionic ligands or with an excess of the N-donor converted the dinuclear core into mononuclear species, which all have an O4/N2 coordination sphere, with a six-coordinate iron atom. Other mononuclear iron(II) complexes of bulky carboxylates have a lower coordination number of 4 or 5.
Supplementary Material
Scheme 3.

Acknowledgement
This work was supported by grant GM032134 from the National Institute of the General Medical Sciences. We thank Dr. Peter Müller for assistance with X-ray crystallography. J.J.K. is the recipient of a National Institute of Health postdoctoral fellowship (F32 GM069236-01). K.L.F. acknowledges the UROP office of MIT for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Van Niekerk JN, Schoening FRL. Acta Crystallogr. 1953;6:227. [Google Scholar]
- [2].Van Niekerk JN, Schoening FRL. Nature. 1953;171:36. [Google Scholar]
- [3].Cotton FA, Murillo CA, Walton RA, editors. Multiple Bonds Between Metal Atoms. 2005. [Google Scholar]
- [4].ConQuest v1.9: ConQuest. Cambridge Crystallographic Data Center; 2007. [Google Scholar]
- [5].Hilderbrand SA, Lippard SJ. Inorg. Chem. 2004;43:5294. doi: 10.1021/ic049649l. [DOI] [PubMed] [Google Scholar]
- [6].Randall CR, Shu L, Chiou Y-M, Hagen KS, Ito M, Kitajima N, Lachicotte RJ, Zang Y, Que L., Jr. Inorg. Chem. 1995;34:1036. [Google Scholar]
- [7].Reisner E, Telser J, Lippard SJ. Inorg. Chem. 2007;46:10754. doi: 10.1021/ic701663j. [DOI] [PubMed] [Google Scholar]
- [8].Chavez FA, Ho RYN, Pink M, Young VG, Jr., Kryatov SV, Rybak-Akimova EV, Andres H, Münck E, Que L, Jr., Tolman WB. Angew. Chem., Int. Ed. 2002;41:149. doi: 10.1002/1521-3773(20020104)41:1<149::aid-anie149>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- [9].Rardin RL, Tolman WB, Lippard SJ. New J. Chem. 1991;15:417. [Google Scholar]
- [10].Lippard SJ. Angew. Chem., Int. Ed. 1988;27:344. [Google Scholar]
- [11].Holm RH, Kennepohl P, Solomon EI. Chem. Rev. 1996;96:2239. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- [12].Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem. Rev. 2000;100:235. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- [13].Du Bois J, Mizoguchi TJ, Lippard SJ. Coord. Chem. Rev. 2000;200-202:443. [Google Scholar]
- [14].Tshuva EY, Lippard SJ. Chem. Rev. 2004;104:987. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- [15].Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew. Chem., Int. Ed. 2001;40:2782. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- [16].Elango N, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Protein Sci. 1997;6:556. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bollinger JM, Jr., Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Science. 1991;253:292. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- [18].Logan DT, Su XD, Åberg A, Regnström K, Hajdu J, Eklund H, Nordlund P. Structure. 1996;4:1053. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- [19].Stubbe J, van der Donk WA. Chem. Rev. 1998;98:705. doi: 10.1021/cr980059c. [DOI] [PubMed] [Google Scholar]
- [20].Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- [21].Lindqvist Y, Huang W, Schneider G, Shanklin J. EMBO J. 1996;15:4081. [PMC free article] [PubMed] [Google Scholar]
- [22].Sazinsky MH, Bard J, Di Donato A, Lippard SJ. J. Biol. Chem. 2004;279:30600. doi: 10.1074/jbc.M400710200. [DOI] [PubMed] [Google Scholar]
- [23].Tolman WB, Que L., Jr. J. Chem. Soc., Dalton Trans. 2002;5:653. [Google Scholar]
- [24].Lee D, Lippard SJ. Inorg. Chem. 2002;41:2704. doi: 10.1021/ic020186y. [DOI] [PubMed] [Google Scholar]
- [25].Hagadorn JR, Que L, Jr., Tolman WB. J. Am. Chem. Soc. 1998;120:13531. doi: 10.1021/ja983333t. [DOI] [PubMed] [Google Scholar]
- [26].Yoon S, Kelly AE, Lippard SJ. Polyhedron. 2004;23:2805. [Google Scholar]
- [27].Yoon S, Lippard SJ. J. Am. Chem. Soc. 2004;126:16692. doi: 10.1021/ja044489y. [DOI] [PubMed] [Google Scholar]
- [28].Yoon S, Lippard SJ. J. Am. Chem. Soc. 2005;127:8386. doi: 10.1021/ja0512531. [DOI] [PubMed] [Google Scholar]
- [29].Reisner E, Abikoff TC, Lippard SJ. Inorg. Chem. 2007;46:10229. doi: 10.1021/ic7014176. [DOI] [PubMed] [Google Scholar]
- [30].Friedman L, Logullo FM. J. Org. Chem. 1963;28:1549. [Google Scholar]
- [31].Kawada Y, Iwamura H. J. Org. Chem. 1981;46:3357. [Google Scholar]
- [32].Hagen KS. Inorg. Chem. 2000;39:5867. doi: 10.1021/ic000444w. [DOI] [PubMed] [Google Scholar]
- [33].Kent TA. WMOSS v2.5. Mössbauer Spectral Analysis Software; 1998. [Google Scholar]
- [34].SMART v5.6: SMART Bruker AXS. Madison, WI: 2000. [Google Scholar]
- [35].Sheldrick GM. SHELXTL00: Program for Refinement of Crystal Structures. University of Göttingen; Göttingen, Germany: 2000. [Google Scholar]
- [36].Sheldrick GM. Acta Crystallogr., Sect. A. 2008;A64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- [37].Sheldrick GM. SADABS: Area-Detector Absorption Correction. University of Göttingen; Göttingen, Germany: 2001. [Google Scholar]
- [38].Spek AL. PLATON: A Multipurpose Crystallographic Tool. Utrecht University; Utrecht, The Netherlands: 2000. [Google Scholar]
- [39].Herold S, Lippard SJ. Inorg. Chem. 1997;36:50. [Google Scholar]
- [40].Zang Y, Jang HG, Chiou Y-M, Hendrich MP, Que L., Jr. Inorg. Chim. Acta. 1993;213:41. [Google Scholar]
- [41].Christoph GG, Koh YB. J. Am. Chem. Soc. 1979;101:1422. [Google Scholar]
- [42].Lee D, Lippard SJ. Inorg. Chim. Acta. 2002;341:1. [Google Scholar]
- [43].Zhao M, Song D, Lippard SJ. Inorg. Chem. 2006;45:6323. doi: 10.1021/ic0602906. [DOI] [PubMed] [Google Scholar]
- [44].Hagadorn JR, Que L, Jr., Tolman WB. Inorg. Chem. 2000;39:6086. doi: 10.1021/ic000531o. [DOI] [PubMed] [Google Scholar]
- [45].Long GJ, Galeazzi G, Russo U, Valle G, Calogero S. Inorg. Chem. 1983;22:507. [Google Scholar]
- [46].Chen K, Zhang YL, Feng MQ, Liu CH. Act. Crystallogr., Sect. E. 2007;63:m2033. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.