

Abstract
Resistance of T cells to activation induced cell death (AICD) is associated with autoimmunity and lymphoproliferation. We found that apigenin (4′,5,7-Trihydroxyflavone), a nonmutagenic dietary flavonoid, augmented both extrinsic and intrinsic pathways of apoptosis in recurrently activated, but not in primarily stimulated, human blood CD4+ T cells. Apigenin potentiated AICD by inhibiting NF-κB activation and suppressing NF-κB-regulated anti-apoptotic molecules, cFLIP, Bcl-xL, Mcl-1, XIAP and IAP, but not Bcl-2. Apigenin suppressed NF-κB translocation to nucleus and inhibited I-κBα phosphorylation and degradation in response to TCR stimulation in re-activated peripheral blood CD4 T cells, as well as in leukemic Jurkat T cell lines. Among the pathways that lead to NF-κB activation upon TCR stimulation, apigenin selectively inhibited PI3K-PKB/Akt, but not PKC-θ activation in the human T cells, and synergized with a PI3K inhibitor to markedly augment AICD. Apigenin also suppressed expression of anti-apoptotic cyclooxygenase 2 (COX-2) protein in activated human T cells, but it did not affect activation of Erk MAPKinase. Thus, in chronically activated human T cells, relatively non-toxic apigenin can suppress anti-apoptotic pathways involving NF-κB activation, and especially cFLIP and COX-2 expression that are important for functioning and maintenance of immune cells in inflammation, autoimmunity and lymphoproliferation.
Keywords: Apoptosis, T cells, Human, Signal Transduction, Autoimmunity
2. INTRODUCTION
Regulated apoptosis programs are critical in maintaining T cell homeostasis after appropriate immune responses [1–4]. Inappropriate proliferation and autoimmunity from recurrent activation of T cells is checked by activation induced cells death (AICD). The major AICD pathway in T cells is thought to be mediated via Fas ligand (FasL) crosslinking Fas death receptor that leads to recruitment of Fas-associated death domain (FADD) protein and subsequent activation of initiator caspase 8 and downstream effector caspases [5–7]. Mutations in Fas/FasL causes lupus-like disease in MRL-lpr and Gld/gld mice, but are rare in classical human SLE [8], and absent in the B/WF1, SNF1 or BXSB mouse models of spontaneous SLE. However, downstream events in the Fas pathway might play a role. Cellular FADD-like IL-1βconverting enzyme inhibitory proteins (cFLIP), which include cFLIPL (long form) and cFLIPS (short form) can inhibit Fas-induced apoptosis by blocking formation of active caspase 8 [6]. Normal T cells downregulate cFLIP after initial activation [7,9,10], but lupus T cells do not [11,12]. In several autoimmune and lymphoproliferative diseases in humans, and in mutant mice, other pathways may intersect to prevent AICD. For instance, abnormal upregulation of COX-2 along with cFLIP in lupus cells [11,13], increased protein kinase B/Akt activity or diminished activity of the inhibitor PTEN in the PI3K-PTEN-PKB/Akt pathway [14], or deficiency of the forkhead transcription factor Foxo3a [15], also result in autoimmunity and lymphoproliferation. The common denominator in these anti-apoptotic pathways is NF-κB activation. Activation of NF-κB signaling pathway in T cells is important for cell survival, proliferation, and effector function [16–21]. TCR plus CD28 engagement eventually induces the canonical NF-κB signaling pathway, which involves phosphorylation-dependent degradation of cytosolic IκBs and subsequent nuclear translocation of p65- and c-Rel-containing NF-κB dimers predominately [18,22,23]. In the earlier steps of TCR signaling, phospholipase PLC-γ1 is activated, which degrades phosphatidylinositol-4-5-biphosphate to generate the lipid second messengers, inositol triphosphate and diacylglycerol. The latter activates protein kinase C (PKC)-θ that phosphorylates CARMA1 (caspase-recruitment domain (CARD)–membrane-associated guanylate kinase (MAGUK) protein 1), which then associates with Bcl10 (B-cell lymphoma 10) and MALT1 (mucosa-associated lymphoid tissue lymphoma translocation protein 1) forming the CBM complex scaffold for activating IKK, leading to NF-κB activation [22,24,25]. TCR plus CD28 signaling also activates the phosphatidylinositol 3-kinase (PI3K) pathway, which generates lipid second messengers, leading to activation of the serine-threonine kinase, protein kinase B (PKB/Akt) that plays a role in the suppression of AICD by inducing NF-κB activation [19]. Recent evidence suggests that Akt cooperates with signals from PKC-θ to induce NF-κB activation in T cells [26], and genetic alteration or drug induced inhibition of PKB/Akt or PKC-θ leads to increased apoptosis of activated T cells [19,20,24,27]. In contrast to AICD, death of activated T cells from deprivation of survival cytokines may not involve PKB [28].
Because strong and irreversible inhibition of NF-κB would be hazardous, relatively innocuous agents that could be used to overcome resistance to AICD in T cells of autoimmune and lymphoproliferative diseases would be of value. Apigenin (4′,5,7-Trihydroxyflavone) is a nontoxic, nonmutagenic dietary flavonoid found in parsley, thyme, peppermint, and herbs like chamomile. Apigenin has been shown to possess anti-carcinogenic properties in vitro, or when administered in vivo by injection [29–34]. In skin cancer cells, apigenin decreases COX-2 transcription/expression, but does not counteract COX-2 enzyme itself, unlike the COX-2 inhibitors, Celecoxib (Celebrex) or Rofecoxib (Vioxx) [35]. Despite suppression of COX-2, apigenin has vasorelaxing, anti-platelet and anti-oxidant properties, which could actually reduce the risk of coronary heart disease and improve endothelial function [36–39]. However, the effect of apigenin on primary non-malignant T cells has not been well defined. Because increased expression of NF-κB dependent genes, such as COX-2, cFLIP and cIAP may cause resistance to AICD in autoimmune T cells of lupus [11,13], we decided herein to study the effect of apigenin on human T cells in the context of AICD.
3. MATERIALS AND METHODS
3.1. Abs and reagents
Apigenin, caspase inhibitor and Nonidet P-40 were purchased from Sigma Chemical (St Louis, MO). Anti-COX-2 polyclonal Ab, antibodies to Bcl-xL, Bcl-2, Mcl-1, IAP-1, IAP-2, XIAP, PARP and pro-caspase 8 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-FLIPL/S mAb (DAVE-2) was purchased from Alexis (San Diego, CA). Anti-PKC-θ and anti-phospho-PKC-θ (anti-phospho-Thr538) Abs, anti-Akt and anti-phospho-Akt Abs (anti-phospho-Ser473), anti-IKK and anti-phospho-IKKα/β Abs (anti-phospho-Ser176/180), anti-I-κBα and anti-phospho- I-κBα Abs (anti-phospho-Ser32) were obtained from Cell Signaling Technology (Danvers, MA) and New England Biolabs (Ipswich, MA). Anti-CD3 (clone UCTH1) and anti-CD28 Ab (clone L293) were purchased from BD biosciences (San Jose, CA). Apigenin was dissolved in DMSO to make a 100 mM stock solution and then diluted in PBS to required concentrations, as described [29,35]. Appropriate dilution of the vehicle (DMSO) was used as a control.
3.2. Cell preparations and short-term T cell lines
Human PBMC were obtained from healthy donors and CD4 T cells were selected from PBMC. To obtain sufficient cells for all the studies and render them susceptible to AICD in vitro, short-term T cell lines were made within a 10–14 day period after one round of stimulation of the CD4 T cells with plate-bound anti-CD3 and anti-CD28 mAb and rIL-2 (20 U/ml), as described [11,40,41]. Human leukemic Jurkat T cell line was grown and maintained in supplemented RPMI 1640. The studies were approved by Northwestern University’s Institutional Review Board for Human Subjects Research.
3.3. Induction and analysis of activation-induced cell death (AICD)
The in vivo AICD process which occurs in repeatedly activated T cells can be mimicked by an in vitro system using TCR-activated T cells cultured in vitro with IL-2 and then restimulated by anti-CD3 or anti-CD3 plus anti-CD28 antibodies [3,7]. Human CD4+ short-term T cell lines that were previously expanded by anti-CD3 and anti-CD28 antibodies and IL-2, were thawed and reactivated by another round of stimulation with anti-CD3 plus anti-CD28 antibodies and then rested for 7–10 days. Human Jurkat T cells fed with fresh medium the day before the experiments. The CD4 T cells (1 × 106/ml), or Jurkat T cells (0.5 × 106/ml) were treated with apigenin for various durations and concentrations (see Results). To determine the effect of apigenin on AICD, the human T cells (2 × 106) were re-stimulated with either plate-bound anti-CD3 or anti-CD3 plus anti-CD28, in presence of 2U/ml rhIL-2, with apigenin or vehicle, for indicated times. Apoptotic cells were detected by staining with annexin V and propidium iodide (PI), using apoptosis detection kit I (BD PharMingen, Mountain View, CA), and specific apoptosis of CD4 T cells gated by flow cytometry, was calculated as (% of experimental apoptosis − % of spontaneous apoptosis)/(100 − % of spontaneous apoptosis). Both early (Annexin+ PI−) and late apoptotic cells (Annexin+ PI+) were included, as described [11,13]. For comparison, apoptosis was also measured in freshly obtained CD4 T cells from human PBMCs that were rested with 20u/ml IL2 overnight, and then primarily stimulated by anti-CD3 in the presence of apigenin for various time points.
3.4. PI3K inhibitor LY294002 treatment CD4 T cells
LY294002 was purchased from Calbiochem (San Diego, CA) and was dissolved in 100% DMSO to stock concentration of 2 × 10−2 M and stored at −20 °C. Fully rested CD4 line T cells (1 × 106/ml) were pre-incubated with various concentration of LY294002 at 37 °C for 30 min, then cells were be transferred to 96 wells plate with apigenin and/or plate-bound anti-CD3 for another 30 hours at 37 °C before analysis apoptosis.
3.5. Analysis of mitochondrial permeability transition (ΔΨm)
Mitochondrial dysfunction was assessed by utilizing rhodamine 123 (Rh123), as described [42]. Cultures were incubated with Rh123 (0.1 μg/ml) for 30 min, cells were harvested and PI was added just before acquiring data by flow cytometry. Disruption of ΔΨm is associated with a loss of Rh123 retention and a decrease in fluorescence, calculated as 100% − (% of control Rh123+ cells − % of experimental Rh123+ cells)/% of control Rh123+ cells. The % of Rh123+ cells in control samples at the very beginning of the experiment was designated as 100%. Dots with minimal light scatter representing debris were gated out, and any PI+ cells were excluded from the calculation.
3.6. Caspase inhibition assay
Anti-CD3 plus CD28 reactivated and then rested CD4 T cells were treated with a caspase inhibitor, z-DEVD.fmk (Sigma), along with DMSO or apigenin for 2 hours and then plated at 2 × 106 in 24-well plates for an additional 24 h. Apoptosis was assessed with annexin V and PI staining.
3.7. NF-κB p65 activation assay
CD4 T cells were harvested at specific times after treatment with reagents, and the nuclear extracts (10μg) were assayed in triplicate for p65 activity using a Nuclear extractor kit and TransAM NF-κB p65 transcription factor assay kit according to the instructions of the manufacturer (Active Motif, Carlsbad, CA). In this assay, nuclear lysates are incubated with plate-bound NF-κB target sequence oligonucleotides, and NF-κB-bound DNA complexes are detected by sandwich ELISA using Abs against the RelA/p65 subunit of NF-κB [19]. The OD450 was read in an ELISA reader (Molecular Devices, Sunnyvale, CA). Values were expressed as the mean ± SEM.
3.8. Western blot analysis
Human CD4+ T cells from lines, or Jurkat T cells were treated with apigenin or vehicle, and stimulated for the indicated times by anti-CD3 or anti-CD3 plus anti-CD28 Abs and the stimulation was stopped with cold wash buffer containing PBS, 5 mM EDTA, and 0.5 mM pervanadate, then cells were lysed in a lysis buffer containing 10mMTris-HCl (pH 7.4); 136 mM NaCl; 1 mM EGTA; 10% glycerol; 50 mM β-glycerophosphate, 2 mM Na3VO4, 10 mM NaF; 1% Nonidet P-40; 10 μg/ml aprotinin; 10 μg/ml leupeptin; 5 μg/ml pepstatin; and 1 mM PMSF for 20 min on ice. Protein content of the lysate was determined with the BCA protein assay kit (Pierce, Rockford, IL), and 50 μg was subjected to SDS-PAGE.
Blots were blocked for 1 h at room temperature in PBS containing 5% milk and 0.1% Tween 20. Membranes were incubated overnight with specific Abs, and then washed three times in PBS containing 0.1% Tween 20, and detected using HRP-conjugated secondary Abs. After three washes in PBS containing 0.1% Tween 20, signals were revealed by an ECL detection system (GE Healthcare, Piscataway, NJ) and visualized by autoradiography. Equal loading of protein in lanes was confirmed in all experiments by stripping and reprobing the blots for either β-actin or total Erk1/2.
3.9. Statistical analysis
Significance (p value < 0.05) was determined by Student’s two-tailed t test. Data are presented as mean ± SEM.
4. RESULTS
4.1. Apigenin potentiates activation-induced cell death in human CD4 T cells
Apigenin has been shown to cause apoptosis in cancer cells, but its effect on normal immune cells has not been defined. We first examined the ability of apigenin to induce apoptosis in short-term primary T cell lines from human peripheral blood which had been previously activated and then rested, as well as human leukemic Jurkat T cell line. Apigenin induced significant apoptosis in human CD4 line T cells (Fig. 1A and 1B) and in Jurkat T cells (Fig. 1C and 1D), in a dose and time dependent manner. Because apigenin by itself had little apoptotic effect on the T cells at 12.5 μM, we next examined whether that low concentration could enhance cell death after T cells were restimulated by TCR cross-linking. The in vivo AICD process can be reproduced in vitro using TCR-activated T cells from short term lines cultured in vitro with IL-2 and then restimulated by the anti-CD3 or anti-CD3 plus anti-CD28 antibodies [3,7]. Herein, the human CD4 T cells from short-term lines were first reactivated with plate-bounded anti-CD3 plus anti-CD28 in the presence of exogenous IL-2 for five days, followed by rest for 7 to 10 days in the presence of low concentration of IL-2 (2 IU/ml). The CD4 T cells were restimulated by anti-CD3, and even at low doses of anti-CD3 stimulation, the addition of 12.5 μM apigenin augmented AICD, as early as 18 hours (p<0.05) (Fig. 1E and 1F). By contrast, apigenin could not augment anti-CD3 induced apoptosis in primarily stimulated fresh human CD4 T cells at the low dose (12.5μM) or even at relative high concentration (25 μM, data not shown) for as long as 48 hours incubation (Fig. 1G). Indeed, primary stimulation with anti-CD3 antibody by itself induced much lower level of apoptosis in the freshly obtained CD4 T cells (Fig. 1G vs. 1F).
Figure 1. Apigenin enhances activation-induced cell death in human CD4 T cells.


A and B. Apigenin (Apn) causes CD4 T cell apoptosis in a dose- and a time-dependent fashion. Short-term line, CD4+ T cells derived from peripheral blood were stimulated once with plate-bounded anti-CD3 (10 μg/ml) plus anti-CD28 (1 μg/ml) for 5 days and then were rested for 7–10 days in the presence of 20 IU/ml of IL-2. The reactivated and then rested CD4+ T cells (1 × 106/ml) were treated with apigenin at indicated concentrations (A), or for various time points at fixed concentrations (B). Specific apoptosis was calculated from flow cytometry of the cells after FITC-conjugated annexin V and PI staining (See Materials and Methods).
C and D. Apigenin causes Jurkat T cell apoptosis in a dose- and a time-dependent fashion. Human Jurkat T cells (0.5 × 106/ml) were treated with apigenin at indicated concentrations (C), or for various time points at fixed concentrations (D), and apoptosis was detected as above.
E and F. Apigenin potentiates activation-induced cell death in re-stimulated human CD4 T cells. Reactivated and then rested CD4+ T cells (1 × 106/ml) from the short-term lines were re-stimulated by anti-CD3 alone in different concentrations (E), or with low-dose anti-CD3 in the presence of DMSO (vehicle control) or 12.5μM apigenin for various time points (F), and apoptosis was detected as above. Apigenin augmented anti-CD3 induce AICD in re-stimulated human T cells as early as 18 hours by about 2 folds (p<0.05). The data represent at least three independent experiments and values are mean ± SEM. * P <0.05; ** P <0.01.
G. Apigenin could not augment apoptosis in freshly obtained human CD4 T cells. Primary CD4 T cells were purified from fresh PBMCs and rested with 20u/ml IL2 overnight, those CD4 T cells (1 × 106/ml) were then stimulated for the first time by anti-CD3 (0.4μg/ml) in the presence of 12.5μM or 25μM Apigenin and 2 U/ml IL2 for various time points, and apoptosis was measured. Apoptosis in fresh human primary CD4 T cells on primary stimulation by anti-CD3 antibody was low, as compared to recurrently activated short-term line cells, and apigenin had no potentiating effect at the low dose (12.5μM) used in Fig. 1F, or ever higher doses (25 μM of Apigenin, data not shown). Experiment was repeated three times and values are mean ± S.D.
4.2. Apigenin-induced activated T cell death involves a caspase-dependent pathway
To determine whether apigenin-induced cell death was contributed by toxic necrosis, in addition to activation of the apoptotic machinery (Fig. 1), we first investigated specific cleavage of the caspase substrate, poly (ADP-ribose) polymerase or PARP [43]. CD4 T cells were treated with various concentrations of apigenin or vehicle for 2 hours followed by low dose anti-CD3 re-stimulation for another 6, 12 or 24 hours. As shown in Figure 2A, PARP (p116) was cleaved in apigenin-treated cells compared with DMSO vehicle control-treated cells (“0”), as demonstrated by reduction of the p116 precursor with simultaneous increase in the p85 cleaved product. Higher concentrations of apigenin were used to detect the changes in blots, as compared to more sensitive flow cytometry (Fig. 1), but apigenin treatment of anti-CD3 re-stimulated cells clearly increased PARP cleavage over anti-CD3 alone (“0”). Apigenin treatment alone, also induced PARP cleavage of the pre-activated and rested T cells, showing that it does not cause death by toxic necrosis, even at the higher concentrations, but it mimics effector caspase activation, as described in anti-CD3 induced T-cell AICD [5–7].
Figure 2. Apigenin induces caspase-dependent cell death in human primary T cells.
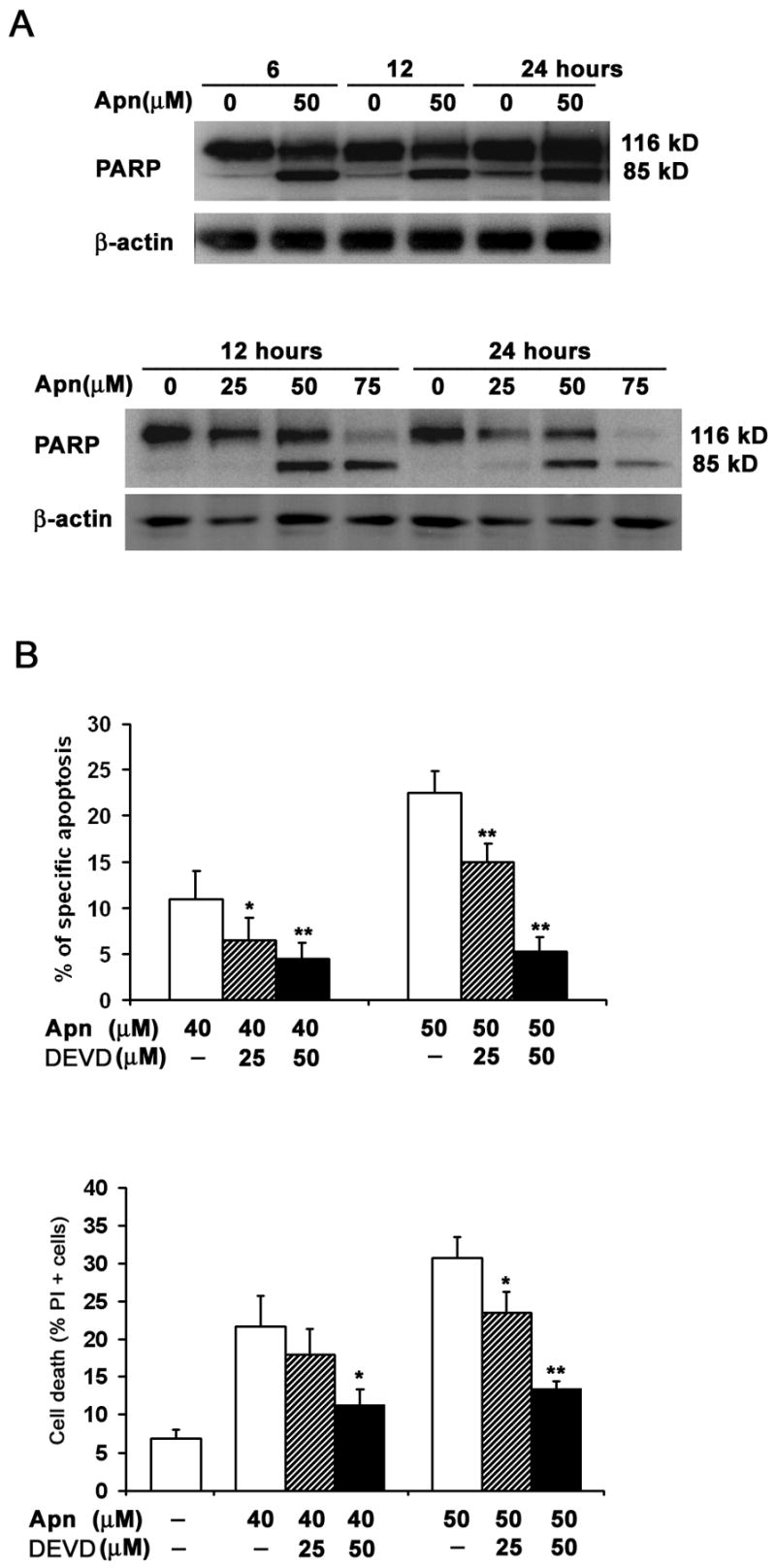
A. Specific cleavage of caspase substrate PARP (poly (ADP-ribose) polymerase) during apigenin-induced apoptosis of CD4 T cells. Reactivated and then rested CD4+ T cells from short-term lines were treated with various concentrations of apigenin for 2 hours before transferring to plates for low-dose anti-CD3 re-stimulation lasting another 6, 12 or 24 hours (Upper Panel); or cultured further without anti-CD3 re-stimulation (Lower Panel). The CD4 T cell lysates were analyzed by immunoblot with anti-PARP antibody. PARP (p116) protein was cleaved to p85 fragment in apigenin-treated cells in contrast to DMSO vehicle control (“0” Apn).
B. Caspase inhibitor (DEVD for z-DEVD.fmk), which preferentially inhibits caspase-3 at the concentrations used, suppressed apigenin-mediated T cell apoptosis when compared with DMSO controls. Reactivated and then rested CD4+ T cells from short-term lines were treated for 24 h with 40μM or 50 μM of apigenin (Apn) alone, or pretreated with 25 μM or 50 μM z-DEVD.fmk for 1 h prior to the addition of apigenin. The percentage of apoptotic cells was determined by annexin V plus PI staining. Specific apoptosis calculated from all Annexin V+ T cells, as described in Methods (Upper Panel) and % of all PI+ dead cells (Lower Panel). Compared to DMSO control (−), z-DEVD.fmk markedly inhibited apigenin-mediated increase in apoptosis. The data represent at least three independent experiments, and values are mean ± SEM. * P <0.05; ** P <0.01.
Because caspase-3 activation is critical in executing final steps of apoptotic cell death, we examined the effect of z-DEVD.fmk, a caspase inhibitor relatively specific for caspase-3 at low doses. As shown in Figure 2B, z-DEVD.fmk markedly inhibited apigenin-mediated increase in activated T-cell apoptosis (Fig. 2B).
4.3. Apigenin-induced activated T cell death occurs via both intrinsic and extrinsic pathways
In the Fas death receptor pathway, caspase-8 is most upstream in the activation cascade of caspases responsible for AICD, and cFLIP proteins play a major inhibitory function against this apoptotic pathway. In the presence of IL-2, activated T cells undergo apoptosis following downregulation of cFLIP proteins [6,7]. We found a decrease in the 55/54-kd caspase-8 proenzyme in re-stimulated CD4 T cells treated with apigenin for 30 hours, indicating that apigenin caused persistent activation of caspase-8 (Fig. 3A), and both cFLIPL and cFLIPS were diminished in presence of apigenin within 3 hours (Fig. 3B). Bcl-2 family members such as Bcl-2, Bcl-xL and Mcl-1 are the major inhibitors in the mitochondrial pathway of apoptosis, and the IAP family proteins are able to inhibit both pathways through their direct binding and inhibiting effector caspases, such as 3, 7, and 9, as reviewed [12]. Apigenin also induced a dose- and time-dependent decrease in levels of Bcl-xL, Mcl-1 expression in CD4 T cells, occurring within 3 hours of re-stimulation, but it did not affect the level of total Bcl-2 (Fig. 3A and 3B). IAP family proteins (IAP-2 and XIAP) also were similarly inhibited by apigenin (Fig. 3A). Addition of apigenin to antiCD3 plus anti-CD28 treatment clearly augmented the above changes over anti-CD3 plus anti-CD28 treatment alone (“0” in Fig. 3A or “DM” in Fig. 3B). For these blots, the T cells had to be re-stimulated with anti-CD3 plus anti-CD28 first to optimally upregulate expression of the anti-apoptotic molecules, so that their reduction by addition of apigenin treatment could be discernible, and again higher concentrations of apigenin were used to detect the changes in proteins in the blots, as compared to more sensitive flow cytometry (Fig. 1).
Figure 3. Apigenin-induced death of activated T cells involves both intrinsic and extrinsic pathways.

A. Apigenin induces a dose-dependent decrease in the levels of Bcl-xL, Mcl-1, IAP family proteins, and pro-Caspase-8, but not Bcl-2 in activated T cells. Human CD4 T cells of short term lines were reactivated and rested, and then treated with various concentrations of apigenin (Apn) starting 2 hours before re-stimulation with anti-CD3 plus anti-CD28 Ab for a total 30 hours. Cell culture was stopped with cold wash buffer and cell lysates were subjected to western blot analysis with antibodies against appropriate apoptotic molecules. Equal loading of protein in lanes was confirmed in all experiments by stripping and reprobing with anti-β-actin antibody. This is a representative of three independent experiments.
B. Apigenin treatment decreases of Bcl-xL, Mcl-1 and FLIP expression in activated CD4 T cells in a time-dependent fashion. CD4 T cells were prepared as in A, and were treated with 50 μM of apigenin (Apn) or DMSO (DM) vehicle for 2 hours before starting re-stimulation by anti-CD3 Ab for various time points. This is a representative of three independent experiments.
C and D. Apigenin-treated primary CD4 T cells exhibit a time-dependent loss of mitochondrial membrane permeability transition. Reactivated and rested human CD4 T cells from short-term lines were treated with 12.5μM (D only) or 50 μM of apigenin for various time points, and then incubated with Rh123 (0.1 μg/ml) for 30 min, harvested, and analyzed by flow cytometry. Compared to control cells, apigenin reduced the % of Rh123+ cells as early as 30 minutes (D, p < 0.01), and the decrease continued at 1, 3, 6, and 24 h of treatment. Disruption of ΔΨm causes loss of Rh123 retention of treated cells vs. controls, which was calculated as described in Materials and Methods. The loss of Rh123 retention is represented in dot plot in C. All data in C and D represent at least three independent experiments and values are mean ± SEM.
E. Apigenin-treated primary CD4 T cells exhibit a dose-dependent loss of mitochondrial membrane permeability transition. Experiment design is the same as in C and D.
We also examined disruption of inner mitochondrial transmembrane potential (ΔΨm) by Rh123 retention over time. Apigenin-treated CD4 T cells from the short-term lines exhibited both dose- and time-dependent loss of ΔΨm, with eventual transition to PI positivity, as illustrated in Fig. 3C – 3E. Compared to control cells, apigenin reduced the mean fluorescence intensity of Rh123 as early as 30 min (p < 0.01), and the reduction continued through 1, 3, 6, and 24 h of treatment (Fig. 3D). These data suggest that mitochondrial dysfunction is an early event in apigenin-induced T cell apoptosis.
4.4. Apigenin induces cell death by suppressing NF-κB activation in human T cells
One of the key signaling events implicated in the prevention of T cell AICD is activation of NF-κB, especially p65/RelA [18,19]. Because NF-κB regulated anti-apoptotic molecules were decreased by apigenin in inducing T cell apoptosis, we asked whether the apigenin was blocking NF-κB activation. Human CD4 T cells that were previously expanded by anti-CD3/CD28, reactivated once and then rested, were pre-incubated with various concentrations of apigenin, before transferring to plates for restimulation with anti-CD3 or with anti-CD3 plus anti-CD28 in the presence of low concentration IL-2 for the time indicated. We used an ELISA-based assay to detect nuclear translocation of NF-κB, as measured by DNA-binding NF-κB complexes in nuclear lysates [19]. As expected, higher levels of NF-κB-bound DNA complexes were detected with the nuclear extract of T cells stimulated by anti-CD3 plus anti-CD28 versus anti-CD3 alone, but apigenin caused a dose dependent decrease of NF-κB activity under both conditions (Fig. 4A). We also found that 40 or 50 μM of apigenin inhibited NF-κB activity well below basal level for extended periods in the restimulated T cells (Fig. 4B, data not shown). Specificity of the assay was confirmed using competing oligonucleotides containing canonical NF-κB binding sites (Fig. 4C).
Figure 4. Apigenin inhibits NF-κB activation and nuclear translocation in TCR-stimulated human primary CD4+ T cells.

A. Apigenin causes a dose-dependent decrease of NF-κB activity in activated human primary CD4 T cells. Rested CD4 line T cells were pre-incubated with 25 μM or 40 μM of apigenin for 2 hours before beginning restimulation with anti-CD3 or with anti-CD3 plus anti-CD28 in presence of low concentration IL-2 for a total of 24 hours. Nuclear NF-κB binding complexes were detected by ELISA. Values are expressed as the mean ± SEM for samples determined in triplicate from three independent experiments. ** P <0.01.
B. Apigenin causes a time-dependent decrease of NF-κB activity in activated human primary CD4 T cells, and at 40 μM concentration (Apn 40), apigenin causes inhibition of NF-κB activity well below basal level by 24 hours in the re-stimulated CD4 T cells. Similar results were obtained with 50 μM concentration of apigenin (data not shown). Experimental design was same as in A.
C. The specificity of the assay was confirmed using competing oligonucleotides containing canonical NF-κB binding sites vs. mutant oligonucleotides that cannot bind to NF-κB.
4.5. Apigenin inhibits IκBα phosphorylation and degradation in activated T cells
To examine whether inhibition by apigenin of TCR/CD3-induced NF-κB activation and nuclear translocation was due to inhibition of IκBα phosphorylation (and subsequent degradation), we incubated the CD4 T cells or Jurkat T cells with 50 μM of apigenin for 1.5 – 2 hours and then transferred them to anti-CD3 or anti-CD3 plus anti-CD28 antibody coated plates for different time periods, and extracts from the T cells were examined by western blot for phosphorylated IκBα. As shown in Fig. 5A and 5B, anti-CD3-induced IκBα phosphorylation was suppressed by apigenin in human CD4+ T cells and Jurkat T cells respectively. It is notable that the primary T cells were repeatedly activated for AICD studies, and therefore, even after “resting” IκBα phosphorylation was detectable in this heterogenous (asynchronous in their resting and activation states) population of primary T cells. Moreover, after anti-CD3 + CD28 restimulation IκBα phosphorylation appeared to diminish in the primary T cells even in the vehicle treated controls, most probably because IκBα levels were reduced rapidly by degradation in T cells within the population that had phosphorylated IκBα at time “0”. To add further to this complexity, IκBα phosphorylation and subsequent degradation and re-synthesis of IκBα in human T cells or synovial fibroblasts shows up and down phase variation causing oscillations in nuclear translocation of NF-κB; and IκBα degradation is detectable at about 45 min of TCR stimulation [44,45]. Similar fluctuations were found in the human T cells here upon further TCR re-stimulation, and consistent with inhibition of phosphorylation of IκBα at subsequent time points, its degradation was also inhibited by apigenin in human primary CD4+ T cells, as detected at 40 min after TCR re-stimulation (Fig. 5C). Overall, the results were much clearer in the case of Jurkat T cells, where anti-CD3 plus anti-CD28 stimulation-induced IκBα phosphorylation was also suppressed by apigenin (Fig. 5B).
Figure 5. Apigenin inhibits IκB phosphorylation in activated T cells.

A. Effect of apigenin on TCR activation-induced phosphorylation of IκBα in human primary T cells. Rested CD4 T cells (2 × 106 cells/well) from short-term lines were treated with 50 μM apigenin for 1.5 – 2 hours before being exposed to plate-coated anti-CD3 plus anti-CD28 for the indicated times. Cytoplasmic extracts were then prepared, fractionated with SDS-PAGE, and electro-transferred to a PVDF membrane, after which a western blot analysis were performed with anti- IκBα and anti-phospho-specific IκBα. Compared to vehicle control (D), apigenin suppressed IκBα phosphorylation in treated cells, detectable at 5 min and almost completely in 40 min. “A” means apigenin, and “D” means DMSO vehicle control. “P+” means positive control from TCR-stimulated Jurkat T cell lysate.
B. Effect of apigenin on TCR activation-induced phosphorylation of IκBα in Jurkat T cells. Jurkat T cells (1 × 106 cells/well) were treated with 50 μM apigenin for 1.5 – 2 hours before beginning stimulation by plate-coated anti-CD3 plus anti-CD28 for the indicated times. Cytoplasmic extracts were then prepared, and western blot analysis was performed as above. Apigenin suppressed IκBα phosphorylation in Jurkat T cells, occurring as early as 5 min and almost completely by 20 min onward.
C. Degradation of total IκBα detectable in TCR-stimulated CD4+ T cells at 40 min was inhibited by apigenin treatment under the same experimental conditions as in A.
The results are representative of three experiments.
4.6. Apigenin does not affect TCR-induced PKC-θ activation
We next investigated whether the apigenin-induced apoptosis in activated human T cells affects PKC-θ phosphorylation, a critical upstream event for TCR stimulation-induced activation of NF-κB pathway. CD4 T cells and Jurkat cells were pre-treated with 50μM apigenin for 1.5 – 2 hours before transferring to plates for activation by anti-CD3 or anti-CD3 plus anti-CD28 for different time periods, and then lysed. Apigenin did not significantly affect levels of either phosphorylated or total PKC-θ in the T cells, either with or without anti-CD3 plus anti-CD28 stimulation (Fig. 6A). Again, it is notable that the primary T cells were repeatedly activated for AICD studies, and therefore, even after “resting” PKC-θ phosphorylation was detectable in this heterogenous (asynchronous in their resting and activation states) population of primary T cells.
Figure 6. Apigenin inhibits PKB/Akt, but not PKC-θ or Erk phosphorylation in TCR activated human T cells.

A. Effect of apigenin on TCR re-stimulation induced activation of PKC-θ (upper panel) and MAPK Erk (lower panel). Short-term CD4 line T cells were rested and treated with 50 μM apigenin for 1.5 – 2 hours, before starting re-stimulation with anti-CD3 plus anti-CD28 for different time periods, and then lysed. Activation of PKC-θ or Erk was determined by immunoblot with antibodies specific for the phosphorylated form of PKC-θ or Erk respectively. Apigenin did not significantly affect the levels of p-PKC-θ or p-Erk or the unphosphorylated total proteins.
B. Effect of apigenin on TCR stimulation-induced Akt activation in short-term line derived, CD4 T cells (2 × 106 cells/well) that were treated with 50 μM apigenin (“A”) or vehicle (“D”) for 1.5 – 2 hours before being transferred to plates coated with anti-CD3 plus anti-CD28 for the indicated times. Whole-cell extracts were prepared and analyzed by western blot analysis with anti-phospho-Akt antibodies. The same membrane was reprobed with an anti-total Akt antibody. “P+” is positive control, as in Figure 5. The results shown are representative of three independent experiments.
C. Effect of apigenin on TCR activation-induced phosphorylation of PKB/Akt in Jurkat T cells. Jurkat T cells (1 × 106 cells/well) were treated with 50 μM apigenin (“A”) or vehicle (“D”) for 1.5 – 2 hours before transfer to plates coated with anti-CD3 plus anti-CD28 for the indicated times. Whole-cell extracts were prepared and analyzed by western blot using anti-phospho-Akt antibody. The same membrane was reprobed with an anti-Akt Ab, and anti-Erk1/2 as a loading control. The results are representative of three independent experiments.
D. PI3K inhibitor LY294002 synergized with apigenin in augmenting anti-CD3 induced AICD in recurrently stimulated human CD4 T cells. CD4 T cells were prepared as in Fig. 1A and B. CD4+ line T cells (1 × 106/ml) were treated with plate-bound anti-CD3 (0.4μg/ml), or apigenin (12.5μM) alone, or anti-CD3 plus apigenin in the presence of LY294002 at indicated concentrations, and after 30 hours, specific apoptosis was measured as described in Fig. 1A and B. LY294002 had an additive effect on apigenin’s potentiation of AICD, in a dose dependent manner. Results represent the mean ± S.D. of three experiments.
4.7. Apigenin suppresses TCR-induced PI3K-PKB/Akt activation pathway
PI3K-dependent activation of PKB/Akt upon TCR/CD3 plus CD28 costimulation is also critical in preventing AICD by activating NF-κB, and inhibition of Akt potentiates AICD [19]. Treatment with apigenin during re-stimulation of the CD4 line T cells resulted in a time-dependent decrease in the levels of phosphorylated (activated) Akt (Fig. 6B). However, the activity of Erk1/2, a MAPK that plays a role in proliferation of activated T cells, was not affected by apigenin treatment of the CD4 line T cells (Fig. 6A). Apigenin also inhibited PKB/Akt phosphorylation in leukemic Jurkat T cells (Fig 6C), and caused their apoptosis (Figure 1).
4.8. PI3K inhibitor LY294002 synergizes with apigenin’s potentiating effect on AICD of human CD4 T cells
Since we found that apigenin induces T cell apoptosis by suppressing TCR- stimulation induced PKB/Akt activation, we tested whether the PI3K inhibitor LY294002 would have an additive effect on AICD of re-stimulated CD4 T cells in the presence of low dose Apigenin. We pre-incubated CD4 T cells with various concentrations of LY294002 at 37 °C for 30 min to inhibit PI3K/Akt signaling, and then exposed the cells to anti-CD3 and/or apigenin for another 30 hours. The results show that LY294002 could enhance apigenin plus anti-CD3 induced cell death in human CD4 T cells in the dose dependent manner, but could not enhance the level of apoptosis in the presence of anti-CD3 or apigenin treatment alone (Fig. 6D). Interestingly, the level of augmented AICD reached by the combination of low-dose apigenin with LY294002 treatment was significantly higher than that achieved with the highest doses of LY294002 alone on the anti-CD3 restimulated T cells, indicating that apigenin was acting by additional mechanisms besides PI3K/Akt inhibition, as discussed below.
4.9. Apigenin suppresses another NF-kB regulated anti-apoptotic and inflammatory gene product in human T cells
In addition to its anti-apoptotic function in some cancer cells, COX-2 is a major inflammatory mediator, and the promoter region of its gene has an NF-κB binding site [46]. To examine whether apigenin suppressed expression of COX-2 protein upon TCR stimulation, we pretreated CD4 T cells with apigenin and transferred the cells to plates for anti-CD3, or anti-CD3 plus anti-CD28 stimulation, in the presence of apigenin. Apigenin suppressed COX-2 expression in the activated T cells (Fig. 7).
Figure 7. Apigenin suppresses another NF-κB-regulated anti-apoptotic and inflammatory gene product in T cells.

Rested short-term line CD4 T cells (2 × 106 cells/well) were treated with various concentrations of apigenin for 2 hours before being exposed to plate-coated anti-CD3 plus anti-CD28 for the indicated times. Cytoplasmic extracts were then prepared, fractionated with SDS-PAGE, and electrotransferred to a PVDF membrane, after which a western blot analysis was performed with anti-COX2 polyclonal antibody. Apigenin suppresses COX-2 protein expression in a dose- and time-dependent manner. The results are representative of three independent experiments.
5. DISCUSSION
In this study, we show that apigenin causes apoptosis of recurrently activated human T cells, involving the same pathways that have been described for AICD. We hypothesized that apigenin, a non-mutagenic plant flavone, mediates its apoptotic effects on repeatedly activated human T cells through the regulation of the NF-κB signaling pathway. Our findings show that apigenin is a potent suppressor of PKB/Akt activation that is associated with inhibition of NF-κB translocation to the nucleus of activated T cells and diminished expression of NF-κB-regulated anti-apoptotic gene products including Bcl-xL, cFLIPs, XIAP and IAPs. The reduction in expression of the antiapoptotic molecules (Fig. 3A and B) could be due to inhibition of transcription, as well as increased degradation, however, the level of Bcl-2 was not affected. The reduction in expression of Mcl-1 could be due to an NF-κB-independent effect of suppression of PKB/Akt activation by apigenin, as implied in other systems [47]. In AICD of T cells, Fas-apoptosis is mediated by caspase-8 activation, which was increased after apigenin treatment. Active caspase-8 cleaves the Bcl-2 homologue, Bid to tBid that inhibits Bcl-2 in the mitochondrial membrane [2,48]. Apigenin caused reduction of cFLIPs, which prevent Fas-mediated apoptosis in activated T cells by inhibiting caspase-8 at the death inducing signal complex [10]. Apigenin also decreased cellular inhibitors of apoptosis proteins (c-IAPs) that inhibit caspase-3, 7 and 9 [2,48]. Consistent with above, apigenin caused a loss of mitochondrial integrity in T cells that initiates the mitochondrial apoptosis pathway by cytochrome c release [2,48]; and treatment with z-DEVD.fmk, a caspase inhibitor relatively specific for caspase-3 at low doses, markedly inhibited apigenin-mediated increase in activated T-cell apoptosis. Thus, both pathways of apoptosis, the extrinsic (Fas) and intrinsic (mitochondrial), are augmented by apigenin leading to elimination of activated T cells.
PKB/Akt, but not PKC-θ activation upon TCR stimulation was significantly inhibited by apigenin in both normal and malignant human T cells. PI3K-dependent activation of Akt is known to play a role in NF-κB activation in several cell types in addition to T cells [19,49,50]. Akt acts upstream of the IKK complex to increase IKK activation, IκBα phosphorylation and degradation, and consequently release of NF-κB components for nuclear translocation, as shown by the NF-κB activation assay used here. Akt could also act by directly increasing NF-κB phosphorylation and coactivator binding [51–53]. These variations may be due in part to cell type-specific differences in IKK expression [54]. Indeed, while our studies on T cells were in progress, it was shown that in macrophages, apigenin inhibits NF-κB activity by acting on the non-canonical pathway [55]. But the effect of apigenin in T lymphocytes is clearly different from results of that study performed on LPS-activated monocyte/macrophages, where apigenin suppressed NF-κB p65 phosphorylation, but had no effect on IkBα phosphorylation/degradation, or DNA binding activity of NF-κB in nuclear lysates. Thus, the major mechanism for preventing AICD in T lymphocytes involves the classical pathway of NF-κB activation [19], which is shown to be inhibited by apigenin here in our study.
In addition to its direct effect, Akt may cooperate with signals from PKC-θ in order to induce NF-κB activation in T cells [26,51]. Interaction between Akt and CARMA1, or Akt-dependent phosphorylation of Bcl10 may account for one possible link between the two major players in T cell activation and proliferation [56], or another possibility is that 3-phosphoinositide-dependent kinase 1 (PDK1) recruits both PKC θ and scaffold protein CARMA1 to lipid rafts, and PDK1-associated CARMA1 then recruits the Bcl10-MALT1 complex, thereby allowing activation of IKK [57]. Together, these reports suggest that PKB/Akt may also interact with PKC-θ signaling pathway in the promotion activated T cell survival.
In addition to PKB/Akt inhibition other mechanisms are probably involved in the augmenting effect of apigenin on AICD of recurrently activated T cells, because a substantial increase was seen over and above that reached with high doses of the PKB/Akt inhibitor, Ly294002 alone (Fig. 6D). We found that apigenin was also able to suppress anti-CD3 induced cyclooxygenase 2 (COX-2) protein expression in human T cells. Over-expression of COX-2 is important for survival and function of recurrently activated, autoimmune inflammatory cells in lupus, but COX-2 gene expression is only partially regulated by NF-κB, and apigenin could downregulate COX-2 at the post-transcriptional level [11–13,58]. Similarly, the anti-apoptotic molecules cFLIPL and cFLIPS were rapidly diminished in presence of apigenin within 3 hours, again suggesting post-transcriptional mechanisms, in addition to NF-κB dependent pathway (Fig. 3B). Therefore, low doses of apigenin could augment AICD of T cells by additional mechanisms, but why that augmenting effect is manifested only when low-dose apigenin is combined the PI3K-Akt inhibitor, Ly294002, along with anti-CD3 restimulation, remains to be worked out in the future.
COX-2 and associated molecules are critical target/s for developing benign, non-mutagenic, steroid-sparing drugs for autoimmune inflammatory disease therapy [59,60]. In preliminary studies, we have found that apigenin is a potent inhibitor of lupus autoimmunity (Kang, Ecklund, and Datta, in preparation). Remarkably, unlike the currently used COX-2 inhibitors, apigenin has vasorelaxing, anti-platelet and anti-oxidant properties, which reduces the risk of coronary disease and improves endothelial function. Obviously improved and relatively harmless, NF-κB and COX-2 inhibitors will be of value not only in lupus therapy, but also for suppressing inflammation in other autoimmune rheumatic diseases, such as rheumatoid arthritis and osteoarthritis, and in prevention of cancers over-expressing COX-2 (colon, breast). Although apigenin is found in commonly consumed herbs, and vegetable, diet is insufficient for bioavailable therapeutic levels due to first pass metabolism in gut and liver. This bioavailability problem might be overcome in the future, as has been the case with similar drugs.
Acknowledgments
This work was supported by NIH grants (R37–AR39157 and RO1–AI41985) to SKD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 1998;280:243–48. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- 2.Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol Rev. 2003;193:70–81. doi: 10.1034/j.1600-065x.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 3.Lenardo MJ. Molecular regulation of T lymphocyte homeostasis in the healthy and diseased immune system. Immunol Res. 2003;27:387–98. doi: 10.1385/IR:27:2-3:387. [DOI] [PubMed] [Google Scholar]
- 4.Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–87. doi: 10.1146/annurev.immunol.22.012703.104554. [DOI] [PubMed] [Google Scholar]
- 5.Ju S-T, Panka DJ, Cui H, Ettinger R, el-Khatib M, Sherr DH, et al. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–48. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 6.Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol. 2001;1:50–58. doi: 10.1038/35095508. [DOI] [PubMed] [Google Scholar]
- 7.Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–23. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- 8.Vaishnaw AK, Toubi E, Ohsako S, Drappa J, Buys S, Estrada J, et al. The spectrum of apoptotic defects and clinical manifestations, including systemic lupus erythematosus, in humans with CD95 (Fas/APO-1) mutations. Arthritis Rheum. 1999;42:1833–42. doi: 10.1002/1529-0131(199909)42:9<1833::AID-ANR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 9.Kirchhoff S, Muller WW, Li-Weber M, Krammer PH. Up-regulation of c-FLIPshort and reduction of activation-induced cell death in CD28-costimulated human T cells. Eur J Immunol. 2000;30:2765–74. doi: 10.1002/1521-4141(200010)30:10<2765::AID-IMMU2765>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 10.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–95. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 11.Xu L, Zhang L, Yi Y, Kang HK, Datta SK. Human lupus T cells resist inactivation and escape death by upregulating COX-2. Nat Med. 2004;10:411–15. doi: 10.1038/nm1005. [DOI] [PubMed] [Google Scholar]
- 12.Datta SK, Zhang L, Xu L. T-helper cell intrinsic defects in lupus that break peripheral tolerance to nuclear autoantigens. J Mol Med. 2005;83:267–78. doi: 10.1007/s00109-004-0624-2. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Bertucci AM, Smith KA, Xu L, Datta SK. Hyperexpression of cyclooxygenase 2 in the lupus immune system and effect of cyclooxygenase 2 inhibitor diet therapy in a murine model of systemic lupus erythematosus. Arthritis Rheum. 2007;56:4132–41. doi: 10.1002/art.23054. [DOI] [PubMed] [Google Scholar]
- 14.Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/− mice. Science. 1999;285:2122–25. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- 15.Lin L, Hron JD, Peng SL. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004;21:203–13. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 16.Zheng Y, Vig M, Lyons J, Van Parijs L, Beg AA. Combined deficiency of p50 and cRel in CD4+ T cells reveals an essential requirement for nuclear factor-κB in regulating mature T cell survival and in vivo function. J Exp Med. 2003;197:861–74. doi: 10.1084/jem.20021610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mora AL, Corn RA, Stanic AK, Goenka S, Aronica M, Stanley S, et al. Antiapoptotic function of NF-κB in T lymphocytes is influenced by their differentiation status: roles of Fas, c-FLIP, and Bcl-xL. Cell Death Differ. 2003;10:1032–44. doi: 10.1038/sj.cdd.4401257. [DOI] [PubMed] [Google Scholar]
- 18.Mittal A, Papa S, Franzoso G, Sen R. NF-κB-dependent regulation of the timing of activation-induced cell death of T lymphocytes. J Immunol. 2006;176:2183–89. doi: 10.4049/jimmunol.176.4.2183. [DOI] [PubMed] [Google Scholar]
- 19.Jones RG, Saibil SD, Pun JM, Elford AR, Bonnard M, Pellegrini M, et al. NF-κB couples protein kinase B/Akt signaling to distinct survival pathways and the regulation of lymphocyte homeostasis in vivo. J Immunol. 2005;175:3790–99. doi: 10.4049/jimmunol.175.6.3790. [DOI] [PubMed] [Google Scholar]
- 20.Saibil SD, Jones RG, Deenick EK, Liadis N, Elford AR, Vainberg MG, et al. CD4+ and CD8+ T cell survival is regulated differentially by protein kinase Cθ, c-Rel, and protein kinase B. J Immunol. 2007;178:2932–39. doi: 10.4049/jimmunol.178.5.2932. [DOI] [PubMed] [Google Scholar]
- 21.Kerstan A, Hunig T. Cutting Edge: Distinct TCR- and CD28-Derived Signals Regulate CD95L, Bcl-xL, and the Survival of Primary T Cells. J Immunol. 2004;172:1341–45. doi: 10.4049/jimmunol.172.3.1341. [DOI] [PubMed] [Google Scholar]
- 22.Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor κB. Immunity. 2006;25:701–15. doi: 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3:221–27. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 24.Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, et al. PKC-θ is required for TCR-induced NF-κB activation in mature but not immature T lymphocytes. Nature. 2000;404:402–7. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- 25.Coudronniere N, Villalba M, Englund N, Altman A. NF-κB activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-θ. Proc Natl Acad Sci USA. 2000;97:3394–9. doi: 10.1073/pnas.060028097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauer B, Krumbock N, Fresser F, Hochholdinger F, Spitaler M, Simm A, et al. Complex formation and cooperation of protein kinase C θ and Akt1/protein kinase B α in the NF-κB transactivation cascade in Jurkat T cells. J Biol Chem. 2001;276:31627–34. doi: 10.1074/jbc.M103098200. [DOI] [PubMed] [Google Scholar]
- 27.Manicassamy S, Sun Z. The critical role of protein kinase C-θ in Fas/Fas ligand-mediated apoptosis. J Immunol. 2007;178:312–19. doi: 10.4049/jimmunol.178.1.312. [DOI] [PubMed] [Google Scholar]
- 28.Scheel-Toellner D, Wang K, Henriquez NV, Webb PR, Craddock R, Pilling D, et al. Cytokine-mediated inhibition of apoptosis in non-transformed T cells and neutrophils can be dissociated from protein kinase B activation. Eur J Immunol. 2002;32:486–93. doi: 10.1002/1521-4141(200202)32:2<486::AID-IMMU486>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 29.McVean M, Weinberg WC, Pelling JC. A p21(waf1)-independent pathway for inhibitory phosphorylation of cyclin-dependent kinase p34(cdc2) and concomitant G(2)/M arrest by the chemopreventive flavonoid apigenin. Mol Carcinog. 2002;33:36–43. doi: 10.1002/mc.10016. [DOI] [PubMed] [Google Scholar]
- 30.Wang IK, Lin-Shiau SY, Lin JK. Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of caspase-9 and caspase-3 in leukaemia HL-60 cells. EurJ Cancer. 1999;35:1517–25. [PubMed] [Google Scholar]
- 31.Vargo MA, Voss OH, Poustka F, Cardounel AJ, Grotewold E, Doseff AI. Apigenin-induced-apoptosis is mediated by the activation of PKCδ and caspases in leukemia cells. Biochem Pharmacol. 2006;72:681–92. doi: 10.1016/j.bcp.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Fotsis T, Pepper MS, Aktas E, Breit S, Rasku S, Adlercreutz H, et al. Flavonoids, dietary-derived inhibitors of cell proliferation and in vitro angiogenesis. Cancer Res. 1997;57:2916–21. [PubMed] [Google Scholar]
- 33.Yin F, Giuliano AE, Law RE, Van Herle AJ. Apigenin inhibits growth and induces G2/M arrest by modulating cyclin-CDK regulators and ERK MAP kinase activation in breast carcinoma cells. Anticancer Res. 2001;21:413–20. [PubMed] [Google Scholar]
- 34.Chen D, Daniel KG, Chen MS, Kuhn DJ, Landis-Piwowar KR, Dou QP. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem Pharmacol. 2005;69:1421–32. doi: 10.1016/j.bcp.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 35.Van Dross RT, Hong X, Pelling JC. Inhibition of TPA-induced cyclooxygenase-2 (COX-2) expression by apigenin through downregulation of Akt signal transduction in human keratinocytes. Mol Carcinog. 2005;44:83–91. doi: 10.1002/mc.20123. [DOI] [PubMed] [Google Scholar]
- 36.Woodman OL, Chan E. Vascular and anti-oxidant actions of flavonols and flavones. Clin Exp Pharmacol Physiol. 2004;31:786–90. doi: 10.1111/j.1440-1681.2004.04072.x. [DOI] [PubMed] [Google Scholar]
- 37.Olszanecki R, Gebska A, Kozlovski VI, Gryglewski RJ. Flavonoids and nitric oxide synthase. J Physiol Pharmacol. 2002;53:571–84. [PubMed] [Google Scholar]
- 38.Zhang YH, Park YS, Kim TJ, Fang LH, Ahn HY, Hong JT, et al. Endothelium-dependent vasorelaxant and antiproliferative effects of apigenin. General Pharmacol. 2000;35:341–47. doi: 10.1016/s0306-3623(02)00113-1. [DOI] [PubMed] [Google Scholar]
- 39.Guerrero JA, Lozano ML, Castillo J, Benavente-Garcia O, Vicente V, Rivera J. Flavonoids inhibit platelet function through binding to the thromboxane A2 receptor. J Thrombosis & Haemostasis. 2005;3:369–76. doi: 10.1111/j.1538-7836.2004.01099.x. [DOI] [PubMed] [Google Scholar]
- 40.Desai-Mehta A, Mao C, Rajagopalan S, Robinson T, Datta SK. Structure and specificity of T-cell receptors expressed by pathogenic anti-DNA autoantibody-inducing T cells in human lupus. J Clin Invest. 1995;95:531–41. doi: 10.1172/JCI117695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi Y, McNerney M, Datta SK. Regulatory defects in Cbl and mitogen-activated protein kinase (extracellular signal-related kinase) pathways cause persistent hyperexpression of CD40 ligand in human lupus T cells. J Immunol. 2000;165:6627–34. doi: 10.4049/jimmunol.165.11.6627. [DOI] [PubMed] [Google Scholar]
- 42.Pagliari LJ, Perlman H, Liu H, Pope RM. Macrophages require constitutive NF-κB activation to maintain A1 expression and mitochondrial homeostasis. Mol Cell Biol. 2000;20:8855–65. doi: 10.1128/mcb.20.23.8855-8865.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Germain M, Affar EB, D’Amours D, Dixit VM, Salvesen GS, Poirier GG. Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. J Biol Chem. 1999;274:28379–84. doi: 10.1074/jbc.274.40.28379. [DOI] [PubMed] [Google Scholar]
- 44.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IκB-NF-κB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–45. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 45.Bai S, Liu H, Chen KH, Eksarko P, Perlman H, Moore TL, et al. NF-κB-regulated expression of cellular FLIP protects rheumatoid arthritis synovial fibroblasts from tumor necrosis factor alpha-mediated apoptosis. Arthritis Rheum. 2004;50:3844–55. doi: 10.1002/art.20680. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor-κB and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem. 1995;270:31315–20. doi: 10.1074/jbc.270.52.31315. [DOI] [PubMed] [Google Scholar]
- 47.Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages. Role of Mcl-1, independent of nuclear factor (NF)-κB, Bad, or caspase activation. J Exp Med. 2001;194:113–26. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109:S97–S107. doi: 10.1016/s0092-8674(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 49.Jones RG, Elford AR, Parsons MJ, Wu L, Krawczyk CM, Yeh WC, et al. CD28-dependent activation of protein kinase B/Akt blocks Fas-mediated apoptosis by preventing death-inducing signaling complex assembly. J Exp Med. 2002;196:335–48. doi: 10.1084/jem.20020307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kane LP, Andres PG, Howland KC, Abbas AK, Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-γ but not TH2 cytokines. Nat Immunol. 2001;2:37–44. doi: 10.1038/83144. [DOI] [PubMed] [Google Scholar]
- 51.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 52.Romashkova JA, Makarov SS. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 53.Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS, Jr, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-κB. Mol Cell Biol. 2000;20:1626–38. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gustin JA, Ozes ON, Akca H, Pincheira R, Mayo LD, Li Q, et al. Cell type-specific expression of the IκB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-κB activation. J Biol Chem. 2004;279:1615–20. doi: 10.1074/jbc.M306976200. [DOI] [PubMed] [Google Scholar]
- 55.Nicholas C, Batra S, Vargo MA, Voss OH, Gavrilin MA, Wewers MD, et al. Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-κB through the suppression of p65 phosphorylation. J Immunol. 2007;179:7121–7. doi: 10.4049/jimmunol.179.10.7121. [DOI] [PubMed] [Google Scholar]
- 56.Narayan P, Holt B, Tosti R, Kane LP. CARMA1 is required for Akt-mediated NF-κB activation in T cells. Mol Cell Biol. 2006;26:2327–36. doi: 10.1128/MCB.26.6.2327-2336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee KY, D’Acquisto F, Hayden MS, Shim JH, Ghosh S. PDK1 nucleates T cell receptor-induced signaling complex for NF-κB activation. Science. 2005;308:114–18. doi: 10.1126/science.1107107. [DOI] [PubMed] [Google Scholar]
- 58.Tong X, Van Dross RT, Abu-Yousif A, Morrison AR, Pelling JC. Apigenin prevents UVB-induced cyclooxygenase 2 expression: coupled mRNA stabilization and translational inhibition. Mol Cell Biol. 2007;27:283–96. doi: 10.1128/MCB.01282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turini ME, DuBois RN. Cyclooxygenase-2: a therapeutic target. Annu Rev Med. 2002;53:35–57. doi: 10.1146/annurev.med.53.082901.103952. [DOI] [PubMed] [Google Scholar]
- 60.Flower RJ. The development of COX2 inhibitors. Nat Rev Drug Discov. 2003;2:179–91. doi: 10.1038/nrd1034. [DOI] [PubMed] [Google Scholar]