

Abstract
Kaposi’s sarcoma-associated herpesvirus (KSHV) is associated with three different human malignancies including Kaposi’s sarcoma (KS), primary effusion lymphoma and multicentric Castleman’s disease. The KS lesion is of endothelial cell in origin and is highly dependent on autocrine and paracrine factors for survival and growth. In this study, we demonstrate that KSHV infection of endothelial cells induces the activation of the pro-survival PI3K/Akt/mTOR pathway. KSHV infection of endothelial cells augmented cell survival in the presence of apoptotic inducers, including etoposide and staurosporine, and under conditions of serum deprivation. We found that KSHV infection of endothelial cells also increased the ability of these cells to form an in vitro tubular network under conditions of stress and growth factor deprivation. Finally, we show that the NF-κB and PI3K pathways are also required for endothelial tubular network formation. Collectively, these results suggest that KSHV infection of endothelial cells modulates cell signaling pathways and induces cell survival and angiogenesis, thereby contributing to the pathogenesis induced by KSHV.
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) is a gammaherpesvirus first identified from Kaposi’s sarcoma (KS) biopsies (1). Since its discovery, KSHV has been found in all epidemiological forms of KS, and viral genomic DNA is present in AIDS-associated KS as well as HIV-negative classic and transplant-associated KS (2). KSHV is also linked to two lymphoproliferative diseases: primary effusion lymphomas (PELs) and multicentric Castleman’s disease (MCD)(3, 4).
Angiogenesis is the formation of new blood vessels from a pre-existing microvascular network. Angiogenesis is important for many normal physiological processes such as organ development and wound healing (5). Many tumors usurp this process and angiogenesis is central for tumor growth and metastasis.
KS is a highly inflammatory and angiogenic vascular tumor with characteristic spindle cells that are of endothelial origin (6). The KS lesion has been shown to express high levels of vascular endothelial growth factor (VEGF) and fibroblast growth factor (bFGF/FGF-2), which are necessary for the maintenance of the lesion (7). In addition, KS-derived cells were found to constitutively release matrix metalloproteinase 9 (MMP-9) (8). Recent studies have shown that KSHV infection of endothelial cells in vitro induces the secretion of angiopoietin 2, MMP-2 and MMP-9 (9-12). Furthermore, anti-angiogenic therapies are currently being used for treatment of KS (13).
The angiogenic potential of several genes of KSHV has also been investigated. KSHV G protein-coupled receptor (vGPCR) is a homolog of human IL-8 receptor, and can induce constitutive, ligand-independent signaling activity. Signaling by vGPCR results in elaboration of many mitogenic and angiogenic cytokines that are vital to KSHV biology and KSHV-driven malignancies. vIL6, a homolog of human IL-6 has also been implicated in the development of tumorigenesis and angiogenesis (14). Our recent studies have shown that the K1 protein of KSHV induces the secretion of VEGF, MMP-9, and also enhances angiogenesis and tumor size in vivo (15).
The PI3K/Akt signaling pathway plays an important role in cell growth and survival. PI3K is a heterodimer composed of a catalytic subunit (p110) and an adaptor/regulatory subunit (p85), which are activated by receptor activation. PI3K activation leads to Akt activation and phosphorylation on residues Threonine 308 and Serine 473 (16, 17). Akt is a critical regulator of PI3K-mediated cell survival, and it phosphorylates and inactivates several pro-apoptotic proteins including members of the forkhead family of transcription factors (FKHR/FOXO) and glycogen synthase kinase-3 beta (18, 19). Activated Akt also enhances protein synthesis through increasing the phosphorylation of the mammalian target of rapamycin (mTOR) (20). As a serine/threonine protein kinase, mTOR has been shown to regulate cell growth, proliferation, motility, survival, protein synthesis, and gene transcription (21, 22). Akt can activate mTOR in different ways. First, Akt can directly phosphorylate and activate mTOR (23). Second, it can activate mTOR through phosphorylation and inhibition of tuberous sclerosis complex 2 (TSC 2), which is a negative regulator of mTOR (24, 25). Third, Akt can modulate mTOR activity through the regulation of cellular ATP levels which leads to the inactivation of AMP-activated protein kinase (AMPK) and TSC 2 (26).
We have previously shown that the KSHV K1 protein activates the PI3K/Akt/mTOR pathway in B cells and endothelial cells (15, 27). Similarly, KSHV vGPCR has also been found to upregulate the PI3K/Akt/mTOR pathway (28-30). These studies were performed with individual KSHV viral genes, and in this manuscript we examined the role of PI3K/Akt/mTOR activation on endothelial cell survival in the context of the whole virus.
Here, we report that KSHV infection of endothelial cells enhances angiogenesis, activates the PI3K/Akt/mTOR pathway, and inactivates AMPK. We find that this activation of the PI3K/Akt cell survival pathway in endothelial cells infected by KSHV confers a survival advantage and protects infected cells from apoptosis.
Materials and Methods
rKSHV.219 Production
Vero cells containing latent rKSHV.219 (KSHV-Vero) and a recombinant baculovirus KSHV Orf50 (BacK50) were kindly provided by Dr. Jeffrey Vieira (31). rKSHV.219 expresses green fluorescent protein (GFP) from the EF-1α promoter, and puromycin resistance gene as a selectable marker (32). The insect SF9 cells were grown in Grace’s insect media (Gibco) supplied with 10% fetal bovine serum (FBS) at 28°C in a 5% CO2 incubator. Three days post BacK50 infection, the baculovirus-containing supernatant was separated from SF9 cells by centrifugation (3500 rpm for 10 min). KSHV-Vero cells were infected with BacK50 and treated with 1mM sodium butyrate. At 65-72 hours post infection, media was harvested and cells were removed by centrifugation (3500 rpm for 10 min). Supernatants were passed through a 0.45μm filter.
Infection of Endothelial cells
Primary human umbilical vein endothelial cells (HUVEC) from Clonetics were cultured in sterile endothelial growth medium (EGM-2, Clonetics) with 10% FBS and were maintained at 37°C in a 5% CO2 environment. Immortalized HUVEC were made by infecting primary HUVEC with a hTERT-encoding retrovirus that also expressed the hygromycin resistance gene as previously described (32). rKSHV.219 produced from KSHV-Vero cells was used to infect immortalized HUVEC in the presence of 0.8mg/ml polybrene. After three days, the cells were selected in media containing 0.5μg/ml puromycin for about two weeks to select for stable KSHV-HUVEC.
Serum-Starvation Assay
1×105 primary immortalized HUVEC and KSHV-HUVEC were plated in each well of a 6-well plate and cultured in EGM-2 with 10% FBS at 37°C. After 24 hours, cells were washed twice with PBS and cultured in endothelial basic media, EBM-2 (Clonetics) with no serum or supplements in a 37°C incubator for up to 5 days. The images were taken with a Zeiss Axiovert 200 inverted fluorescence microscope. The number of cells in ten different fields were counted and averaged after trypan blue staining.
Caspase-3 Activation Assay and PARP-1 Activity Assay
1×106 cells HUVEC and KSHV-HUVEC were seeded in 100mm dishes. After 24 hours, the cells were washed twice with PBS, and then treated with EBM-2 media containing one of the following: DMSO vehicle control, 500 ng/ml TNFα (Sigma), 100 μM etoposide (Sigma) or 20nM staurosporine (Sigma) for 24 hours (33, 34). The cells were harvested and a Caspase-3 activation assay was performed using the ApoAlert® Caspase-3 Fluorescent Assay kit (Clontech). A PARP-1 activity assay was also performed using the Universal Colorimetric PARP Assay kit (Trevigen). In addition, the Caspase-3 inhibitor, Z-VAD-FMK (Sigma), was used in the Caspase-3 activation and PARP-1 assays. In this case cells were grown in EGM-2 complete media with 10%FBS and treated with one of the following: DMSO vehicle control, 100 μM etoposide, 100 μM etoposide plus 50 μM Z-VAD-FMK (35), 20 nM staurosporine, or 20 nM staurosporine plus 50 μM Z-VAD-FMK (35) for 24 hours. Caspase-3 and PARP-1 activity was measured in the cells using the ApoAlert® Caspase-3 Fluorescent Assay kit and the Universal Colorimetric PARP-1 Assay kit, respectively.
Western Blots
HUVEC and KSHV-HUVEC were seeded and grown in EGM-2 media, and after 48 hours, the cells were washed with ice-cold PBS containing 1 mmol/L Na3VO4 and protease inhibitor cocktail (Roche, Mannheim, Germany). Cells were lysed in Triton/NP40 lysis buffer. Western blots were performed with the following antibodies: rabbit phospho-(Tyrosine) p85 PI3K binding motif antibody (1:2000 dilution), rabbit phospho-Akt (Ser473) antibody (1:1000 dilution), rabbit phospho-mTOR (Ser2448) antibody (1:2000 dilution) and phospho-AMPK (Thr172) antibody (1:1000 dilution). All of these antibodies were from Cell Signaling Technology (Beverly, MA).
Tubule Formation Assays
A KSHV-negative lymphoma cell line, BJAB, and the body-cavity-based lymphoma cell line BCBL-1 were grown in RPMI media (Cellgro) with 10% FBS. Cells were washed with PBS twice, and then incubated in serum-free plain RPMI media for 48 hours at 37°C. The conditional media from BJAB and BCBL-1 cells was harvested by centrifugation at 3500 rpm for 10 min. Growth factor-reduced Matrigel (BD Biosciences) was added to the wells of a pre-chilled 24-well plate (0.25ml per well). The plate was placed in a 37°C incubator for 30 min and the matrigel was allowed to solidify. To determine whether the cells could form tubules in B cell conditioned media, 3×104 immortalized HUVEC and KSHV-HUVEC were resuspended in either 1ml plain RPMI media or conditioned media from BJAB or BCBL-1 cells, and were then plated on the solidified matrigel. The plate was incubated in a 37°C incubator for different time points and images were taken with a Nikon T200 fluorescence microscope. To determine whether the cells could form tubules in the absence of VEGF and FGF, the cells were resuspended in VEGF minus EGM-2 media or FGF minus EGM-2 media, plated on the solidified matrigel and incubated at 37°C for upto four days. For the VEGF antibody experiments, HUVEC and KSHV-HUVEC were resuspended in EBM-2 media or EBM-2 media containing 20μg/ml VEGF antibody (Sigma) (36). The cells were then seeded on matrigel and incubated at 37°C incubator for16 hours. Images were taken with a Nikon T200 fluorescence microscope.
For drug treatment experiments, 3×104 immortalized HUVEC and KSHV-HUVEC treated with EBM-2 media overnight were resuspended in 1ml EGM-2 media containing either 50nM Rapamycin (mTOR inhibitor, Calbiochem) (37), 150μM Ciglitazone (AMPK activator, Calbiochem), 2.5μM SU6656 (Src inhibitor, Sigma) (38), 20μM Piceatannol (Syk inhibitor, Calbiochem) (39), 5μM Bay11-7085 (NF-κB inhibitor, Calbiochem) (40) or 20μM LY294002 (PI3K inhibitor, Calbiochem) (41). The cells were then plated on the solidified matrigel. The plate was put in a 37°C incubator for 4 hours. The angiogenic index was calculated as the number of branch points in a field for a particular treatment. Five different fields were counted for each treatment and the number of branch points was averaged.
Results
KSHV infection of endothelial cells induces activation of the PI3K/Akt/mTOR pathway and inactivation of AMPK
In order to obtain KSHV infected endothelial cells, HUVEC were infected with a recombinant KSHV virus containing a green fluorescent protein (GFP) expression cassette and a puromycin expression cassette (31). After selection with 0.5μg/ml puromycin for several weeks, we obtained a 100% KSHV-infected stable HUVEC cell line, which is denoted as KSHV-HUVEC.
In order to determine whether the KSHV infected endothelial cells displayed an activated PI3K/Akt/mTOR pathway, equal micrograms of HUVEC and KSHV-HUVEC cell lysate were loaded on SDS-PAGE and subjected to western blot analysis with antibodies directed against components of the PI3K/Akt pathway. KSHV infection of HUVEC resulted in increased phosphorylation of the p85 sub-unit of PI3K (Fig. 1), indicative of its activation. We also found that there was increased phosphorylation of Akt on Serine 473, concomitant with its activation (Fig.1). mTOR, a downstream target of Akt also appeared to be more activated in KSHV-infected HUVEC compared to uninfected HUVEC, as evidenced by phosphorylation of serine residue 2448. Interestingly, AMPK, a negative regulator of mTOR, showed decreased phosphorylation on threonine residue 172 in KSHV-infected HUVEC (Fig. 1), suggesting that AMPK is less activated in KSHV-infected HUVEC compared to uninfected HUVEC. Thus, the net effect of KSHV infection of endothelial cells is activation of the PI3K/Akt/mTOR pathway and inactivation of AMPK (Fig. 1).
Figure 1. KSHV infection induces activation of the PI3K/Akt/mTOR pathway.
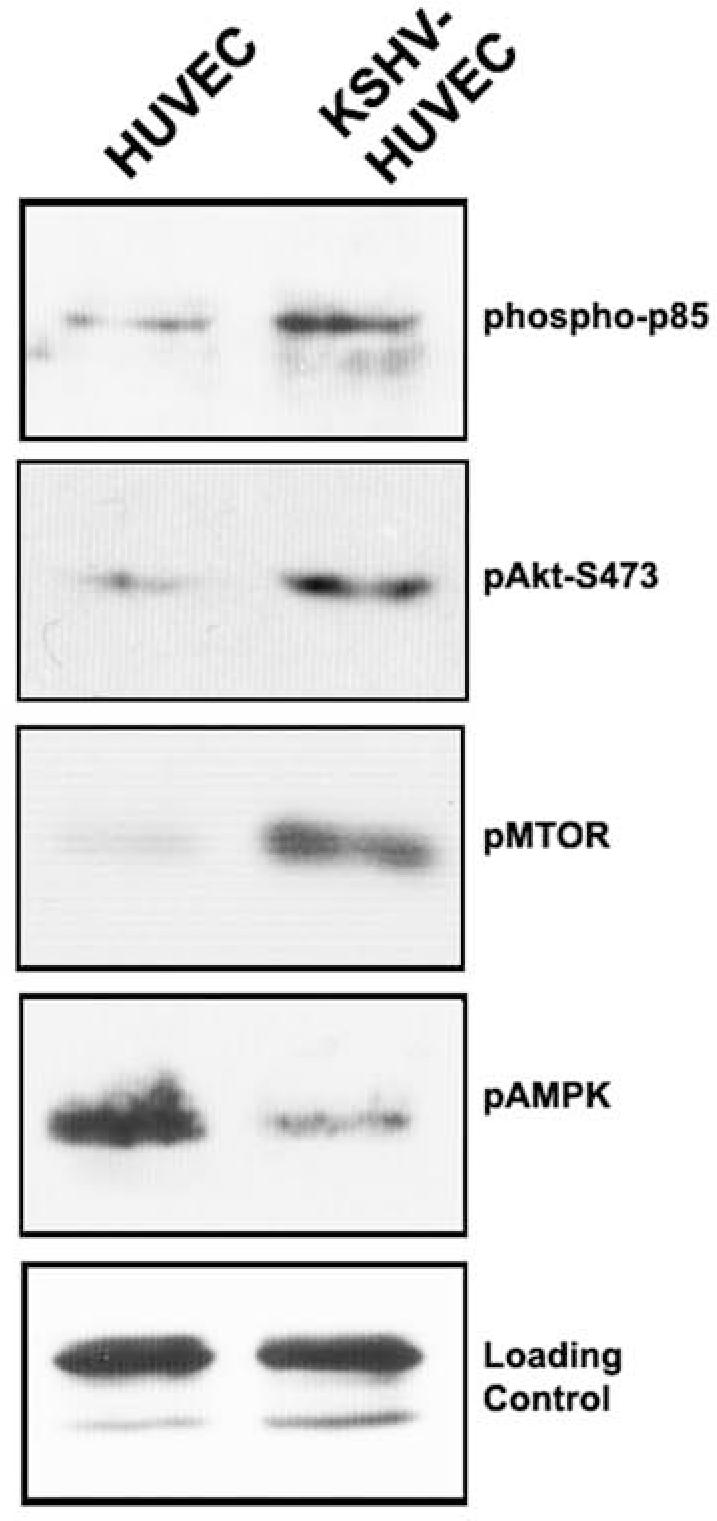
HUVEC and KSHV-HUVEC were harvested 48 hours post-seeding, and equal micrograms of cell lysates were subjected to SDS-PAGE. The gel was transferred to nitrocellulose and western blots were performed with the indicated antibodies. KSHV infection of endothelial cells increased phosphorylation of the p85 sub-unit of PI3K, phosphorylation of Akt, and mTOR. We also found that KSHV-HUVEC displayed a decrease in the phosphorylation of AMPK.
KSHV infection augments endothelial cell survival upon serum deprivation
Since we observed that the PI3K/Akt pathway was activated in KSHV-HUVEC and because this pathway has been associated with cell survival (42, 43), we determined whether the KSHV-infected cells were more resistant to apoptosis induced by serum starvation. We subjected uninfected HUVEC versus KSHV-infected HUVEC to serum starvation using EBM-2 media without serum or cytokine supplements for various time-points. Twenty-four hours post-serum starvation there was no significant difference between the two cell lines, but by 48 hours, some of the uninfected HUVEC died and most of the uninfected HUVEC appeared to be undergoing apoptosis. In contrast, the KSHV-HUVEC appeared healthy (Fig. 2). By 72 hours, more than 60% of the uninfected HUVEC died and had come off the plate. At this time-point, most of the KSHV-infected HUVEC still appeared healthy and were attached to the dish. At 96 hours (day 4) there were only a few surviving uninfected HUVEC compared to KSHV-HUVEC. By 120 hours (day 5), almost all of the uninfected HUVEC died. In contrast, about 40% of KSHV-HUVEC remained alive at day 5 and maintained normal endothelial cell morphology and behavior (Fig 2). These results have been repeated four times. Our results suggest that KSHV infection protects endothelial cells under serum starvation conditions.
Figure 2. KSHV infection confers endothelial cell survival upon serum deprivation.
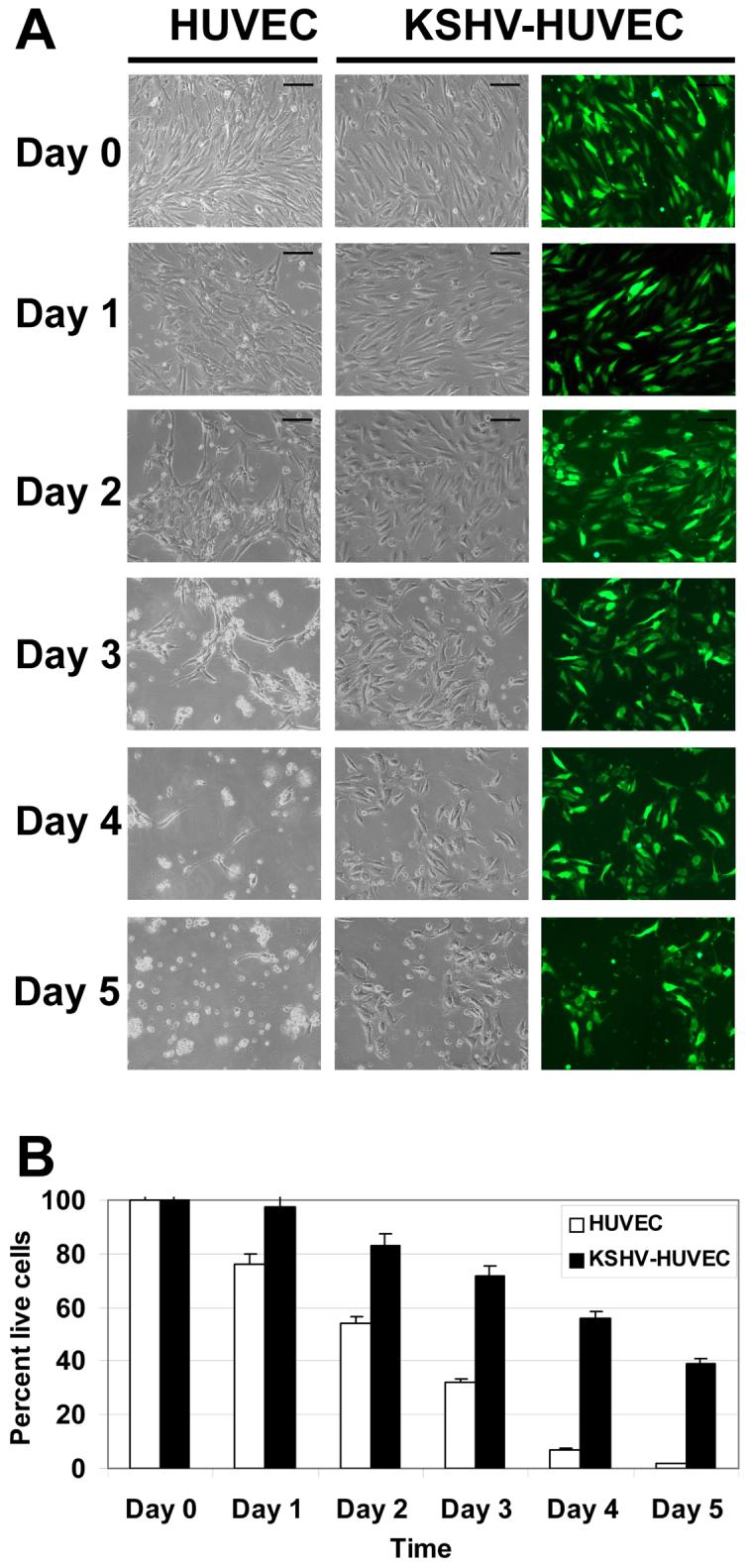
HUVEC and KSHV-HUVEC were grown in serum-free media for five days. (A) Images of the HUVEC and KSHV-HUVEC cells were taken under bright field microscopy. Additionally images of the KSHV-HUVEC (which express GFP) were taken under fluorescence microscopy. Time-points included were 24, 48, 72, 96 and 120 hours post-serum starvation. Images shown were taken at 4X magnification. (B) Graph of the percent of live cells at different time points. The number of live cells in ten different fields were counted and averaged.
KSHV infection suppresses etoposide and staurosporine-induced apoptosis
Next, we sort to determine the ability of uninfected versus KSHV-infected HUVEC to counteract various apoptotic stimuli. HUVEC and KSHV-HUVEC were treated with DMSO (control), or apoptosis inducers including 500ng/ml TNF-α, 100 μM etoposide or 100 nM staurosporine for 24 hours. Caspase-3 is a cysteine protease that has been identified as a key mediator of cell apoptosis and its activation is often used as a hallmark of apoptosis (44, 45). The HUVEC and KSHV-HUVEC cells were then harvested and lysed. Equal micrograms of cell lysates were used to perform Caspase-3 activation assays using a colorimetric assay that measures the cleavage of the Caspase-3 peptide substrate, DEVD-7-amino-4-trifluoromethyl coumarin (DEVD-AFC) resulting in AFC, which could be detected at an emission of 505 nm.
Etoposide inhibits DNA synthesis (46) and staurosporine is a relatively non-selective inhibitor of several protein kinases including protein kinase C and protein kinase A (47, 48). Both of these drugs have previously been shown to induce cellular apoptosis (33, 34). As shown in Fig. 3A, with the treatment of 100 μM etoposide, the activity of Caspase-3 in the uninfected HUVEC was more than twice that of the KSHV-HUVEC, suggesting that KSHV infection of endothelial cells confers a protective effect against etoposide-induced apoptosis. Similarly, activation of Caspase-3 in staurosporine-treated KSHV-HUVEC was lower than that seen in staurosporine-treated uninfected HUVEC (Fig. 3A). Together these data suggest that KSHV infection inhibits endothelial cell apoptosis induced by either etoposide or staurosporine.
Figure 3. KSHV infection suppresses apoptosis induced by etoposide and staurosporine.

(A) HUVEC and KSHV-HUVEC were treated with endothelial media, EBM-2, containing 500 ng/ml TNFα, 100 μM etoposide or 20nM staurosporine for 24 hours. Cells were then harvested and Caspase-3 activity was measured using the Caspase-3 substrate, DEVD-AFC. Etoposide and staurosporine, but not TNFα, induced Caspase-3 activation in the HUVEC and KSHV-HUVEC. However, Caspase-3 activity induced in KSHV-infected HUVEC was lower than that induced in uninfected HUVEC. (B) HUVEC and KSHV-HUVEC were incubated in EGM-2 media containing 10%FBS, with either 100 μM etoposide, 100 μM etoposide plus 50μM Z-VAD-FMK, 20nM staurosporine, or 20nM staurosporine plus 50μM Z-VAD-FMK for 24 hours. Cells were then harvested and Caspase-3 activity was measured as above. Z-VAD-FMK, a Caspase-3 inhibitor, dramatically suppressed Caspase-3 activity induced by etoposide and staurosporine in HUVEC and KSHV-HUVEC. (C) HUVEC and KSHV-HUVEC were incubated in EBM-2 media, containing either 100μM etoposide or 20nM staurosporine for 24 hours. Cells were then harvested and PARP-1 activity was measured using a Colorimetric PARP Assay kit (Trevigen). KSHV-HUVEC treated with etoposide maintained higher PARP-1 activity compared to uninfected HUVEC. D) HUVEC and KSHV-HUVEC were incubated in EGM-2 media containing 10%FBS, with either 100 μM etoposide, 100 μM etoposide plus 50μM Z-VAD-FMK, 20nM staurosporine, or 20nM staurosporine plus 50μM Z-VAD-FMK for 24 hours. Cells were then harvested and PARP-1 activity was measured as above. Z-VAD-FMK dramatically increased PARP-1 activity in HUVEC and KSHV-HUVEC stimulated with etoposide and staurosporine.
Interestingly, TNF-α treatment of either HUVEC or KSHV-HUVEC did not seem to induce much apoptosis in either cell line as measured by Caspase-3 activity (Fig. 3A). This has been previously reported for TNF-α exposure of HUVEC (49). A20, a TNF-α inducible early response Zinc finger protein, has been previously shown to downregulate NF-κB signaling in HUVEC and also inhibit apoptosis of endothelial cells induced by TNF-α (50, 51). Thus, endothelial cells appear to be resistant to the effects of TNF-α.
Further, to determine whether the apoptosis we observed with etoposide and staurosporine was Caspase-3-dependent, we performed Caspase-3 activity assays with HUVEC and KSHV-HUVEC in the presence of the Caspase-3 inhibitor, Z-VAD-FMK. As shown in Fig. 3B, apoptosis induced by both etoposide and staurosporine was Caspase-3 dependent since almost no apoptosis was induced in either HUVEC or KSHV-HUVEC with etoposide or staurosporine in the presence of the Caspase-3 inhibitor, Z-VAD-FMK (Fig. 3B).
We also performed poly (ADP-ribose) polymerase (PARP-1) activity assays to measure apoptosis. PARP-1 catalyzes the NAD-dependent addition of poly ADP-ribose to adjacent nuclear proteins (52). During apoptosis, PARP-1 is specifically cleaved from its enzymatically active form to an inactive form, and the cleavage of PARP has been demonstrated to be a reliable marker for apoptosis in a wide variety of cell types (53-56). We performed a PARP-1 activity assay using lysates from HUVEC or KSHV-HUVEC treated with etoposide or staurosporine. Higher PARP-1 activity corresponds to lower apoptosis and conversely, lower PARP-1 activity corresponds to a higher degree of apoptosis. We found that the PARP-1 activity in HUVEC treated with etoposide or staurosporine was lower than the PARP-1 activity seen in KSHVHUVEC treated with these same drugs (Fig. 3C). This is consistent with the Caspase-3 activity data shown in Fig. 3A. Finally, we also performed the PARP-1 activity assay in the presence of the Caspase-3 inhibitor, Z-VAD-FMK (Fig. 3D). The presence of Z-VAD-FMK greatly increased PARP-1 activity in the etoposide and staurosporine treated HUVEC and KSHV-HUVEC. In summary, our data indicates that Caspase-3-mediated apoptosis by etoposide and staurosporine is much reduced in KSHV-infected HUVEC as compared to uninfected HUVEC.
KSHV-infected endothelial cells form tubular networks under conditions of cell stress
The tubule formation assay is an in vitro assay for monitoring the formation of capillary-like tubules by endothelial cells and is used to model and measure cell angiogenesis (57). We used growth factor-reduced matrigel as the basement matrix to induce endothelial cell tubule formation under various conditions. Unlike standard matrigel, growth factor-reduced matrigel contains significantly lower levels of stimulatory cytokines and growth factors which allowed us to compare infected and uninfected HUVEC without the complications of high levels of cytokines in the media. HUVEC and KSHV-HUVEC were plated on matrigel and allowed to form tubule networks. We found that the KSHV-infected HUVEC formed more tubule structures than uninfected HUVEC within a 24-hour period (Fig. 4A).
Figure 4. Tubule formation assay for HUVEC and KSHV-HUVEC.
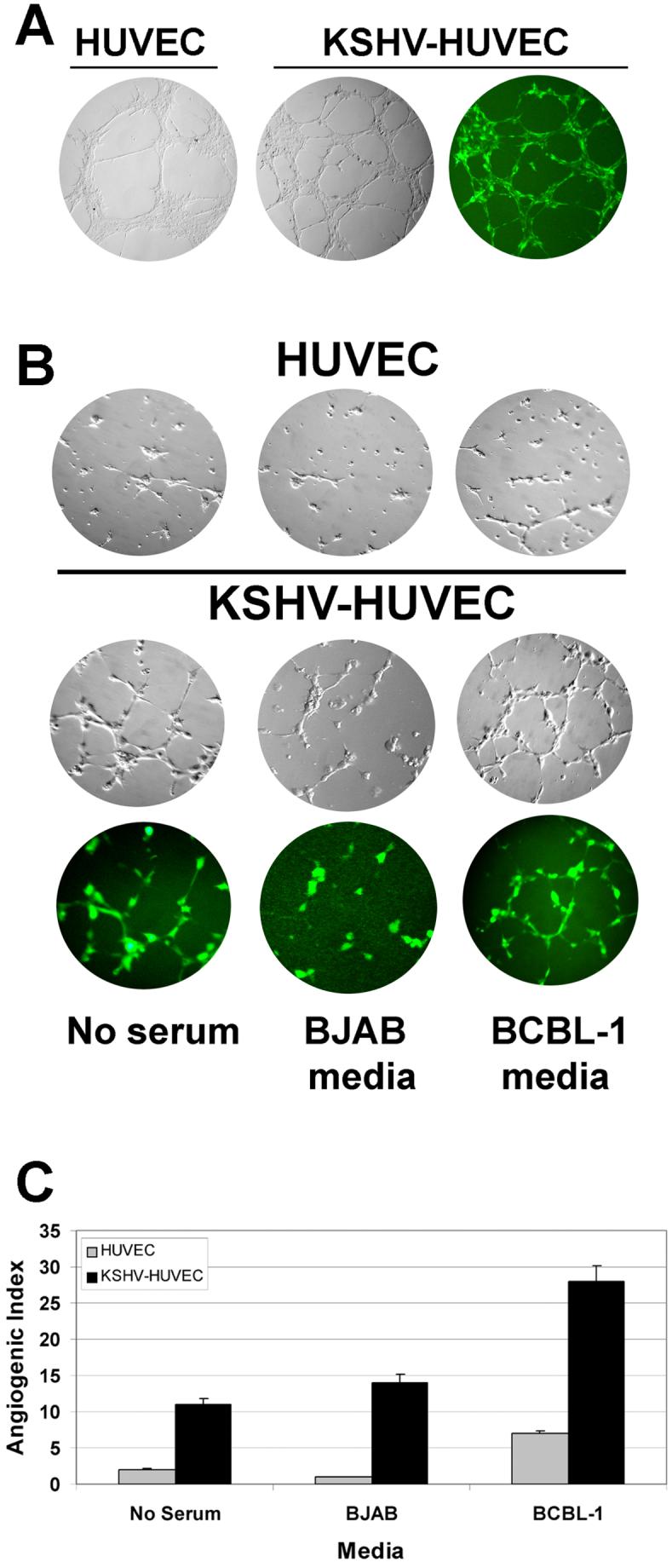
(A) HUVEC and KSHV-HUVEC were plated on growth factor reduced matrigel in the presence of media with serum for 24 hours and allowed to form capillary-like tubules. (B) KSHV-negative BJAB and KSHV-positive BCBL-1 cells were incubated in plain RPMI media without serum for 48 hours at 37°C, and then the conditioned media from both cell lines were harvested for tubule formation assays. HUVEC and KSHV-HUVEC were seeded on matrigel to form capillary-like tubules in the presence of media without serum, or conditioned media from BJAB or BCBL-1 cells. Images of the HUVEC and KSHV-HUVEC cells were taken under bright field microscopy. Additionally images of the KSHV-HUVEC (which express GFP) were taken under fluorescence microscopy. KSHV-HUVEC cells were capable of forming more tubules than the uninfected HUVEC under all three conditions. (C) Depicted is the angiogenic index, which reflects the number of branch points formed under each condition. Shown is the average number of branch points in five different fields per sample.
KSHV is associated with KS, a tumor of endothelial origin, and primary effusion lymphoma (PEL), a tumor of B cell origin. We wanted to investigate whether media from KSHV-positive B cell lymphomas could sustain endothelial tubule formation of KSHV-infected HUVEC. BJAB is a KSHV-negative B cell lymphoma and BCBL-1 is a KSHV-positive B cell lymphoma. We cultured the two cell lines in plain media without serum for 48 hours and then harvested the media for endothelial tubule formation assays, to see whether HUVEC and KSHV-HUVEC could form tubules in the presence of the conditioned media from these two B cells. We found that uninfected HUVEC could not form any tubule networks in either plain media without serum or the conditioned media collected from BJAB cells (Figure 4B and 4C). In contrast, KSHV-HUVEC formed a branching network of tubules to a significantly greater extent than uninfected HUVEC in BJAB conditioned media (Figure 4B and 4C). Furthermore, the conditioned media from the KSHV positive PEL (BCBL-1) amply supported capillary-like tubule formation of KSHV-HUVEC (Fig. 4B and 4C). The KSHV-HUVEC tubules formed in the conditioned BCBL-1 media were also more intact compared with KSHV-HUVEC tubules formed in either plain media without serum, or the conditioned media from BJAB cells. The uninfected HUVEC did form a couple of tubular structure in the presence of BCBL-1 cells but to a significantly lesser degree than KSHV-HUVEC. This suggests that KSHV-positive BCBL-1 cells secret pro-angiogenic cytokines and growth factors that enhance the angiogenesis associated with endothelial cells, and to a greater degree with KSHV-infected endothelial cells.
KSHV-infected endothelial cells form tubules in the absence of exogenous VEGF and bFGF
VEGF and bFGF are two important growth factors involved in endothelial cell proliferation and angiogenesis. Many groups have reported that infection of endothelial cells with KSHV results in increased production of VEGF (58, 59). We have also found that KSHV-HUVEC secreted more VEGF than uninfected HUVEC (data not shown). We performed tubule formation assays in the absence of VEGF or FGF to determine if deprivation of these growth factors affects endothelial cell tubule formation over a four-day time course. We found that at 24 hours after seeding the cells on matrigel, both HUVEC and KSHV-HUVEC formed networks of tubules in complete EGM-2 media, VEGF-minus EGM-2 media and bFGF-minus EGM-2 media (Fig 5A). By day 2, some of the tubules formed by HUVEC and KSHV-HUVEC in complete media were broken, but most of them were maintained very well, and there was no significant difference between the two cell lines (Fig 5A). However, in the VEGF-minus EGM-2 media and the FGF-minus EGM-2 media, there were a significantly greater number of uninfected HUVEC tubules that were broken. In contrast, more intact tubules could still be visualized in the KSHV-HUVEC cells incubated in VEGF-minus EGM-2 media or FGF-minus EGM-2 media (Fig. 5A). By the third day, most of the uninfected HUVEC died and almost all of the tubules were disintegrated. In contrast, we still found some intact tubular structures in the KSHV-HUVEC sample (Fig. 5A). Finally, on day 4, almost all of the uninfected HUVEC were dead but a proportion of the KSHV-HUVEC cells were still expressing GFP and formed tubules to some extent (Fig. 5A). Thus, KSHV infection of HUVEC confers a survival advantage and helps maintain the endothelial tubular network under conditions of growth factor deprivation.
Figure 5. KSHV infection of endothelial cells allows tubule formation in the absence of VEGF and bFGF.

(A) Tubule formation assays were performed with HUVEC and KSHV-HUVEC in endothelial cell complete media, media without VEGF, or media without bFGF for four days. Images of the HUVEC and KSHV-HUVEC cells were taken under bright field microscopy. Additionally images of the KSHV-HUVEC (which express GFP) were taken under fluorescence microscopy. The following time-points were taken: 24 hours (panel A), 48 hours (panel B), 72 hours (panel C), and 96 hours (panel D) hours. Images shown were taken at 10X magnification. The tubules formed by KSHV-HUVEC appeared to be maintained on matrigel for a longer period of time compared with those formed by HUVEC. (B) Tubule formation assays were performed with HUVEC and KSHV-HUVEC in serum-free EBM-2 media, with or without anti-VEGF antibody. Images were taken after 16 hours at 4X magnification. Uninfected HUVEC could not form tubules in the presence of the blocking VEGF antibody. Although overall numbers of tubules was greatly reduced, KSHV-infected HUVEC could still form a few tubules in the presence of the blocking antibody.
Since we and other have previously reported that KSHV infection of endothelial cells results in VEGF upregulation, we next determined whether VEGF production by KSHV-HUVEC contributes to the increased angiogenesis seen with these cells. Hence, we performed the above tubule assays with HUVEC and KSHV-HUVEC in serum-free EBM-2 media containing a blocking anti-VEGF antibody (20μg/ml). We found that in the presence of the anti-VEGF antibody there were hardly any tubules formed by the uninfected KSHV-HUVEC (Fig. 5B). Although significantly inhibited, the KSHV-infected HUVEC still managed to form a very small number of tubules in the presence of the VEGF blocking antibody (Fig. 5B), suggesting that KSHV-HUVEC continuously secrete more VEGF and all of it is not completely blocked by the antibody.
NF-κB and PI3K pathways are essential for tubule formation by KSHV-HUVEC
We next determined the cellular pathways that are important for KSHV angiogenesis in vitro. We used a series of drug inhibitors to treat the endothelial cells in our tubule assays to determine which signaling pathways may play a key role in endothelial cell tubule formation and angiogenesis. In the presence of DMSO vehicle control, 2.5μM SU6656 (Src inhibitor) (38), or 20μM Piceatannol (Syk inhibitor) (39), both HUVEC and KSHV-HUVEC formed tubule networks (Fig 6A), implying that the Src and Syk signal transduction pathways are not necessary for HUVEC or KSHV-HUVEC angiogenesis in vitro under these conditions.
Figure 6. NF-κB and PI3K pathways are essential for tubule formation by KSHV-HUVEC.
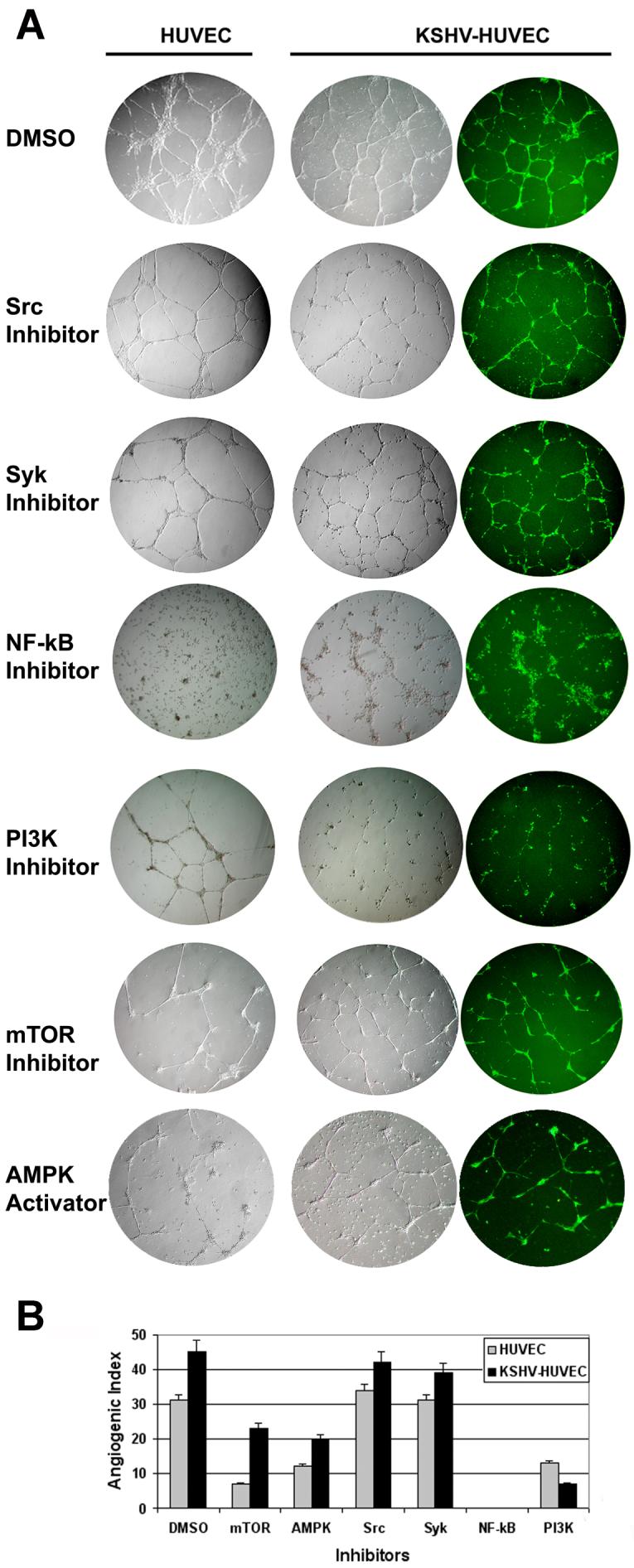
(A) The tubule formation assays were performed on matrigel with HUVEC and KSHV-HUVEC in EBM-2 media containing 50nM Rapamycin (mTOR inhibitor), 150μM Ciglitazone (AMPK activator), 2.5μM SU6656 (Src inhibitor), 20μM Piceatannol (Syk inhibitor), 5μM Bay11-7085 (NF-κB inhibitor) or 20μM LY294002 (PI3K inhibitor). Images of the HUVEC and KSHV-HUVEC cells under bright field were taken at 24, 48, 72 and 96 hours. KSHV-HUVEC were also imaged under fluorescence microscopy. Images shown were taken at 4X magnification. (B) Depicted is the angiogenic index, which reflects the number of branch points formed under each condition. Shown is the average number of branch points in five different fields per sample.
We also tested the importance of AMPK and mTOR signaling in tubule formation (Fig 6A). We found that activation of AMPK by 150 μM Ciglitazone or inhibition of mTOR by 50 nM rapamycin completely disintegrated the HUVEC cells and reduced the number of tubules formed by the KSHV-HUVEC (Fig. 6A and B). The same was true when we used 1 μM rapamycin (data not shown).
Interestingly, 5μM Bay11-7085 (NF-κB inhibitor) treatment killed the HUVEC and KSHV-HUVEC and did not support tubule formation in either cell type (Fig 6A and B). Furthermore, 20μM LY294002 (PI3K inhibitor) treatment resulted in a modest effect on the tubule formation of uninfected HUVEC, although most of the cells appeared unhealthy. In contrast, most of the KSHV-HUVEC died and could not form intact tubules with LY294002 treatment (Fig 6A and B). These data suggest that the KSHV-HUVEC cells are more sensitive to inhibition of the PI3K signaling pathway than uninfected HUVEC. Thus, the NF-κB, PI3K, mTOR and AMPK proteins are important for the viability and angiogenic properties of endothelial cells.
Discussion
In order to proliferate in an uncontrolled fashion, many cancer cells not only bypass cell cycle check-points but also inhibit apoptotic pathways thereby ensuring their survival (see review by Bocchetta and Carbone (60)). KSHV is associated with three different malignancies in the human population. In this report we have evaluated the ability of KSHV to protect endothelial cells from different apoptotic stimuli.
It is well established that the PI3K/Akt signaling pathway regulates cell proliferation and cell survival (42). Here we report that KSHV infection of endothelial cells induces activation of the PI3K and Akt kinases compared to uninfected cells. KSHV infected cells also show a decreased activation of AMPK and an increase in phosphorylation and activation of mTOR. Previous studies have shown that two KSHV oncoproteins, vGPCR and K1, activate the PI3K/Akt/mTOR signaling pathway in B cells and endothelial cells, and immortalize primary HUVEC (15, 27, 29, 30).
Since activation of the PI3K/Akt pathway has been linked to cell survival events, we investigated whether the KSHV-infected cells had a cell survival advantage over uninfected cells under conditions of cell stress. We found that the KSHV-infected HUVEC were protected from serum starvation for a longer period of time compared to uninfected HUVEC and a significantly higher proportion of the KSHV-infected cells were alive five days post-serum starvation compared to uninfected cells. Furthermore, the KSHV-HUVEC were more resistant to etoposide- and staurosporine-induced cell death compared to uninfected HUVEC as measured by Caspase-3 and PARP-1 activity assays.
We also evaluated the ability of KSHV-infected cells to mediate angiogenesis under suboptimal conditions using an in vitro tubule formation assays. In all experiments, KSHV-infected cells could form more tubules in the absence of serum, or in conditioned spent media from KSHV-negative BJAB cells and KSHV-positive BCBL-1 cells, than uninfected HUVEC. This is likely due to the fact that several KSHV viral proteins have been shown to increase VEGF expression and secretion (29, 61) and hence KSHV-infected cells may be less dependent on external growth factors than uninfected cells. Interestingly, in the presence of conditioned media from BCBL-1 cells (and no exogenous growth factor addition), both uninfected HUVEC and KSHV-infected HUVEC could form tubules with the latter forming a significantly higher number of tubules. KSHV-infected PEL have previously been shown to secrete cytokines and growth factors such as VEGF and IL-6 (14, 37, 62) in the supernatant that likely facilitates this endothelial tubule formation.
Additionally, the KSHV-infected HUVEC could also form more tubules than uninfected cells when either VEGF or bFGF was removed from the endothelial cell media or when a blocking VEGF antibody was added to the media. Moreover, the KSHV-HUVEC maintained their tubule structures in the absence of VEGF or bFGF for a longer period of time (upto 4 days) compared to uninfected cells. It has previously been shown that KS lesions have high-levels of expression of VEGF and bFGF (9-12, 63) and this suggests that VEGF secreted from infected cells contributes to endothelial cell survival and angiogenesis.
Finally, it appears that the PI3K and NF-kB pathways are critical for KSHV-induced tubule formation. We performed tubule formation assays in the presence of inhibitors of Syk, Src, PI3K, mTOR, AMPK, and NF-kB. The Src and Syk inhibitors did not show any effect on the ability of the KSHV-HUVEC or uninfected HUVEC to form tubules. However, inhibitors of PI3K (LY294002), NF-kB (Bay11-7085), mTOR (Rapamycin) and AMPK (Ciglitazone) either inhibited or reduced the ability of KSHV-HUVEC from forming tubules. NF-kB inhibition completely obliterated the KSHV-HUVEC and uninfected HUVEC from forming tubules suggesting that this pathway is essential for angiogenesis. Interestingly, the PI3K inhibitor LY294002 reduced the ability of uninfected HUVEC to form tubules, but the KSHV-infected HUVEC were much more susceptible to PI3K inhibition suggesting that these cells are highly dependent on PI3K activation. Weinstein and colleagues originally proposed the concept of oncogene addition in which cancer cells often rely on the activation of a specific signaling pathway. They suggest that inhibition of this specific pathway leads to preferential death of the cancer cell over a normal cell (64). A similar situation may be occurring in the KSHV-infected endothelial cells where they are more sensitive to PI3K inhibition than normal cells. This implies that therapeutic targets that inhibit members of the PI3K signaling pathway may be beneficial in the clinic against KS. Indeed, sirolimus (Rapamycin) which targets mTOR, the downstream effector of the PI3K and Akt kinases, has been shown to be efficacious against KS (37, 65). Further, we have recently published that Rapamycin is also effective against PEL and can inhibit PEL cell growth in vitro and tumor formation in immunocompromised mice (37, 65). The data presented here demonstrates that rapamycin inhibition of mTOR reduces the number of tubules formed by the KSHV infected HUVEC by fifty percent. Further, inhibition of the upstream kinase PI3K using LY294002, completely inhibited KSHV-HUVEC tubule formation. This suggests that inhibitors of the PI3K/Akt/mTOR pathway may prove efficacious against a broad spectrum of KSHV-associated cancers, including PEL and KS.
Taken together, our results provide compelling evidence that KSHV infection activates the PI3K/Akt/mTOR signaling pathway in endothelial cells and confers a cell survival and angiogenic advantage to these cells.
Acknowledgements
We thank members of the Damania and Dittmer labs for helpful discussions. This work was supported by NIH grants CA096500 and HL083469, and American Heart Association grant number 0640041N to BD. BD is a Leukemia & Lymphoma Society Scholar and Burroughs Welcome Fund Investigator in Infectious Disease.
Abbreviations
- KSHV/HHV-8
Kaposi’s sarcoma-associated herpesvirus
- VEGF
Vascular endothelial growth factor
- PI3K
phosphatidyl-inositol-3′-OH-kinase
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–9. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Dupin N, Grandadam M, Calvez V, et al. Herpesvirus-like DNA sequences in patients with Mediterranean Kaposi’s sarcoma. Lancet. 1995;345:761–2. doi: 10.1016/s0140-6736(95)90642-8. [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS- related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–91. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 4.Soulier J, Grollet L, Oksenhendler E, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86:1276–80. [PubMed] [Google Scholar]
- 5.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 6.Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol Biol Rev. 2003;67:175–212. doi: 10.1128/MMBR.67.2.175-212.2003., table of contents.
- 7.Samaniego F, Markham PD, Gendelman R, et al. Vascular endothelial growth factor and basic fibroblast growth factor present in Kaposi’s sarcoma (KS) are induced by inflammatory cytokines and synergize to promote vascular permeability and KS lesion development. Am J Pathol. 1998;152:1433–43. [PMC free article] [PubMed] [Google Scholar]
- 8.Blankaert D, Simonart T, Van Vooren JP, et al. Constitutive release of metalloproteinase-9 (92-kd type IV collagenase) by Kaposi’s sarcoma cells. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18:203–9. doi: 10.1097/00042560-199807010-00002. [DOI] [PubMed] [Google Scholar]
- 9.Qian LW, Xie J, Ye F, Gao SJ. Kaposi’s sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J Virol. 2007;81:7001–10. doi: 10.1128/JVI.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konstantinopoulos PA, Sullivan RJ, Karamouzis MV, Dezube BJ. Investigational agents for treatment of AIDS-related Kaposi’s sarcoma. Expert Opin Investig Drugs. 2007;16:495–504. doi: 10.1517/13543784.16.4.495. [DOI] [PubMed] [Google Scholar]
- 11.Ye FC, Blackbourn DJ, Mengel M, et al. Kaposi’s sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J Virol. 2007;81:3980–91. doi: 10.1128/JVI.02089-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sadagopan S, Sharma-Walia N, Veettil MV, et al. Kaposi’s sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J Virol. 2007;81:3949–68. doi: 10.1128/JVI.02333-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Little RF, Yarchoan R. Treatment of gammaherpesvirus-related neoplastic disorders in the immunosuppressed host. Semin Hematol. 2003;40:163–71. doi: 10.1053/shem.2003.50016. [DOI] [PubMed] [Google Scholar]
- 14.Aoki Y, Tosato G. Role of vascular endothelial growth factor/vascular permeability factor in the pathogenesis of Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphomas. Blood. 1999;94:4247–54. [PubMed] [Google Scholar]
- 15.Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 2006;66:3658–66. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- 16.Anderson KE, Lipp P, Bootman M, et al. DAPP1 undergoes a PI 3-kinase-dependent cycle of plasma-membrane recruitment and endocytosis upon cell stimulation. Curr Biol. 2000;10:1403–12. doi: 10.1016/s0960-9822(00)00794-6. [DOI] [PubMed] [Google Scholar]
- 17.Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol. 2000;57:652–8. [PubMed] [Google Scholar]
- 18.Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–10. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudek H, Datta SR, Franke TF, et al. Regulation of neuronal survival by the serinethreonine protein kinase Akt. Science. 1997;275:661–5. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 20.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–13. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beevers CS, Li F, Liu L, Huang S. Curcumin inhibits the mammalian target of rapamycin-mediated signaling pathways in cancer cells. Int J Cancer. 2006;119:757–64. doi: 10.1002/ijc.21932. [DOI] [PubMed] [Google Scholar]
- 22.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 23.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(Pt 2):427–31. [PMC free article] [PubMed] [Google Scholar]
- 24.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 25.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–65. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 26.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–9. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 27.Tomlinson CC, Damania B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol. 2004;78:1918–27. doi: 10.1128/JVI.78.4.1918-1927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sodhi A, Montaner S, Patel V, et al. Akt plays a central role in sarcomagenesis induced by Kaposi’s sarcoma herpesvirus-encoded G protein-coupled receptor. Proc Natl Acad Sci U S A. 2004;101:4821–6. doi: 10.1073/pnas.0400835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bais C, Van Geelen A, Eroles P, et al. Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR. Cancer Cell. 2003;3:131–43. doi: 10.1016/s1535-6108(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 30.Sodhi A, Chaisuparat R, Hu J, et al. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell. 2006;10:133–43. doi: 10.1016/j.ccr.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 31.Vieira J, O’Hearn PM. Use of the red fluorescent protein as a marker of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325:225–40. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 32.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 33.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vinci MC, Visentin B, Cusinato F, Nardelli GB, Trevisi L, Luciani S. Effect of vascular endothelial growth factor and epidermal growth factor on iatrogenic apoptosis in human endothelial cells. Biochem Pharmacol. 2004;67:277–84. doi: 10.1016/j.bcp.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 35.Kirby JE, Nekorchuk DM. Bartonella-associated endothelial proliferation depends on inhibition of apoptosis. Proc Natl Acad Sci U S A. 2002;99:4656–61. doi: 10.1073/pnas.072292699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Binetruy-Tournaire R, Demangel C, Malavaud B, et al. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. Embo J. 2000;19:1525–33. doi: 10.1093/emboj/19.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sin SH, Roy D, Wang L, et al. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood. 2007;109:2165–73. doi: 10.1182/blood-2006-06-028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lannutti BJ, Blake N, Gandhi MJ, Reems JA, Drachman JG. Induction of polyploidization in leukemic cell lines and primary bone marrow by Src kinase inhibitor SU6656. Blood. 2005;105:3875–8. doi: 10.1182/blood-2004-10-3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lang ML, Chen YW, Shen L, et al. IgA Fc receptor (FcalphaR) cross-linking recruits tyrosine kinases, phosphoinositide kinases and serine/threonine kinases to glycolipid rafts. Biochem J. 2002;364:517–25. doi: 10.1042/BJ20011696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell. 2007;18:14–23. doi: 10.1091/mbc.E06-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dawson CW, Tramountanis G, Eliopoulos AG, Young LS. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem. 2003;278:3694–704. doi: 10.1074/jbc.M209840200. [DOI] [PubMed] [Google Scholar]
- 42.Carpenter CL, Cantley LC. Phosphoinositide 3-kinase and the regulation of cell growth. Biochim Biophys Acta. 1996;1288:M11–6. doi: 10.1016/0304-419x(96)00018-2. [DOI] [PubMed] [Google Scholar]
- 43.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–98. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 44.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 45.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 46.van Maanen JM, Retel J, de Vries J, Pinedo HM. Mechanism of action of antitumor drug etoposide: a review. J Natl Cancer Inst. 1988;80:1526–33. doi: 10.1093/jnci/80.19.1526. [DOI] [PubMed] [Google Scholar]
- 47.Matsumoto H, Sasaki Y. Staurosporine, a protein kinase C inhibitor interferes with proliferation of arterial smooth muscle cells. Biochem Biophys Res Commun. 1989;158:105–9. doi: 10.1016/s0006-291x(89)80183-4. [DOI] [PubMed] [Google Scholar]
- 48.Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem Biophys Res Commun. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 49.Pohlman TH, Harlan JM. Human endothelial cell response to lipopolysaccharide, interleukin-1, and tumor necrosis factor is regulated by protein synthesis. Cell Immunol. 1989;119:41–52. doi: 10.1016/0008-8749(89)90222-0. [DOI] [PubMed] [Google Scholar]
- 50.Dixit NM, Layden-Almer JE, Layden TJ, Perelson AS. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature. 2004;432:922–4. doi: 10.1038/nature03153. [DOI] [PubMed] [Google Scholar]
- 51.Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem. 1996;271:18068–73. doi: 10.1074/jbc.271.30.18068. [DOI] [PubMed] [Google Scholar]
- 52.Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–8. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 53.Tewari M, Quan LT, O’Rourke K, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–9. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 54.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–85. [PubMed] [Google Scholar]
- 55.Casciola-Rosen L, Nicholson DW, Chong T, et al. Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J Exp Med. 1996;183:1957–64. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duriez PJ, Shah GM. Cleavage of poly(ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochem Cell Biol. 1997;75:337–49. [PubMed] [Google Scholar]
- 57.Staton CA, Stribbling SM, Tazzyman S, Hughes R, Brown NJ, Lewis CE. Current methods for assaying angiogenesis in vitro and in vivo. Int J Exp Pathol. 2004;85:233–48. doi: 10.1111/j.0959-9673.2004.00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masood R, Cesarman E, Smith DL, Gill PS, Flore O. Human herpesvirus-8-transformed endothelial cells have functionally activated vascular endothelial growth factor/vascular endothelial growth factor receptor. Am J Pathol. 2002;160:23–9. doi: 10.1016/S0002-9440(10)64344-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mutlu AD, Cavallin LE, Vincent L, et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell. 2007;11:245–58. doi: 10.1016/j.ccr.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bocchetta M, Carbone M. Epidemiology and molecular pathology at crossroads to establish causation: molecular mechanisms of malignant transformation. Oncogene. 2004;23:6484–91. doi: 10.1038/sj.onc.1207855. [DOI] [PubMed] [Google Scholar]
- 61.Wang L, Wakisaka N, Tomlinson CC, DeWire S, Krall S, Pagano JS, Damania B. The Kaposi’s Sarcoma-Associated Herpesvirus (KSHV/HHV8) K1 Protein Induces Expression of Angiogenic and Invasion Factors. Cancer Research. 2004 doi: 10.1158/0008-5472.can-03-3653. [DOI] [PubMed] [Google Scholar]
- 62.Asou H, Said JW, Yang R, et al. Mechanisms of growth control of Kaposi’s sarcoma-associated herpes virus-associated primary effusion lymphoma cells. Blood. 1998;91:2475–81. [PubMed] [Google Scholar]
- 63.Carroll PA, Kenerson HL, Yeung RS, Lagunoff M. Latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J Virol. 2006;80:10802–12. doi: 10.1128/JVI.00673-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 65.Stallone G, Schena A, Infante B, et al. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N Engl J Med. 2005;352:1317–23. doi: 10.1056/NEJMoa042831. [DOI] [PubMed] [Google Scholar]