

Abstract
Evidence is mounting in support of the inoculum effect (i.e., slow killing at large initial inocula [CFUo]) for numerous antimicrobials against a variety of pathogens. Our objectives were to (i) determine the impact of the CFUo of Pseudomonas aeruginosa on ceftazidime activity and (ii) to develop and validate a pharmacokinetic/pharmacodynamic (PKPD) mathematical model accommodating a range of CFUo. Time-kill experiments using ceftazidime at seven concentrations up to 128 mg/liter (MIC, 2 mg/liter) were performed in duplicate against P. aeruginosa PAO1 at five CFUo from 105 to 109 CFU/ml. Samples were collected over 24 h and fit by candidate models in NONMEM VI and S-ADAPT 1.55 (all data were comodeled). External model qualification integrated data from eight previously published studies. Ceftazidime displayed approximately 3 to 4 log10 CFU/ml net killing at 106.2 CFUo and concentrations of 4 mg/liter (or higher), less than 1.6 log10 CFU/ml killing at 107.3 CFUo, and no killing at 108.0 CFUo for concentrations up to 128 mg/liter. The proposed mechanism-based model successfully described the inoculum effect and the concentration-independent lag time of killing. The mean generation time was 28.3 min. The effect of an autolysin was assumed to inhibit successful replication. Ceftazidime concentrations of 0.294 mg/liter stimulated the autolysin effect by 50%. The model was predictive in the internal cross-validation and had excellent in silico predictive performance for published studies of P. aeruginosa ATCC 27853 for various CFUo. The proposed PKPD model successfully described and predicted the pronounced inoculum effect of ceftazidime in vitro and integrated data from eight literature studies to support translation from time-kill experiments to in vitro infection models.
Pseudomonas aeruginosa is an opportunistic, gram-negative pathogen responsible for high morbidity and mortality (19). P. aeruginosa has multiple mechanisms of resistance to antibiotics, including efflux pumps, the enzymatic degradation of antibiotics by, e.g., beta-lactamases, and target structure alteration (19, 25, 45). Due to its remarkable ability to resist killing by antibiotics (45), many P. aeruginosa isolates from nosocomial infections are multidrug resistant. The proportion of ceftazidime-resistant P. aeruginosa isolates from intensive care units increased from approximately 14% in 1997 to 24% in 2003 in the United States (23).
Infections with a high bacterial density at the initiation of antibiotic therapy may present a therapeutic problem, including a higher risk for the emergence of resistance due to the larger number of bacteria present and the higher probability of having at least one resistant bacterial cell within a large initial inoculum (CFUo) (32). The probability of the emergence of resistance may be substantially increased at high CFUo, as the amplification of resistant subpopulations has been demonstrated to occur secondarily to low-intensity antimicrobial exposure (60). The inoculum effect was first described by Kirby (34) in vitro for penicillin activity against staphylococci. Subsequently, many studies assessed the effect of CFUo on the MIC (6, 16). Mouton et al. (47, 48) derived the relationship between MIC and CFUo if the growth rate and maximal killing rate constant are independent of the CFUo. This effect of the CFUo on the MIC needs to be distinguished from the inoculum effect in our time-kill study, since we assessed the effect of the CFUo on the rate of bacterial killing. Importantly, high CFUo are associated with increased mortality and attenuate antibiotic effects in animal infection models (6, 20, 57).
Potential mechanistic explanations for the inoculum effect of beta-lactams include the breakdown of beta-lactams by beta-lactamases, cell-to-cell communication, and the differential expression of penicillin-binding proteins (PBPs) at a high bacterial density (14, 57). In P. aeruginosa, cell-to-cell communication is known to be mediated by the release of freely diffusible signal molecules such as two N-acylhomoserine lactones and the Pseudomonas quinolone signal molecule (2-heptyl-3-hydroxy-4-quinolone) (29, 54). High concentrations of the N-butyryl-l-homoserine lactone signal molecule induce the expression of MexAB-OprM in P. aeruginosa (40, 53), for which ceftazidime is a substrate (5, 19, 41), and the mexAB-oprM operon has its highest expression during the mid-stationary-growth phase (40). These signal molecules are known to affect the expression of several hundred genes in P. aeruginosa (54).
Mathematical models that can describe a slower bacterial killing rate at high CFUo have not been published. A pharmacokinetic/pharmacodynamic (PKPD) model that can describe the inoculum effect of P. aeruginosa may support the optimization of dosage regimens and generate hypotheses on how to minimize the emergence of resistance for new and established antibiotics.
The objectives of the current study were to (i) study the effect of CFUo on the rate and extent of the killing of P. aeruginosa PAO1 by ceftazidime in vitro, (ii) develop a mechanism-based, mathematical model that can accommodate a range of CFUo, and (iii) qualify this model by integrating literature results for in vitro PD models with P. aeruginosa PAO1 and P. aeruginosa ATCC 27853 for ceftazidime monotherapy.
(This work was presented in part at the 2008 Annual Meeting of the Population Approach Group in Australia & New Zealand, Dunedin, New Zealand, and at the 2007 Annual Meeting of the American Association of Pharmaceutical Scientists, San Diego, CA.)
MATERIALS AND METHODS
Bacterial isolates, media, and susceptibility testing.
A genetically characterized, clinical isolate of P. aeruginosa, PAO1, obtained from the R. E. W. Hancock Laboratory at the University of British Columbia, Vancouver, Canada, was used for all experiments (58). Luria-Bertani (LB) broth (Difco Laboratories, Detroit, MI) growth medium was supplemented with calcium (25 mg/liter) and magnesium (12.5 mg/liter). This medium was used for MIC determination, time-kill experiments, and preparing antibiotic stock solutions. LB agar (Difco Laboratories, Detroit, MI) was used for growing fresh bacteria. Ceftazidime was obtained from Sigma-Aldrich, St. Louis, MO. A fresh stock solution was prepared in the morning of each experimental run by dissolving ceftazidime in LB broth. The MIC was determined by broth microdilution in 96-well plates by following Clinical Laboratory Standards Institute guidelines (12).
Time-kill experiments.
P. aeruginosa PAO1 time-kill experiments were performed at CFUo of 106, 107, and 108 CFU/ml using a previously described methodology (65). Additionally, CFUo of 105 and 109 CFU/ml were studied for external model qualification (see below). Ceftazidime concentrations ranged from 0 to 128 mg/liter (0 to 64 times the MIC). Briefly, fresh bacterial colonies of PAO1 were grown for approximately 18 h for each experiment. Each run contained a growth control. Bacteria were suspended in normal saline, and bacterial concentrations were standardized using a spectrophotometer at 620 nm. An appropriate amount of bacteria was diluted in prewarmed LB broth to achieve the desired CFUo in a final volume of 20 ml LB broth. These suspensions were placed into a water bath with constant shaking at 37°C for approximately 45 min before an appropriate volume of the (diluted) ceftazidime stock solution was added to achieve the desired ceftazidime concentration. For all curves, actual viable counts were determined within less than 10 min before dosing, and growth during this time period was ignored. All experiments were performed in duplicate.
Broth samples were taken immediately before the addition of ceftazidime and at 0.5, 1, 2, 4, 8, and 24 h. As we found no killing from 0 to 8 h for the 107 CFUo, samples at 37 and 48 h additionally were obtained for this CFUo. Samples were serially diluted between 10- and 100,000-fold to minimize antibiotic carryover. Colony counts were determined as described by Tsuji et al. (65).
Descriptive modeling of the inoculum effect.
Our initial PD modeling analysis aimed at characterizing the growth and killing of the susceptible population. For this initial analysis, the data points that showed the regrowth of the resistant population were excluded (see below). This descriptive modeling approach used different PD parameter estimates at each CFUo. Bacterial growth was described by a first-order growth rate constant (kg). A logistic growth model with the maximum population size (CFUmax) was used. The drug was assumed to inhibit growth (model A) by an inhibitory Hill-type model. The IC50 is the drug concentration inhibiting growth rate by 50%. The maximum extent of inhibition was assumed to be 1, assuming that ceftazidime only inhibits growth and does not stimulate death. At high drug concentrations, the maximum rate constant of bacterial loss is the first-order natural death rate constant kd. This model has been previously described by other authors (11, 15, 26, 44):
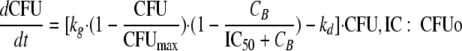 |
(1) |
The initial condition (IC) denotes the value of the state variable at time 0 h. For all models describing time-kill experiments, the ceftazidime concentration in broth (CB) was assumed to degrade with a fixed half-life of 45.9 h according to data from Viaene et al. (69). To account for lag time prior to the onset of bacterial killing, a biophase model (model B) was used to describe the diffusion of drug from broth to the target site (56). Model B can describe a slow onset of killing, with the delay becoming shorter at high drug concentrations. Equation 1 also applies to model B when the biophase concentration (Cbio) is used instead of CB to describe the drug effect. The keq is the equilibration rate constant between the drug concentration in broth and that in the biophase:
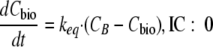 |
(2) |
Mechanism-based model for lag time of killing.
A new model (model C) was developed that describes the slow onset of bacterial killing as a system property that is independent of drug concentration. The slow onset was assumed to be caused by a delay between ceftazidime binding to PBPs and the depletion of cell wall constituents that are required for bacterial growth (Fig. 1). Ceftazidime was assumed to inhibit the synthesis of the cell wall (CW) and, thus, inhibit the growth rate:
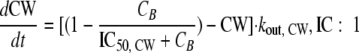 |
(3) |
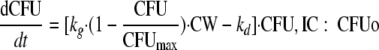 |
(4) |
The synthesis of the cell wall is expressed as a fraction of the baseline value. The IC50, CW is the drug concentration in broth that inhibits 50% of cell wall synthesis, and kout, CW is the first-order rate constant of the turnover of cell wall constituents. All other parameters are described above. As models A, B, and C were not intended to be used for simulations that include the resistant bacterial population, observations that showed the regrowth of the resistant population were excluded during estimations. These were the observations at 24 h for the 106 CFUo and the 37- and 48-h samples for the 107 CFUo. As the inoculum effect manifested in our study during the first 8 h, the predominant population that is susceptible at low CFUo showed the inoculum effect. Therefore, the exclusion of data points showing the regrowth of the resistant population seems justifiable to keep model C simple, as model C was intended only to show the mechanism-based model feature of the concentration-independent lag time of killing. Importantly, all observations were included during the fittings of the mechanism-based model described below.
FIG. 1.

Model for growth and bacterial killing of one subpopulation (model C). Symbols are defined in Table 1.
Mechanism-based model for the inoculum effect. (i) Subpopulation model.
As most PD parameter estimates changed systematically with CFUo for model C, a mechanism-based model (model D) that can describe the inoculum effect and make predictions for other inocula was developed. This model (Fig. 2) included one genotypically susceptible population (Fig. 2, top) and one genotypically resistant population (Fig. 2, bottom) that share all parameters except the maximum stimulation of autolysin activity (Smax).
FIG. 2.

Mechanism-based model for ceftazidime against P. aeruginosa that describes the phenotypic tolerance at high initial inocula by cell-to-cell communication via signal molecules (model D). Symbols are defined in Table 2.
(ii) Life cycle model for bacterial replication.
For both populations, the bacterial life cycle was simplified to consist of two states. State 1 (susceptible bacteria [S1]) occurs at the beginning of the cycle immediately after doubling, and state 2 (S2) occurs immediately before doubling. The first-order transition (k12) from state 1 to state 2 determines the mean generation time (MTT12), since the doubling was assumed to be very fast (k21 was fixed to 50 h−1). The rate of growth is decreased by the inhibition term Inhk12 at high concentrations of signal molecules and/or of ceftazidime. The differential equations for the susceptible population are the following:
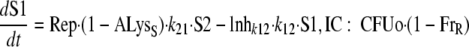 |
(5) |
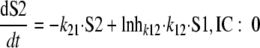 |
(6) |
In the absence of drug, the replication efficiency factor (Rep) approaches 2, indicating a 100% probability of successful doubling. The sum of all viable bacteria (SRALL) and Rep are described by the following equations (R terms denote the resistant population):
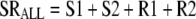 |
(7) |
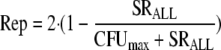 |
(8) |
(iii) Effects of antibiotic and signal molecules.
The autolysin activity (susceptible population [ALysS]) is stimulated by ceftazidime, which binds to PBPs (StimDrug,S). Autolysin activity was assumed to decrease the probability for successful replication (indicated by [1 − ALysS] in equation 5).
High signal molecule concentrations (CSign1) were assumed to cause an inoculum effect by stimulating the loss of autolysin activity (maximum stimulation is indicated as Smax, loss):
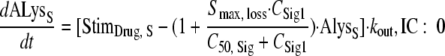 |
(9) |
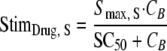 |
(10) |
For this parameterization, the maximum value of ALysS is Smax, S, and an Smax, S of 1 indicates that successful replication can be completely inhibited at high drug concentrations. The SC50 is the drug concentration at which the input of autolysin effect is half maximally stimulated. The Smax, loss parameter describes the maximum extent of the inoculum effect at high signal molecule concentrations. Freely diffusible signal molecules (54) were assumed to be synthesized and released by all viable bacteria that are included in SRALL, as shown in the following equations:
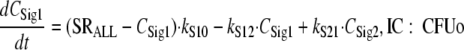 |
(11) |
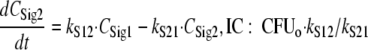 |
(12) |
To illustrate the inoculum effect, we simulated the probability of successful replication for the susceptible population based on the steady-state solution of equation 9 for a range of CFUo. It was assumed that the signal molecule concentration (CSig1) is at its initial condition, CFUo. The drug effect was described by equation 10. The probability of successful replication equals 1 − ALysS in equation 5. A probability of 100% indicates that all replication steps are successful (perfect growth), 50% indicates that every second replication (doubling) is successful and results in net stasis, and a probability of 0% indicates that no cells replicate successfully (fastest possible killing).
The concentration of signal molecules in the central compartment CSig1 was assumed to slow down the rate of bacterial replication as observed by Diggle et al. (18) for the Pseudomonas quinolone signaling molecule. CSig1 was assumed to equilibrate with CSig2. If both signal molecules and drug in broth (CB) were present, the generation time was assumed to be prolonged by the presence of both:
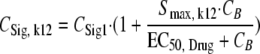 |
(13) |
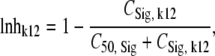 |
(14) |
CSig, k12 is the effective signal molecule concentration that exceeds CSig1 due to the presence of drug concentrations. Since CSig1 and the stimulation term in equation 13 are multiplied, CSig1 and CB both inhibit the rate of bacterial replication.
The differential equations for the genotypically resistant population were the same as that for the susceptible population (the enabling equations are available on request). As only limited data were available on the response of the resistant population, we shared as many parameters as possible between both populations following the rule of parsimony. Models with different MTT12, SC50, and Smax values for both populations (see equations 9 and 10) were considered. The fraction of resistant bacteria (FrR) in the CFUo was estimated.
(iv) Internal model qualification.
Internal model qualification was performed by case deletion diagnostics and by a leave-one-inoculum-out test. To assess the influence of any combination of ceftazidime concentration and CFUo studied on the parameter estimates, model parameters were reestimated based on datasets that lacked all replicates of each of the 18 combinations of CFUo and drug concentration.
As a challenging method of internal model qualification, all data at one CFUo (except the growth control of the 106 CFUo) were deleted from the data set, parameters were reestimated, and the data of the CFUo not used during reestimation were predicted and compared to the observations. This was repeated for all inocula. As the 106 CFUo contained unique information about the regrowth of the resistant population, data from a preliminary run with CFUo of 105 (and 109) CFU/ml additionally were included in this leave-one-inoculum-out test.
External model qualification. (i) In-house time-kill data.
A pilot experiment on the inoculum effect of ceftazidime against PAO1 at concentrations of 0 and 128 mg/liter and CFUo of 105, 106, 108, and 109 CFU ml−1 was performed in duplicate. The CFU-versus-time profiles of this experiment were simulated using model D and were compared to the observations. As this pilot experiment included only one high drug concentration, it would not have added much new information to the main data set. However, this pilot study contained four CFUo within and outside the range of CFUo studied in the main experiment and is therefore suitable for an external validation of the model. The data of this pilot study were not used during estimation.
(ii) Literature data on in vitro PD models and time-kill data.
Based on a systematic literature search, we included all available in vitro studies of ceftazidime monotherapy against P. aeruginosa strains PAO1 and ATCC 27853 that assessed the time course of bacterial killing. These data comprised eight published studies (1, 2, 9, 28, 43, 46, 49, 55, 61) on these two strains that were used to assess the applicability and translation of model D to more sophisticated in vitro experiments and to assess strain-to-strain differences. Literature data from both in vitro PD models and time-kill experiments were digitized by GetData Graph Digitizer (version 2.23; by S. Fedorov, Russia) and Grab It! XP (Build 10; Datatrend Software, Inc., Raleigh, NC). The average times and log CFU counts from both programs were used.
Data from Henrichfreise et al. (28) on ceftazidime against PAO1 were simulated based on unmodified parameter estimates of model D. In a second step, selected model parameters were reestimated using these data to refine the parameter estimates.
(iii) External model qualification using literature data on P. aeruginosa ATCC 27853.
Data from in vitro PD models and time-kill experiments from Barclay et al. (1, 2) and Shalit et al. (55), as well as the growth control data (but no antibiotic containing regimens) from Cappelletty et al. (9), Mouton and den Hollander (46), and McGrath et al. (43) on P. aeruginosa ATCC 27853, were used to estimate all model parameters except the rate constants for signal molecules, CFUmax, and SC50. Different generation times (MTT12) were estimated for these studies, since the growth rate differed considerably between studies. If no filter was used in the in vitro system and no adjustment of the bacterial counts was made as described by Keil and Wiedemann (33), mathematical equations were adjusted for the washout of bacteria due to the efflux of broth. CFU-versus-time profiles for the ceftazidime containing monotherapy regimens of Cappelletty et al. (9), Mouton and den Hollander (46), and McGrath et al. (43) and the time-kill data on ceftazidime from Tam et al. (61) were simulated and compared to the observations. After this external qualification procedure, model parameters for P. aeruginosa ATCC 27853 were reestimated and reported based on all available data (except the Mouton and den Hollander study).
(iv) Computation.
Nonlinear mixed-effects modeling in NONMEM VI (level 1.2; NONMEM Project Group, Icon Development Solutions, Ellicott City, MD) (4) was used. The ADVAN9 differential equation solver was applied for all modeling. The log-both-sides method (10, 51) was used to fit CFU counts, and an additive error model on the log scale was applied. As the variability between each experimental run was very small and curves at the same experimental condition were reproducible, no other variability terms were included in the NONMEM analysis.
Model D additionally was estimated in parallelized S-ADAPT (version 1.55) by a full population PD analysis with between-subject variability on every model parameter to confirm the NONMEM results. This population PD analysis used the importance-sampling Monte-Carlo parametric expectation-maximization method (pmethod = 8 in S-ADAPT) (3) and was implemented on a parallelized computer cluster. WinNonlin Professional (version 5.0; Pharsight Corp., Mountain View, CA) was used for statistical and graphical analysis.
RESULTS
Time-kill experiments.
The MIC of ceftazidime against P. aeruginosa PAO1 was 2.0 mg/liter, which is consistent with other studies (28). Figure 3 shows the observed CFU-versus-time profiles for the 106, 107, and 108 CFUo and the population predictions from model D. The onset of bacterial killing showed a lag time of approximately 1 to 2 h for the 106 CFUo and of approximately 4 h for the 107 CFUo. For the 106 CFUo, ceftazidime concentrations of 4 mg/liter and higher achieved a net bacterial killing of 3 to 4 log10 at 8 and 24 h. Concentrations of up to 64 mg/liter achieved 1.6 log10 of killing or less for the 107 CFUo, and less than 0.3 log10 of net killing was seen, at all concentrations, for the 108 CFUo.
FIG. 3.

Time-kill data (symbols) and model fitting from NONMEM with model D (lines) for ceftazidime against P. aeruginosa at various initial inocula: 106 (A), 107 (B), and 108 CFU/ml (C) (only the 107 inoculum was studied up to 48 h).
Empirical models for the inoculum effect.
A logistic growth model with ceftazidime inhibiting growth (model A) captured the plateau, growth control, and the general killing pattern. As expected, model A could not describe the lag time of killing. The biophase model (model B) could describe the delay of killing at low concentrations. As the lag time of killing depends on the drug concentration for the biophase model, curve fits of model B at high drug concentrations showed misfits similar to those of model A. The objective function was better by 103 for model C relative to model A and by 79 for model C relative to model B.
Mechanism-based model for lag time of killing.
Since the lag time of killing was independent of drug concentration (Fig. 3A and B), this delay was assumed to be caused by the depletion of cell wall constituents, which are required for bacterial replication. The new proposed model (model C) showed precise and unbiased curve fits for the susceptible population at all three CFUo (Fig. 4A), and Table 1 lists the parameter estimates. Relative standard errors were below 15%, except for IC50, CW at 107 and 108 CFUo. Importantly, model C required different parameter estimates at the three inocula, and these estimates varied systematically with the CFUo (Table 1).
FIG. 4.

Observed versus fitted bacterial counts from NONMEM. (A) Fittings of model C (data from the regrowth of the resistant population were excluded from analysis); (B) fittings of model D (using data at log CFUo of 6, 7, and 8); (C) predictions of model D during the leave-one-inoculum-out procedure (data from a preliminary run at log CFUo of 5 and 9 are included); (D) external predictions of a preliminary run at concentrations of 0 and 128 mg/liter.
TABLE 1.
Parameter estimates of the descriptive model (model C)
| Parameter | Variable | Unit | CFUo estimate (%SE) | Range from case deletion |
|---|---|---|---|---|
| Log10 (initial inoculum) | Log10 (CFUo) | 106, 6.13 (0.3); | 6.12-6.14; | |
| 107, 7.35 (0.4); | 7.27-7.36; | |||
| 108, 8.01 (0.2) | 7.95-8.03 | |||
| Maximum population size | Log (CFUmax) | 9.17 (0.4) | 9.16-9.20 | |
| First-order growth rate constant | kg | h−1 | 106, 2.55 (4.3); 107, 0.567 (8.3); 108, 0.319 (8.5) | 2.46-2.68, 0.541-0.876, 0.309-0.575 |
| First-order rate constant for natural death | kd | h−1 | 106, 1.36 (6.8); 107, 0.0925 (14.4); 108, fixed to 0a | 1.27-1.49, 0.0823-0.113, fixed to 0 |
| Drug concn causing 50% of maximum inhibition | IC50, CW | mg/liter | 106, 0.428 (10.9); 107, 0.337 (16.9); 108, 0.185 (23.2) | 0.360-0.472, 0.195-0.386, 0.0732-0.303 |
| MTT for turnover of cell wall | MTTCW = 1/kout, CW | h | 0.985 (13) | 0.863-1.11 |
The parameter was estimated to be 0.001 but was fixed at zero, as this small estimate caused a convergence problem. This did not alter the objective function.
Mechanism-based model for the inoculum effect.
Although model C explained all features of the data for the three studied inocula, it is difficult to predict the phenotypic tolerance of PAO1 at inocula other than the inocula studied in models A, B, and C. Therefore, a mechanism-based model was developed. Model D (Fig. 2) assumes that all viable bacteria synthesize signal molecules that are freely diffusible. A high signal molecule concentration was assumed to cause a loss of the autolysin effect and to slow the rate of bacterial replication.
Model D could describe all data (including the regrowth of resistant bacteria) for all three CFUo (Fig. 3 and 4B). Parameter estimates are shown in Table 2, and the relative standard errors were below 20% for all except two parameters. The estimates from S-ADAPT and NONMEM were well comparable (Table 2), suggesting that the estimated model parameter values were robust. The estimate for EC50, drug was higher in S-ADAPT, potentially due to the slightly larger estimate for maximum population size in S-ADAPT.
TABLE 2.
Estimates of model D for P. aeruginosa PAO1 and ATCC 27853
| Parameter | Symbol | Unit | Estimate for P. aeruginosa PAO1
|
Estimate for P. aeruginosa ATCC 27853 (NONMEM) | ||
|---|---|---|---|---|---|---|
| Estimate (%SE) by
|
Range from case deletionf (NONMEM) | |||||
| NONMEM | S-ADAPT | |||||
| Parameters describing bacterial growth and inoculum size | ||||||
| Log10 IC for 106 CFU/ml, 107 CFU/ml, and 108 CFU/ml | Log10 CFUo | 6.01 (1.7), 7.37 (1.1), 8.10 (0.6) | 6.06 (0.4), 7.37 (0.1), 8.07 (0.1) | 5.99-6.04, 7.35-7.38, 8.08-8.11 | Read from observed CFU data at 0 h | |
| Log10 fraction of resistant population at time zero | Log10 FrR | −3.54 (4.8) | −4.05 (2.8) | −4.09-−3.27 | −3.15 | |
| Generation time at low CFUo | MTT12 = k12−1 | min | 28.3 (7.3) | 31.9 (1.3) | 23.4-34.3 | 24.8f |
| Doubling rate constant | k21 | h−1 | 50 (fixed)a | 50 (fixed)a | 50 (fixed)a | |
| Maximum population size | Log10 CFUmax | 9.78 (5.3) | 10.1 (7.4) | 8.98-10.3 | 9.06 | |
| Parameters describing phenotypic change due to signal molecules | ||||||
| MTT for elimination and synthesis | MTTS10 = kS10−1 | min | 2.33 (40.8) | 1.90 (77) | 1.58-14.7 | 2.33 (fixed) |
| MTT for transfer from central to peripheral compartment | MTTS12 = kS12−1 | min | 1 (fixed)b | 1 (fixed)b | 1 (fixed) | |
| MTT for transfer from peripheral to central compartment | MTTS21 = kS21−1 | h | 24 (fixed)c | 24 (fixed)c | 24 (fixed) | |
| Parameters describing effects of drug and signal molecules | ||||||
| Maximum stimulation of autolysin effect for susceptible population | Smax, S | 1 (fixed)e | 1 (fixed)e | 1 (fixed) | ||
| Maximum stimulation of autolysin effect for resistant population | Smax, R | 0.560 (1.3) | 0.541 (4.1) | 0.542-0.571 | 0.526 | |
| Drug concn causing 50% stimulation of autolysin effect | SC50 | mg/liter | 0.294 (9.3) | 0.262 (15.3) | 0.237-0.381 | 0.294 (fixed)h |
| Ratio of elimination rate constant of autolysin effect and k12 | kout/k12 | 0.438 (20.3) | 0.521 (3.3) | 0.333-0.573 | 1.57 | |
| Log10 of signal molecule concn at 50% of maximum effect | Log10C50, Sig | 7.24 (1.4) | 7.25 (0.6) | 7.16-7.63 | 7.60 | |
| Maximum stimulation of loss of autolysin effect by signal molecules | Smax, loss | 1.18 (7.9) | 1.24 (6.1) | 1.09-1.90 | 0.630 | |
| Ceftazidime concn causing 50% of stimulation for prolonging generation time | EC50, drug | mg/liter | 35.3 (41.9) | 320 (32.2) | 15.7-78.4 | |
| Maximum stimulation of prolonging generation time | Smax, k12 | 10 (fixed)d | 10 (fixed)d | 0 (fixed)g | ||
| SD of residual error on log10 scale | σ | 0.224 (8.5) | 0.186 (5.7) | 0.208-0.228 | 0.378 | |
Assumed to be very fast (i.e., not rate limiting).
As there is a very limited amount of information on this parameter available in the current data set, the onset of the signal molecule effect was assumed to be very fast according to data from starvation experiments and since cell-to-cell communication is tightly controlled.
The parameter was estimated to be greater than 48 h. As the sensitivity of bacterial count curves toward this parameter was low, it was fixed to 24 h.
The parameter was fixed to 10, since there was only limited information available in the data set.
The parameter was estimated to be between 0.99 and 1 and therefore was fixed to 1.
Estimate for the McGrath study. For the in vitro model studies, the estimated MTT12 was 24.0 min for the study by Barclay et al. (1) and 78.2 min for the study by Cappelletty et al. (9). For the time-kill experiments by Shalit et al. (55), Barclay et al. (2), and Tam et al. (61), the estimated MTT12 was 36.6 min.
The estimate was fixed to zero, since the modeling of the in vitro PD model data on PAO1 and on ATCC 27853 resulted in values very close to zero, suggesting no effect.
The SC50 was fixed to the estimate for PAO1, since the MIC for both strains was the same and since ceftazidime concentrations for strain ATCC 27853 were almost fivefold higher than the SC50, which does not support a precise estimation of SC50.
The objective function was 388 points worse (P < 0.0001) for model D (and the same Smax) with different SC50 compared to the model with different Smax (Table 2). A model with both SC50 and Smax being different improved the objective function insignificantly (by 0.8 points) compared to model D and yielded similar estimates for SC50s (0.269 mg/liter for the susceptible and 0.308 mg/liter for the resistant population). A model with different Smax and different MTT12 improved the objective function by 6.8 (P = 0.0091) compared to model D (Table 2); however, the MTT12 was slightly shorter for the resistant population (25.8 and 30.8 min, respectively). As a faster growth of the resistant population is physiologically implausible, we selected model D with different Smax and the same SC50 and MTT12 as the final model.
Several parameters were fixed to specific values (Table 2) to improve the estimation of the remaining model parameters, since our original data set did not contain sufficient information for a sound estimation of all model parameters. The Smax, S was estimated as 0.999 and eventually was fixed to 1. We fixed each of these parameters to a range of values and compared the estimates for the remaining parameters and the curve fits. The final choices of fixed estimates provided indistinguishably good curve fits compared to those of models when all parameters were estimated. More importantly, the choice of parameter values did not cause a noticeable change for the other parameter estimates. The only exception is that the estimate for EC50, drug is conditional on the choice of Smax, k12.
Internal model qualification.
Case deletion diagnostics (Table 2) showed narrow ranges of reestimated parameter values for most parameters. The 5 to 95% percentile intervals of 1,000 runs (results not shown) when four combinations of CFUo and drug concentration were left out randomly were comparable to the ranges of parameter estimates from the case deletion test (Table 2).
Figure 4C shows the population predicted and observed CFU counts during the leave-one-inoculum-out test of model D. The data of the 108 and 109 CFUo and the lag time of killing at all CFUo were predicted very well. There was a minor underprediction of the extent of killing between 24 and 48 h for the 107 CFUo. Up to 8 h, CFU counts were very well predicted for the 105 and 106 CFUo. Slight differences in the extent of regrowth at 24 h between these two low CFUo caused minor mispredictions of the CFU counts at 24 h for these two CFUo. The parameter estimation of model D was robust in all of the reestimation procedures described above.
External model qualification using in-house and literature data.
External model qualification based on in-house data (Fig. 4D) showed that the model could predict CFU-versus-time profiles for CFUo outside the studied range (106 to 108 CFU/ml). The external qualification based on data of P. aeruginosa PAO1 from Henrichfreise et al. (28) revealed that model D and the parameter estimates shown in Table 2 captured the time course of killing and the regrowth of the total population well, given that this is an extrapolation from a static time-kill experiment to a dynamic in vitro PD model (Fig. 5).
FIG. 5.

Observed total and resistant population (triangles indicate a resistant population determined on agar plates containing 8 mg/liter ceftazidime), predicted (using the parameter estimates shown in Table 2), and fitted bacterial counts (lines) of the in vitro PD model study by Henrichfreise et al. (28) for model D.
Predictions for the resistant population qualitatively showed the general trend. However, predictions for the resistant population were biased, since the CFUo of the resistant population was smaller, and since this population grew in the presence of high ceftazidime concentrations between 9 and 16 h with a net growth half-life of approximately 25 min. Parameters that were different between the two types of experiments were identified by reestimation and changes in objective function. The CFUo from the reestimation of parameter values for PAO1 in the one-compartment in vitro PD model was 107.56 CFU/ml, and estimates for the other parameters (original estimates) were −7.33 (−3.54) for log10 FrR, 18.0 min (28.3 min) for MTT12, 0 (10, both fixed) for Smax, k12, 0.469 (0.560) for Smax, R, and 107.96 (107.24) for C50, Sig of the resistant population (C50, Sig for the susceptible population was fixed at 107.24). All other parameters had the same estimates as those shown in Table 2. Curve fits after reestimation were unbiased and precise for the total population and were acceptable for the resistant population (Fig. 5).
Model D could describe the data for strain P. aeruginosa ATCC 27853 from an in vitro PD model and time-kill experiments of six of seven studies very well. For the study by Mouton et al. (46), in silico mispredictions were observed that improved if the growth half-life was faster (results not shown). These datasets were split into an estimation data set (Fig. 6A) and a prediction data set (Fig. 6B). Curve fits for the estimation data set were precise and unbiased (Fig. 6C) with the exception of those from the study by Mouton et al. (46). More importantly, the model was able to provide unbiased and precise predictions across a range of CFUo and studies for the ATCC 27853 strain. None of the data shown in Fig. 6B were used during the estimation of parameters used to generate this plot. Curve fits after the reestimation of the model parameters based on all data for strain P. aeruginosa ATCC 27853 were more precise for all seven studies (data not shown). Table 2 lists the final parameter values when all data, except for that of the study by Mouton et al. (46), were used for estimation.
FIG. 6.

External model qualification of model D (Fig. 2) integrating literature data on P. aeruginosa ATCC 27853 from seven studies. (A) Estimation data set (including data from Barcley et al. [1, 2], Cappelletty et al. [9], McGrath et al. [43], Shalit et al. [55], and Mouton et al. [46]); (B) in silico predictions for time-kill data from Tam et al. (61) at 108 CFUo and in vitro PD model data from Cappelletty et al. (9) for various continuous-infusion (CI) and intermittent-dosage regimens, data from McGrath et al. (43), and data from Mouton et al. (46); (C) observed versus predicted counts and observed versus fitted bacterial counts, with in silico predictions from Mouton et al. (46) shown separately.
DISCUSSION
This study presents a mechanism-based mathematical model that could describe and predict the growth and killing of P. aeruginosa PAO1 across a wide range of CFUo in vitro. The mathematical model also predicted the time course of the antimicrobial effect of ceftazidime on P. aeruginosa ATCC 27853 across various CFUo from six previously published studies, even though this specific isolate was not studied in our time-kill experiments. The model was extensively qualified by internal and external model qualification procedures.
Our study showed a pronounced decrease in the rate of bacterial killing of P. aeruginosa PAO1 by ceftazidime at a high CFUo. We are not aware of PKPD models that can describe the slower bacterial killing at high CFUo, with the exception of our models for colistin (8) and tobramycin (7). The inoculum effect has been shown for several antibiotics against various pathogens, including ceftazidime against P. aeruginosa (6, 13, 16, 22, 31, 34, 61). Most studies assessed the effect of CFUo on the MIC. This type of inoculum effect differs from the inoculum effect in our time-kill study. The current study explored the impact of CFUo on the rate of killing, and the inoculum effect was defined as a lower rate of killing at high CFUo, as this definition also accounts for the time course of bacterial killing. Although the clinical significance of the inoculum effect is unknown, animal infection models have shown for beta-lactams and other anti-infectives that a high CFUo or the delayed treatment of infections may greatly increase mortality or attenuate antiinfective effects (6, 20, 30, 57). Lee et al. (35) showed in a murine infection model with Staphylococcus aureus that up to 72-fold-higher vancomycin doses are required to achieve a static effect at the 107 CFUo than at the 105 CFUo.
As one potential reason for the inoculum effect, increased beta-lactamase production at high CFUo may occur due to the increased breakdown of the cell wall fragments of lysed bacteria. Several beta-lactam antibiotics (ceftazidime, ceftriaxone, cefotaxime, and aztreonam) have demonstrated an inoculum effect despite their stability against beta-lactamases (21). The breakdown of ceftazidime by beta-lactamases from P. aeruginosa (31, 50, 59) is relatively slow due to a low maximal velocity of hydrolysis.
Another potential reason for the inoculum effect is quorum-sensing mechanisms involved in cell-to-cell communication mediated via freely diffusible signal molecules in P. aeruginosa. Stevens et al. (57) proposed that the inoculum effect for beta-lactams was caused by a gradual decrease in the expression of selected PBPs in Streptococcus pyogenes at later growth phases. Ceftazidime has a high binding affinity to PBP3 and PBP1a for P. aeruginosa PAO1 (17, 52). The binding of ceftazidime to PBPs causes a disturbance in cell wall synthesis and, ultimately, cell death (17, 24). Liao and Hancock (38) showed that the expression of PBP3 is downregulated at stationary phase. As ceftazidime causes killing primarily by binding to PBP3, this down-regulation may be an explanation for the inoculum effect of ceftazidime. Finally, another potential reason for the inoculum effect is that the expression of autolysins changes during growth phases. Li et al. (37) showed that the major autolysin in P. aeruginosa PAO1 is a 26-kDa murein hydrolase whose expression has its peak during the mid-exponential growth phase and declines thereafter.
This study proposes two new mathematical models. For model C (Fig. 1), the lag time of bacterial killing is determined by the rate of the turnover of cell wall constituents. As this turnover is a system property, the lag time of killing is independent of antibiotic concentration. The delay between the binding of ceftazidime to PBP3 and cellular death is consistent with data from the literature (27, 39, 62, 63). Previous studies used exponential functions to describe the functional adaptation or lag time of growth or killing (36, 42, 49, 64). Model C described the growth and killing of the susceptible population at each CFUo well (Fig. 4) and accounts for the lag time of killing better than the biophase model. However, model C required different PD parameter estimates for most parameters at each CFUo that varied systematically and significantly with the CFUo (Table 1).
Therefore, a mechanism-based model was created that could describe the time course of bacterial growth and killing at all studied concentrations and CFUo very well (Fig. 3 and 4B). The model included a susceptible and a genotypically resistant population and a new growth model resembling the cellular life cycle. The latter allows one to separate between the rate of bacterial replication (determined by the mean generation time [MTT12 = 1/k12]) and the efficiency of replication (i.e., the probability of successful doubling). This model is more flexible than the logistic (68, 70) or saturable growth model (44), as the life cycle model allows the drug effect to modify the probability of successful growth, the rate of growth, or both. Stasis, for example, can result either from a 50% probability of successful doubling or from a long generation time. Our estimate for the mean generation time of 28.3 min in LB broth was similar to the median growth half-life of approximately 32 min (equivalent to a mean generation time of 46 min) for P. aeruginosa in vitro based on the studies reviewed by Czock and Keller (15), given the relatively large variability of growth rates in vitro from different studies in the literature (Fig. 6A).
Ceftazidime was assumed to bind to PBPs, which stimulate the autolysin effect, and the turnover of the autolysin effect caused the lag time of killing (Fig. 2). For model D, the inoculum effect was caused by signal molecules that are synthesized by all viable bacteria. The rate constant (kS10) for synthesis and the loss of signal molecules was fast, producing a rapid onset of the inoculum effect. The Smax, loss parameter describes the maximum stimulation of loss of the autolysin effect and therefore characterizes the extent of the inoculum effect at high signal molecule concentrations. Figure 7 illustrates the inoculum effect as an increase in the probability of successful replication for the susceptible population at high CFUo. In our model, this increase is caused by the stimulation of the loss of the autolysin effect at high signal molecule concentrations.
FIG. 7.

Probability of successful replication (doubling) as a function of the initial inoculum for a range of constant ceftazidime concentrations (the simulation was based on parameter estimates from NONMEM); a probability of 100% represents perfect growth, 50% represents net stasis due to every second doubling being successful, and 0% represents the fastest possible killing; see Materials and Methods for details.
Literature data from starvation experiments (66, 67) in slow- and nongrowing pneumococci show that bacteria can rapidly adapt to their environment (within minutes) under amino acid starvation, and that such starvation progressively depletes autolysin. In agreement with these data, the onset of the inoculum effect was estimated to be fast in model D (Table 2).
Jumbe et al. (32) studied the impact of CFUo on the probability of isolating resistant mutants after 24 h of levofloxacin treatment in mice, and they found that a substantially higher drug exposure was required to suppress resistance at 24 h for the high (108) CFUo compared to the low (107) CFUo. The high CFUo exceeded the inverse of the mutation frequency, which caused a high probability of having at least one resistant cell in the initial inoculum, whereas no resistant mutants were found in the drug containing samples for their low CFUo. Therefore, the stochastic process of whether or not the resistant mutants were present in their CFUo caused the inoculum effect of the net killing at 24 h in their study.
This type of inoculum effect also may have been present in our study and was accounted for in our model equations. However, the resistant subpopulation comprised 10−3.54 of CFUo in our study (Table 2). If the stochastic process described above primarily caused the inoculum effect in our study, one would expect about 3 log10 net killing during the first 8 h at high ceftazidime concentrations for the 107 and 108 CFUo in Fig. 3. However, we found no net killing or slight growth at all ceftazidime concentrations during the first 8 h at these CFUo. Therefore, the inoculum effect in our study was a property of the predominant population that was susceptible to ceftazidime at low CFUo. Our inoculum effect model (model D) described the inoculum effect as a change of the effective killing rate that depends on the signal molecule concentration and therefore on CFUo (Fig. 7). In contrast, the stochastic process described by Jumbe et al. (32) affects the initial condition of the differential equation for the resistant population, which is a different type of inoculum effect.
Models D and C were robust in case deletion diagnostics (Tables 1 and 2), and model D also was robust in 1,000 internal cross-validation runs (not performed for model C) based on randomly drawn datasets from a nonparametric bootstrap. The model also yielded reasonably precise and unbiased predictions if all data at one CFUo were left out and bacterial counts were simulated (Fig. 4C). Model D yielded precise and unbiased predictions when the bacterial counts of a preliminary time-kill experiment were predicted for CFUo of 105 to 109 CFU/ml (Fig. 4D). The mispredictions shown in Fig. 4C and D were primarily at 24 h and came from a slight difference in the extent of regrowth for the data at the 105 and 106 CFUo. Both the case deletion diagnostics and especially the leave-one-inoculum-out procedure (Fig. 4) showed that the model was robust, and parameters were well estimable after leaving out a significant portion of the data set. This suggested that the model is not overparameterized and that the chosen fixed parameter estimates (Table 2) were reasonable. Also, the consistency of the results in NONMEM and S-ADAPT and the low standard errors in both programs suggested that model D was not overparameterized.
The mean generation time was 28.3 min at low signal molecule concentrations, and the SC50 for the stimulation of the autolysin effect of 0.294 mg/liter was in the range of the IC50s of ceftazidime for binding to PBPs in PAO1 (17) (IC50s are 0.1 mg/liter for PBP3, 0.2 mg/liter for PBP1a, and 5 mg/liter for PBP1b). As pointed out by Hayes and Orr (27), these IC50s and the estimated SC50 for the stimulation of the autolysin effect are not identical, since additional steps between PBP binding and cellular death are involved.
Bacterial counts simulated from model D yielded reasonable predictions even before the reestimation of parameters for the data from Henrichfreise et al. (28). The reestimation of model parameters for this in vitro PD model showed that bacterial replication was faster in this model than in our time-kill experiment, and curve fits were excellent for the total population and reasonable for the resistant population (Fig. 5). The resistant population in the Henrichfreise study was small and might not have been present in the CFUo of our time-kill experiments.
An extensive external model qualification based on literature data for another strain, P. aeruginosa ATCC 27853, showed that the model was robust and could be applied to strains other than PAO1. A relatively small estimation data set from the literature was used to reestimate specific model parameters for the ATCC strain (Fig. 6A). Simulations from model D for other experiments using the ATCC strain revealed an excellent predictive performance of this model across a range in CFUo and different dosage regimens for six of the seven studies (Fig. 6B and C). This is a challenging method of model qualification, since bacterial counts were predicted in silico without using any of the observations shown in Fig. 6B. In silico predictions for the ATCC strain studied by Mouton and den Hollander (46, 49) showed some bias, since the rate of bacterial killing (approximately 4 log10 in 3 h for the continuous and intermittent infusions) was notably faster than the net growth rate, and the lag time of killing was short (Fig. 6). These mispredictions improved if a faster growth half-life was used. Further studies are required to resolve the causes of these in silico mispredictions.
PD parameters were estimated based on all data (except that of the study by Mouton and den Hollander) shown in Fig. 6. The generation time of the ATCC strains varied considerably between experiments. The ATCC strain was estimated to show a smaller extent of the inoculum effect, as the Smax, loss was 0.630 for the ATCC strain and 1.18 for PAO1. The inoculum effect started at higher CFUo for the ATCC strain, since its C50, Sig was 2.3-fold higher than that of PAO1. An important feature of model D is that the maximum rate of bacterial death is the same as the maximum rate of growth, which is in agreement with experiments for slow-growing bacteria by Tuomanen (66). Consequently, this model predicts that the optimal duration of therapy is longer if bacteria turn over more slowly. Further clinical studies are required to assess the optimal duration of therapy.
One potential limitation of our experiment is that ceftazidime concentrations were not measured. Eng et al. (22) found a substantial inoculum effect of ceftazidime in vitro for P. aeruginosa ATCC 27853 but no significant inactivation of ceftazidime at 6 h using a CFUo of 107.7 CFU/ml. Degradation is 8% at 18 h and 40% at 48 h. This slow degradation in broth is similar to the degradation half-life of approximately 45.9 h of ceftazidime at 37°C in water, as reported by Viaene et al. (69). If drug degradation caused the inoculum effect in our study, it would be unusual to observe most of the net killing between 12 and 48 h for the 107 CFUo (Fig. 3B). Henrichfreise et al. (28) studied ceftazidime against P. aeruginosa PAO1 in vitro and found no degradation of ceftazidime. Overall, the degradation of ceftazidime in broth seems unlikely to have caused the inoculum effect in our experiment.
In summary, this study proposed a mechanism-based model that can describe and predict the pronounced inoculum effect of P. aeruginosa PAO1 and P. aeruginosa ATCC 27853 in vitro. This model was robust and yielded accurate predictions in extensive internal and external qualification procedures that included data from eight literature studies of these two strains. This model may be used as the basis for models that account for differential binding to several PBPs for combination therapy with beta-lactams. These in vitro results suggest that the effects of ceftazidime monotherapy for infections at high CFUo are attenuated due to the inoculum effect. However, more animal and clinical data are needed for the clinical significance of the inoculum effect and its impact on the emergence of resistance.
Acknowledgments
We thank Robert E. Hancock and Manjeet Bains for providing the PAO1 strain as a gift. We thank Cornelia Landersdorfer for discussions about this project and a critical review of the manuscript.
J.B. was supported by a postdoctoral fellowship from Johnson & Johnson.
Footnotes
Published ahead of print on 13 October 2008.
REFERENCES
- 1.Barclay, M. L., E. J. Begg, S. T. Chambers, and D. R. Boswell. 1995. Improved efficacy with nonsimultaneous administration of first doses of gentamicin and ceftazidime in vitro. Antimicrob. Agents Chemother. 39:132-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barclay, M. L., E. J. Begg, S. T. Chambers, and B. A. Peddie. 1996. The effect of aminoglycoside-induced adaptive resistance on the antibacterial activity of other antibiotics against Pseudomonas aeruginosa in vitro. J. Antimicrob. Chemother. 38:853-858. [DOI] [PubMed] [Google Scholar]
- 3.Bauer, R. J. 2007. S-ADAPT/MCPEM user's guide (version 1.55). Pharmacokinetic, Pharmacodynamic and Population Data Analysis, Berkeley, CA.
- 4.Beal, S. L., L. B. Sheiner, and A. J. Boeckmann. 2006. NONMEM users guides (1989-2006). Icon Development Solutions, Ellicott City, MD.
- 5.Bratu, S., J. Gupta, and J. Quale. 2006. Expression of the las and rhl quorum-sensing systems in clinical isolates of Pseudomonas aeruginosa does not correlate with efflux pump expression or antimicrobial resistance. J. Antimicrob. Chemother. 58:1250-1253. [DOI] [PubMed] [Google Scholar]
- 6.Brook, I. 1989. Inoculum effect. Rev. Infect. Dis. 11:361-368. [DOI] [PubMed] [Google Scholar]
- 7.Bulitta, J. B., N. S. Ly, B. T. Tsuji, W. J. Jusko, and A. Forrest. 2008. Mechanism-based models for growth and killing of pseudomonas aeruginosa by tobramycin to quantify and predict the inoculum effect, abstr. 01. Abstr. Am. Conf. Pharmacometrics. http://mosaicnj.com/acop/pdfs/1_Bulitta.pdf.
- 8.Bulitta, J. B., J. C. Yang, B. T. Tsuji, W. J. Jusko, and A. Forrest. 2007. Mechanistic PK/PD models for the inoculum effect (over 5 orders of magnitude) of colistin and ceftazidime against Pseudomonas aeruginosa (PA), abstr. 1829. Abstr. Am. Assoc. Pharm. Sci.
- 9.Cappelletty, D. M., S. L. Kang, S. M. Palmer, and M. J. Rybak. 1995. Pharmacodynamics of ceftazidime administered as continuous infusion or intermittent bolus alone and in combination with single daily-dose amikacin against Pseudomonas aeruginosa in an in vitro infection model. Antimicrob. Agents Chemother. 39:1797-1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll, R. J., and D. Ruppert. 1988. Transformation and weighting in regression. Chapman and Hall, New York, NY.
- 11.Chung, P., P. J. McNamara, J. J. Campion, and M. E. Evans. 2006. Mechanism-based pharmacodynamic models of fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:2957-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clinical and Laboratory Standards Institute. 2007. Performance standards for antimicrobial susceptibility testing: 17th informational supplement, vol. 27. Clinical and Laboratory Standards Institute, Wayne, PA.
- 13.Corrado, M. L., S. H. Landesman, and C. E. Cherubin. 1980. Influence of inoculum size on activity of cefoperazone, cefotaxime, moxalactam, piperacillin, and N-formimidoyl thienamycin (MK0787) against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 18:893-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Craig, W. A., S. M. Bhavnani, and P. G. Ambrose. 2004. The inoculum effect: fact or artifact? Diagn. Microbiol. Infect. Dis. 50:229-230. [DOI] [PubMed] [Google Scholar]
- 15.Czock, D., and F. Keller. 2007. Mechanism-based pharmacokinetic-pharmacodynamic modeling of antimicrobial drug effects. J. Pharmacokinet. Pharmacodyn. 34:727-751. [DOI] [PubMed] [Google Scholar]
- 16.Davey, P. G., and M. Barza. 1987. The inoculum effect with gram-negative bacteria in vitro and in vivo. J. Antimicrob. Chemother. 20:639-644. [DOI] [PubMed] [Google Scholar]
- 17.Davies, T. A., M. G. Page, W. Shang, T. Andrew, M. Kania, and K. Bush. 2007. Binding of ceftobiprole and comparators to the penicillin-binding proteins of Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumoniae. Antimicrob. Agents Chemother. 51:2621-2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diggle, S. P., K. Winzer, S. R. Chhabra, K. E. Worrall, M. Camara, and P. Williams. 2003. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol. Microbiol. 50:29-43. [DOI] [PubMed] [Google Scholar]
- 19.Driscoll, J. A., S. L. Brody, and M. H. Kollef. 2007. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67:351-368. [DOI] [PubMed] [Google Scholar]
- 20.Eagle, H. 1949. The effect of the size of the inoculum and the age of the infection on the curative dose of penicillin in experimental infections with streptococci, pneumococci, and Treponema pallidum. J. Exp. Med. 90:595-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eng, R. H., C. Cherubin, S. M. Smith, and F. Buccini. 1985. Inoculum effect of beta-lactam antibiotics on Enterobacteriaceae. Antimicrob. Agents Chemother. 28:601-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eng, R. H., S. M. Smith, and C. Cherubin. 1984. Inoculum effect of new beta-lactam antibiotics on Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 26:42-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaynes, R., and J. R. Edwards. 2005. Overview of nosocomial infections caused by gram-negative bacilli. Clin. Infect. Dis. 41:848-854. [DOI] [PubMed] [Google Scholar]
- 24.Goodman, L. S., A. Gilman, L. L. Brunton, J. S. Lazo, and K. L. Parker. 2006. Goodman & Gilman's the pharmacological basis of therapeutics, 11th ed. McGraw-Hill, New York, NY.
- 25.Grossi, P., and D. Dalla Gasperina. 2006. Treatment of Pseudomonas aeruginosa infection in critically ill patients. Expert Rev. Anti Infect. Ther. 4:639-662. [DOI] [PubMed] [Google Scholar]
- 26.Gumbo, T., A. Louie, M. R. Deziel, L. M. Parsons, M. Salfinger, and G. L. Drusano. 2004. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J. Infect. Dis. 190:1642-1651. [DOI] [PubMed] [Google Scholar]
- 27.Hayes, M. V., and D. C. Orr. 1983. Mode of action of ceftazidime: affinity for the penicillin-binding proteins of Escherichia coli K12, Pseudomonas aeruginosa and Staphylococcus aureus. J. Antimicrob. Chemother. 12:119-126. [DOI] [PubMed] [Google Scholar]
- 28.Henrichfreise, B., I. Wiegand, I. Luhmer-Becker, and B. Wiedemann. 2007. Development of resistance in wild-type and hypermutable Pseudomonas aeruginosa strains exposed to clinical pharmacokinetic profiles of meropenem and ceftazidime simulated in vitro. Antimicrob. Agents Chemother. 51:3642-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heurlier, K., V. Denervaud, and D. Haas. 2006. Impact of quorum sensing on fitness of Pseudomonas aeruginosa. Int. J. Med. Microbiol. 296:93-102. [DOI] [PubMed] [Google Scholar]
- 30.Hope, W. W., G. L. Drusano, C. B. Moore, A. Sharp, A. Louie, T. J. Walsh, D. W. Denning, and P. A. Warn. 2007. Effect of neutropenia and treatment delay on the response to antifungal agents in experimental disseminated candidiasis. Antimicrob. Agents Chemother. 51:285-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson, C. C., L. Livornese, M. J. Gold, P. G. Pitsakis, S. Taylor, and M. E. Levison. 1995. Activity of cefepime against ceftazidime-resistant gram-negative bacilli using low and high inocula. J. Antimicrob. Chemother. 35:765-773. [DOI] [PubMed] [Google Scholar]
- 32.Jumbe, N., A. Louie, R. Leary, W. Liu, M. R. Deziel, V. H. Tam, R. Bachhawat, C. Freeman, J. B. Kahn, K. Bush, M. N. Dudley, M. H. Miller, and G. L. Drusano. 2003. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Investig. 112:275-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keil, S., and B. Wiedemann. 1995. Mathematical corrections for bacterial loss in pharmacodynamic in vitro dilution models. Antimicrob. Agents Chemother. 39:1054-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirby, W. M. 1945. Bacteriostatic and lytic actions of penicillin on sensitive and resistant staphylococci. J. Clin. Investig. 24:165-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee, D., Y. Murakami, T. Stamstad, K. Marchillo, J. Ashbeck, D. R. Andes, and W. A. Craig. 2007. Abstr. 47th Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-37.
- 36.Li, R. C. 1996. Simultaneous pharmacodynamic analysis of the lag and bactericidal phases exhibited by beta-lactams against Escherichia coli. Antimicrob. Agents Chemother. 40:2306-2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li, Z., A. J. Clarke, and T. J. Beveridge. 1996. A major autolysin of Pseudomonas aeruginosa: subcellular distribution, potential role in cell growth and division and secretion in surface membrane vesicles. J. Bacteriol. 178:2479-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao, X., and R. E. Hancock. 1997. Identification of a penicillin-binding protein 3 homolog, PBP3x, in Pseudomonas aeruginosa: gene cloning and growth phase-dependent expression. J. Bacteriol. 179:1490-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livermore, D. M. 1987. Radiolabelling of penicillin-binding proteins (PBPs) in intact Pseudomonas aeruginosa cells: consequences of beta-lactamase activity by PBP-5. J. Antimicrob. Chemother. 19:733-742. [DOI] [PubMed] [Google Scholar]
- 40.Maseda, H., I. Sawada, K. Saito, H. Uchiyama, T. Nakae, and N. Nomura. 2004. Enhancement of the mexAB-oprM efflux pump expression by a quorum-sensing autoinducer and its cancellation by a regulator, MexT, of the mexEF-oprN efflux pump operon in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 48:1320-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masuda, N., E. Sakagawa, S. Ohya, N. Gotoh, H. Tsujimoto, and T. Nishino. 2000. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44:3322-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattie, H., L. C. Zhang, E. van Strijen, B. R. Sekh, and A. E. Douwes-Idema. 1997. Pharmacokinetic and pharmacodynamic models of the antistaphylococcal effects of meropenem and cloxacillin in vitro and in experimental infection. Antimicrob. Agents Chemother. 41:2083-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGrath, B. J., E. M. Bailey, K. C. Lamp, and M. J. Rybak. 1992. Pharmacodynamics of once-daily amikacin in various combinations with cefepime, aztreonam, and ceftazidime against Pseudomonas aeruginosa in an in vitro infection model. Antimicrob. Agents Chemother. 36:2741-2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meagher, A. K., A. Forrest, A. Dalhoff, H. Stass, and J. J. Schentag. 2004. Novel pharmacokinetic-pharmacodynamic model for prediction of outcomes with an extended-release formulation of ciprofloxacin. Antimicrob. Agents Chemother. 48:2061-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mesaros, N., P. Nordmann, P. Plesiat, M. Roussel-Delvallez, J. Van Eldere, Y. Glupczynski, Y. Van Laethem, F. Jacobs, P. Lebecque, A. Malfroot, P. M. Tulkens, and F. Van Bambeke. 2007. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infect. 13:560-578. [DOI] [PubMed] [Google Scholar]
- 46.Mouton, J. W., and J. G. den Hollander. 1994. Killing of Pseudomonas aeruginosa during continuous and intermittent infusion of ceftazidime in an in vitro pharmacokinetic model. Antimicrob. Agents Chemother. 38:931-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mouton, J. W., and A. A. Vinks. 2005. Pharmacokinetic/pharmacodynamic modelling of antibacterials in vitro and in vivo using bacterial growth and kill kinetics: the minimum inhibitory concentration versus stationary concentration. Clin. Pharmacokinet. 44:201-210. [DOI] [PubMed] [Google Scholar]
- 48.Mouton, J. W., and A. A. Vinks. 2005. Relationship between minimum inhibitory concentration and stationary concentration revisited: growth rates and minimum bactericidal concentrations. Clin. Pharmacokinet. 44:767-768. [DOI] [PubMed] [Google Scholar]
- 49.Mouton, J. W., A. A. Vinks, and N. C. Punt. 1997. Pharmacokinetic-pharmacodynamic modeling of activity of ceftazidime during continuous and intermittent infusion. Antimicrob. Agents Chemother. 41:733-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nikaido, H., W. Liu, and E. Y. Rosenberg. 1990. Outer membrane permeability and beta-lactamase stability of dipolar ionic cephalosporins containing methoxyimino substituents. Antimicrob. Agents Chemother. 34:337-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oberg, A., and M. Davidian. 2000. Estimating data transformations in nonlinear mixed effects models. Biometrics 56:65-72. [DOI] [PubMed] [Google Scholar]
- 52.Rains, C. P., H. M. Bryson, and D. H. Peters. 1995. Ceftazidime. An update of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 49:577-617. [DOI] [PubMed] [Google Scholar]
- 53.Sawada, I., H. Maseda, T. Nakae, H. Uchiyama, and N. Nomura. 2004. A quorum-sensing autoinducer enhances the mexAB-oprM efflux-pump expression without the MexR-mediated regulation in Pseudomonas aeruginosa. Microbiol. Immunol. 48:435-439. [DOI] [PubMed] [Google Scholar]
- 54.Schuster, M., and E. P. Greenberg. 2006. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int. J. Med. Microbiol. 296:73-81. [DOI] [PubMed] [Google Scholar]
- 55.Shalit, I., D. F. Welch, V. H. San Joaquin, and M. I. Marks. 1985. In vitro antibacterial activities of antibiotics against Pseudomonas aeruginosa in peritoneal dialysis fluid. Antimicrob. Agents Chemother. 27:908-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheiner, L. B., D. R. Stanski, S. Vozeh, R. D. Miller, and J. Ham. 1979. Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. Clin. Pharmacol. Ther. 25:358-371. [DOI] [PubMed] [Google Scholar]
- 57.Stevens, D. L., S. Yan, and A. E. Bryant. 1993. Penicillin-binding protein expression at different growth stages determines penicillin efficacy in vitro and in vivo: an explanation for the inoculum effect. J. Infect. Dis. 167:1401-1405. [DOI] [PubMed] [Google Scholar]
- 58.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 59.Takeda, S., Y. Ishii, K. Hatano, K. Tateda, and K. Yamaguchi. 2007. Stability of FR264205 against AmpC beta-lactamase of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 30:443-445. [DOI] [PubMed] [Google Scholar]
- 60.Tam, V. H., A. Louie, M. R. Deziel, W. Liu, R. Leary, and G. L. Drusano. 2005. Bacterial-population responses to drug-selective pressure: examination of garenoxacin's effect on Pseudomonas aeruginosa. J. Infect. Dis. 192:420-428. [DOI] [PubMed] [Google Scholar]
- 61.Tam, V. H., A. N. Schilling, D. A. Melnick, and E. A. Coyle. 2005. Comparison of beta-lactams in counter-selecting resistance of Pseudomonas aeruginosa. Diagn. Microbiol. Infect. Dis. 52:145-151. [DOI] [PubMed] [Google Scholar]
- 62.Tomasz, A. 1979. From penicillin-binding proteins to the lysis and death of bacteria: a 1979 view. Rev. Infect. Dis. 1:434-467. [DOI] [PubMed] [Google Scholar]
- 63.Tomasz, A. 1986. Penicillin-binding proteins and the antibacterial effectiveness of beta-lactam antibiotics. Rev. Infect. Dis. 8(Suppl. 3):S260-S278. [DOI] [PubMed] [Google Scholar]
- 64.Treyaprasert, W., S. Schmidt, K. H. Rand, U. Suvanakoot, and H. Derendorf. 2007. Pharmacokinetic/pharmacodynamic modeling of in vitro activity of azithromycin against four different bacterial strains. Int. J. Antimicrob. Agents 29:263-270. [DOI] [PubMed] [Google Scholar]
- 65.Tsuji, B. T., C. von Eiff, P. A. Kelchlin, A. Forrest, and P. F. Smith. 2008. Attenuated vancomycin bactericidal activity against Staphylococcus aureus hemB mutants expressing the small-colony-variant phenotype. Antimicrob. Agents Chemother. 52:1533-1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tuomanen, E. 1986. Phenotypic tolerance: the search for beta-lactam antibiotics that kill nongrowing bacteria. Rev. Infect. Dis. 8(Suppl. 3):S279-S291. [DOI] [PubMed] [Google Scholar]
- 67.Tuomanen, E., and A. Tomasz. 1990. Mechanism of phenotypic tolerance of nongrowing pneumococci to beta-lactam antibiotics. Scand. J. Infect. Dis. Suppl. 74:102-112. [PubMed] [Google Scholar]
- 68.Verhulst, P.-F. 1845. Recherches mathématiques sur la loi d'accroissement de la population. Nouv. Mém. Acad. R. Sci. Belles-Lettres Bruxelles 18:1-41. [Google Scholar]
- 69.Viaene, E., H. Chanteux, H. Servais, M. P. Mingeot-Leclercq, and P. M. Tulkens. 2002. Comparative stability studies of antipseudomonal beta-lactams for potential administration through portable elastomeric pumps (home therapy for cystic fibrosis patients) and motor-operated syringes (intensive care units). Antimicrob. Agents Chemother. 46:2327-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yano, Y., T. Oguma, H. Nagata, and S. Sasaki. 1998. Application of logistic growth model to pharmacodynamic analysis of in vitro bactericidal kinetics. J. Pharm. Sci. 87:1177-1183. [DOI] [PubMed] [Google Scholar]