

Abstract
MmeI from Methylophilus methylotrophus belongs to the type II restriction-modification enzymes. It recognizes an asymmetric DNA sequence, 5′-TCCRAC-3′ (R indicates G or A), and cuts both strands at fixed positions downstream of the specific site. This particular feature has been exploited in transcript profiling of complex genomes (using serial analysis of gene expression technology). We have shown previously that the endonucleolytic activity of MmeI is strongly dependent on the presence of S-adenosyl-l-methionine (J. Nakonieczna, J. W. Zmijewski, B. Banecki, and A. J. Podhajska, Mol. Biotechnol. 37:127-135, 2007), which puts MmeI in subtype IIG. The same cofactor is used by MmeI as a methyl group donor for modification of an adenine in the upper strand of the recognition site to N6-methyladenine. Both enzymatic activities reside in a single polypeptide (919 amino acids [aa]), which puts MmeI also in subtype IIC of the restriction-modification systems. Based on a molecular model, generated with the use of bioinformatic tools and validated by site-directed mutagenesis, we were able to localize three functional domains in the structure of the MmeI enzyme: (i) the N-terminal portion containing the endonucleolytic domain with the catalytic Mg2+-binding motif D70-X9-EXK82, characteristic for the PD-(D/E)XK superfamily of nucleases; (ii) a central portion (aa 310 to 610) containing nine sequence motifs conserved among N6-adenine γ-class DNA methyltransferases; (iii) the C-terminal portion (aa 610 to 919) containing a putative target recognition domain. Interestingly, all three domains showed highest similarity to the corresponding elements of type I enzymes rather than to classical type II enzymes. We have found that MmeI variants deficient in restriction activity (D70A, E80A, and K82A) can bind and methylate specific nucleotide sequence. This suggests that domains of MmeI responsible for DNA restriction and modification can act independently. Moreover, we have shown that a single amino acid residue substitution within the putative target recognition domain (S807A) resulted in a MmeI variant with a higher endonucleolytic activity than the wild-type enzyme.
Restriction-modification (RM) systems are composed of two enzymatic entities: a restriction endodeoxyribonuclease (REase) that cleaves DNA usually within short, specific sequences and a methyltransferase (MTase) that modifies the same sequence in order to protect the host genomic DNA against the action of the cognate restriction enzyme. Based on their molecular structure, functional features, and cofactors requirements, RM systems can be divided into four distinct types (49). The most complex are enzymes of types I and III (and some of type IV), which function as multisubunit molecular machines that exhibit very complex mechanism of action involving NTP hydrolysis and DNA translocation (reviewed in reference 4). On the other hand, type II RM systems comprise relatively simple independent REase and MTase enzymes whose properties, in particular recognition of short specific nucleotide sequences (4 to 8 bp), have made them indispensable tools of modern molecular biology (45). To date, almost 4,000 type II REases have been identified by screening various Bacteria, Archaea, and viruses and characterizing them biochemically (50).
DNA MTases of RM systems belong to several different families within the Rossmann fold methyltransferase superfamily (6). Structural conservation is strong across catalytic domains of all DNA MTases, despite sequence divergence between families and frequent fusions with additional, unrelated domains that are involved in, for example, specific recognition of the target DNA sequence (29). Two major families are the m5C MTases, a group of proteins with high mutual sequence similarity and only remote similarity to other MTases (48), and the N-MTases (m6A and m4C MTases), a large and heterogeneous group of enzymes that exhibit very complex mutual relationships (9, 34). The N-MTase family contains not only type II enzymes, but also MTase subunits of type I and type III enzymes. Thus far, evolutionary relationships between various subgroups of N-MTases from different types and subtypes have remained unclear.
Unlike with DNA MTases, amino acid sequences of type II REases usually reveal no significant similarity, unless enzymes with identical substrate specificity (isoschizomers) are considered (for a review, see reference 43). However, crystal structure analysis demonstrated that even in the absence of sequence similarity many type II REases appear to be related, as they possess a common catalytic domain, with an α/β core that serves as a scaffold for the catalytic/Mg2+-binding site with a weakly conserved PD-Xn-(D/E)-X-K motif, where X indicates any residue (63). EcoRI (23) and EcoRV (67) were the first REases in which the so-called PD-(D/E)XK domain was structurally characterized. Since then, it has been found in many other REases as well as in numerous other nucleases acting on DNA and involved in a variety of cellular function in all known organisms (2, 10, 25). Nonetheless, bioinformatics and biochemical analyses revealed that some type II REases possess unrelated catalytic domains from different nuclease superfamilies, including PLD, HNH, GIY-YIG, and half-pipe (reviewed in reference 8). These findings have been recently highlighted by crystal structures of Mg2+-independent enzymes: PLD-like R.BfiI (20) and half-pipe R.PabI (38).
Type II REases have been traditionally classified into several overlapping subtypes, depending on various features (49). However, many type II REases exhibit a combination of these features, which puts them into several different subtypes at the same time. MmeI from a methanol utilizer, Methylophilus methylotrophus, is a bifunctional RM type IIC enzyme. Type IIC enzymes possess a MTase and an REase within the same polypeptide. MmeI recognizes a partially degenerate nucleotide sequence and cleaves both DNA strands within a nonspecific region at a variable distance: 5′-TCCRAC-3′ (N)20/18↓ or 5′-TCCRAC-3′(N)21/19↓ (R = G or A) (5, 17). This particular feature makes MmeI also a member of type IIS and was used to extend abilities of the serial analysis of gene expression technology (62), where MmeI was found to be the best molecular tool among REases (15). There are also other methods of genome analysis that exploit MmeI's potential, e.g., Cap analysis of gene expression (55) and vector integration tag analysis (35). The restriction activity of MmeI depends on AdoMet (40), a property that makes MmeI also a member of the type IIG REases. In addition, MmeI was found to behave similarly to type III systems (37) in that it very poorly cleaves substrates with a single unmodified MmeI site or with several unmodified sites that are in the same orientation. Instead, MmeI strongly prefers substrates with at least two unmodified recognition sites in inverse orientation (J. Nakonieczna, unpublished data). The MTase activity of MmeI modifies adenine within the upper strand of the 5′-TCCRAC-3′ site. MmeI, like many RM enzymes which recognize asymmetric specific sequences, exists in solution as a monomer (60).
The mmeIRM gene was isolated and cloned in Escherichia coli (39). The inferred amino acid sequence of MmeI (919 amino acids [aa]) revealed an expectedly strong similarity to DNA MTases in the central region but no obvious similarity to REases, including other previously characterized enzymes of type IIS. Therefore, we carried out bioinformatic analysis of the entire MmeI amino acid sequence, including generation of a molecular model of the enzyme, to identify the domain in the terminal segments, and we carried out site-directed mutagenesis to validate functional predictions for selected residues, inferred to be important for DNA binding and restriction activity.
MATERIALS AND METHODS
Sequence analyses.
Sequence database searches were carried out with PSI-BLAST (1) with a conservative e-value threshold of 1E-30. Multiple sequence alignments were calculated with MUSCLE (16). Protein structure predictions from sequences were carried out using a new version (http://genesilico.pl/meta2/) of the GeneSilico MetaServer (30), which is a gateway for a variety of bioinformatic methods, in particular those for protein fold recognition (FR) analysis (it attempts to match the query sequence to known protein structures). Alignments between the target sequence (MmeI; GenBank EU616582) and sequences of proteins of known structure reported by the FR methods were compared, evaluated, and ranked by using the PCONS server (32) to identify the preferred modeling template and the consensus alignment.
Molecular modeling.
The alignments between the MmeI amino acid sequence and the structure of the best templates identified by PCONS were used to carry out comparative modeling using the FRankenstein's Monster approach (26, 27), which comprises cycles of local realignments in uncertain regions, building of alternative models and their evaluation, realignment in poorly scored regions, and merging of the best-scoring fragments. Previously we used this approach for successful building of structural models for the type II REases SfiI (12) and MvaI (28), which were later confirmed by crystallographic analyses. For the evaluation of models we used PROQ (65, 66) and a MetaMQAP method recently developed at IIMCB (https://genesilico.pl/toolkit/unimod?method=MetaMQAPII) (44), which allow prediction of the deviations of individual residues in the model from their counterparts in the native structure. Poorly scoring parts of the model were refolded using the de novo modeling method ROSETTA (56). Sequence conservation was mapped onto the molecular structure using COLORADO3D (53).
Bacterial strains, plasmids, phage, and media.
The E. coli strains used in this study, ER2683 [fhuA2 glnV44 endA1 thi-1 Δ(mcrC-mrr) 114::IS10 Δ(lacI-lacA)200/F′ proAB lacIq ΔlacZM15 zzf::mini-Tn10] and ER1992 [endA1 thi-1 glnV44 mcr-67 Δ(mcrC-mrr)114::IS10 Δ(argF-lac)U169 dinD1::MudI1734] (18) were kindly provided by Richard D. Morgan and Elisabeth Raleigh (New England Biolabs, Ipswitch, MA). The E. coli strain MM294 was used for cloning experiments (3). The pTB (Apr) plasmid with a cloned mmeIRM gene (39) was used for site-directed mutagenesis experiments and DNA sequencing. All strains were grown on LB medium (52) at 37°C. The following concentrations of antibiotics were used when necessary: ampicillin (Ap), 50 μg/μl; kanamycin (Kan), 50 μg/μl. SOS induction was assayed by growing bacteria on LB agar (LA) plates (52) supplemented with 5-bromo-4-chloro-3-indolyl-β-d-thiogalactopyranoside (X-Gal; 35 μg/ml). λvir was used to test the activity of MmeI and its mutated variants in vivo. The λvir phage stock was prepared according to a standard protocol (52).
Enzymes, chemicals, and oligonucleotides.
All enzymes were purchased from MBI Fermentas and used according to the manufacturer's protocols. [γ-32P]ATP was provided by the MP Biomedicals. Oligonucleotides were synthesized at the Institute of Biochemistry and Biophysics, Polish Academy of Sciences (Warsaw, Poland).
DNA manipulations.
Plasmid purification from bacterial cells as well as DNA isolation from agarose gels were carried out with the use of A&A Biotechnology kits (Gdynia, Poland) according to the provided instructions. DNA sequencing was carried out at the Institute of Biochemistry and Biophysics, Polish Academy of Sciences (Warsaw, Poland). Site-directed mutagenesis was carried out by PCR according to the QuikChange protocol (Stratagene) using plasmid pTB as a template (MmeI R+ M+). All mutant clones were verified by DNA sequencing. Standard procedures were used for molecular cloning (52).
Purification of wt MmeI enzyme and its mutated variants.
E. coli ER 2683 was transformed with plasmid carrying the wild-type (wt) mmeIRM gene or with plasmid carrying a mutated variant of mmeIRM. At an optical density at 575 nm of 0.5 the culture was supplemented with isopropyl-β-d-thiogalactopyranoside to a concentration of 1 mM. Cells were cultured for another 3 h, harvested, rinsed with STE buffer (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA, 100 mM NaCl), and kept at −20°C. The MmeI enzyme or its mutated variants were purified from the bacteria using a previously described procedure (60). The final enzyme preparations did not contain nonspecific nucleases.
Assay for endonucleolytic activity of MmeI.
The endonuclease activity was tested by incubation of MmeI with 1 μg λ DNA (18 MmeI-specific sites) at 37°C for 1 h in a 30-μl reaction volume in a standard reaction buffer (10 mM HEPES, pH 8.0, 2.5 mM potassium acetate, pH 8.0, 2.5 mM magnesium acetate, pH 8.0, 2 mM dithiothreitol [DTT], 0.04 mM AdoMet). Reaction products were resolved in 0.8% agarose gels. One unit of MmeI endonucleolytic activity was defined as the amount of enzyme required to cleave 1 μg of λ DNA at 37°C for 1 h in a 30-μl reaction volume. Biological MmeI restriction activity was assessed by comparing the titers of the λvir phage on the host of interest relative to a nonrestricting host. The efficiency of plaquing (EOP) of phage was defined as the phage titer on the host of interest divided by the phage titer on a nonrestricting host.
Assay for methylating activity of MmeI.
The DNA modification activities of wt MmeI and its variants were assessed by incubation of the proteins with 1 μg of λ DNA at 37°C for 1 h in a 30-μl reaction volume in the following buffer: 10 mM HEPES, pH 8.0, 2.5 mM potassium acetate, pH 8.0, 0.04 mM AdoMet. Then, the enzyme was heat inactivated (65°C, 20 min). An excess of wt MmeI was then added, along with magnesium acetate to a final concentration of 5 mM, and samples were incubated for another 1 h and resolved on 0.8% agarose gels. One unit of the MmeI modification activity was described as the amount of the enzyme able to modify 1 μg of λ DNA (18 MmeI-specific sites) at 37°C for 1 h so that it became completely resistant to MmeI cleavage.
Electrophoretic mobility shift assay.
A 32P-labeled 146-bp DNA fragment with one centrally located MmeI-specific site was generated as described by us previously (40). An electrophoretic mobility shift assay was performed in 30-μl reaction mixtures containing 64 fmol of a labeled 146-bp DNA fragment and increasing amounts of wt MmeI or its variants in a range from 0 to 5 nM. DNA-MmeI complexes were formed in the following buffer: 10 mM Tris-HCl, pH 7.7, 20% glycerol, 50 mM KCl, 1 mM DTT, and 0.1 mg/ml bovine serum albumin on ice for 10 min. One pmol of unlabeled competitor DNA was used to decrease the nonspecific interaction of MmeI with the 146-bp DNA fragment. The competitor DNA without a MmeI recognition site was a synthetic, high-performance liquid chromatography-purified, 27-bp oligonucleotide (5′-AAAGGATGTGGATGCGTCTCGAGGAAA-3′ and 5′-TCCTCGAGACGCATCCACATCCTT-3′). The obtained complexes were separated under native conditions in 6% standard polyacrylamide gels (acrylamide/bis-acrylamide, 29:1 [wt/wt]) in 0.35× Tris-borate-EDTA buffer (run at 5 V/cm for 2 to 3 h, 8°C). The gels were dried and subjected to autoradiography. Binding of MmeI to DNA was determined densitometrically as the concentration of MmeI required for 50% complex formation (C50).
Chemical modification of MmeI.
Chemical modification of arginine residues was performed in 30 μl of reaction buffer (10 mM HEPES, pH 8.0, 2.5 mM potassium acetate, pH 8.0, 2 mM DTT, 40 μM AdoMet) containing 20 ng of MmeI at room temperature for 5 min. 2,3-Butanedione was used at a concentration of 0.4 mM. In substrate protection experiments, the enzyme was incubated with pUC18 DNA (0 to 60 nM) for 5 min before a modifying agent was added. To initiate DNA cleavage, magnesium acetate (pH 8.0) was added to a concentration of 5 mM. After 30 min of incubation at 37°C, DNA was separated in 1% agarose gels and the decrease of covalently closed circular plasmid DNA was quantified densitometrically.
RESULTS
Sequence analysis of MmeI RM enzyme.
Sequence database searches with PSI-BLAST revealed the expected significant similarity of the central region of the MmeI enzyme (aa residues ∼310 to 610) to the catalytic domains of known DNA N6-adenine MTases. However, no significant similarity of the terminal regions (aa 1 to 310 and 610 to 919) to any protein family was observed. Nonetheless, the multiple sequence alignment of MmeI homologs (all uncharacterized, putative proteins, inferred from genome sequencing projects) revealed the presence of a candidate catalytic Mg2+-binding motif [PD-(D/E)XK] characteristic for many REases, corresponding to D70-X9-EXK82 in the MmeI sequence (Fig. 1A). We carried out a protein FR analysis (see Materials and Methods) to confirm this preliminary prediction for the N-terminal region, as well as to identify potential homologs and predict the structure for the C-terminal region. Since the FR method is designed to identify remote homology and predict structure for domain-size sequence fragments (20 to 500 aa), the MmeI sequence was split into a series of overlapping segments and submitted to the GeneSilico MetaServer (30).
FIG. 1.

(A) Multiple sequence alignment of the MmeI family and structural templates used for modeling. Proteins were named using the REBASE convention. Known crystal structures identified as the best modeling templates were named using their PDB accession codes. Residues conserved in the MmeI family are highlighted. The amino acid residues of MmeI are numbered. Residues studied by mutagenesis in this work are indicated above the alignment by a single-letter code of the amino acid used as a replacement. Structural domains and conserved motifs characteristic of Rossmann fold methyltransferases (in the catalytic domain) are indicated below the alignment. (B) Schematic representation of the structure-function organization of MmeI. Domains responsible for the endonucleolytic activity (PD-D/EXK), DNA methylation (AdoMet-binding/catalytic), and target recognition (TRD) are indicated.
As expected, the FR analysis of the MmeI sequence identified the structures of type I DNA MTase (N6-adenine M.EcoKI HsdM subunit [2ar0], a putative HsdM subunit of an experimentally uncharacterized RM system, BthVORF4518P [2okc]) and type II DNA MTase (N6-adenine M.TaqI [1g38]) as the best structural templates for the region comprising residues 160 to 610 of MmeI (PCONS score, 3.54) (Fig. 1A). Interestingly, the FR alignments for this region revealed not only the AdoMet-binding/catalytic MTase domain containing nine sequence motifs conserved among m6 N-adenine γ-class DNA methyltransferases (residues ∼310 to 610 in MmeI) but also a helical domain characteristic for the N terminus of the HsdM subunit of type I MTases (residues ∼160 to 310 in MmeI). For the N-terminal region of MmeI with the candidate PD-(D/E)XK motif (residues ∼1 to 160), most FR methods failed to report any significant matches to known structures (data not shown). Nonetheless, the HHsearch method (one of the best FR methods according to the recent CASP7 experiment) (57) identified a significant similarity of this region with the experimentally validated PD-(D/E)XK domain within the HsdR subunit of type I REases (pfam04313 family; HHsearch score, 45.5; P = 1.4E-08). Finally, as shown in Fig. 1A, we found that the C-terminal region of MmeI (aa ∼610 to 919) exhibits similarity to the target recognition domains (TRDs) involved in substrate recognition of the type II MTase M.TaqI (1g38; HHsearch P = 1.5E-05; FFAS [21] score, −10.3) and of the HsdS subunit of putative type I enzymes: ORF MJ0130 from Methanocaldococcus jannaschii (1yf2; e.g., mGenTHREADER [36] score, 0.3) and ORF MG438 from Mycoplasma genitalium (1ydx; e.g., FFAS score, −4.96). Importantly, we could find only one copy of the putative TRD in the MmeI amino acid sequence (such as in M.TaqI, which methylates adenosine in the target TCGA), whereas most of the HsdS subunits of type I enzymes comprise two TRDs that effectively recognize two DNA sites in an inverse orientation, separated by a nonspecific sequence of fixed length (24, 42). The putative TRD in the MmeI sequence is followed by a region (aa 820 to 919) comprising three predicted helices, for which we could not detect any obvious relationship to known REases or MTases or to any known protein structures.
Altogether, the results of our FR analysis suggest that type IIS/C/G enzyme MmeI is a fusion protein comprising a tandem arrangement of type I-like domains: a PD-(D/E)XK nuclease domain related to the corresponding domains in HsdR subunits, a HsdM-like module comprising a helical domain and a highly conserved AdoMet-binding/catalytic MTase domain, and a half of the HsdS subunit, presumably responsible for DNA sequence recognition (Fig. 1B). On the other hand, MmeI lacks other domains characteristic of type I RM enzymes, including the ATP-dependent translocase module of the HsdR subunit and C-terminal domains of either HsdM or HsdR that are important for interactions between type I subunits (14).
Construction of MmeI molecular model.
Based on the assumption that the mutual orientation of domains in MmeI is likely to be similar to that in type I RM enzymes, we constructed a preliminary structural model of MmeI using the set of crystal structures identified by the FR analysis. Coordinates are available for download from the FTP server (ftp://genesilico.pl/iamb/models/RM.MmeI/). The following templates from the Protein Data Bank were used: 1gef for the N-terminal PD-(D/E)XK domain, 2ar0 and 2okc for the helical domain and the MTase domain, 1g38 for the MTase domain and the TRD, and 1yf2 and 1ydx for the TRD. Thus, the mutual positions of the helical, MTase, and TRDs in the MmeI model were dictated by the arrangement of these domains in the template structures. The PD-(D/E)XK domain was placed arbitrarily as the N-terminal extension of the helical domain, with the active site facing the potential position of the DNA but without making explicit contacts with the DNA. The preliminary model was optimized by the FRankenstein's Monster method, followed by remodeling of poorly scoring regions and terminal extensions by ROSETTA, exactly as in the protocol used previously (27). The MmeI target DNA molecule was modeled by “mutating” the M.TaqI target from the 1g38 file using HyperChem 7.1 (Hypercube, Inc.) from 5′-CATCGAAC-3′ to 5′-GTCCGACG-3′ (where the target sequence is underlined and the A in boldface indicates a flipped-out methylatable adenine), followed by extension of the cleavable 3′-terminal end by 29 bp in the ideal B-DNA conformation. Finally, the structure of the MmeI-AdoMet-DNA complex was briefly energy minimized using AMBER 8 (11) to remove steric clashes between the protein and DNA.
Figure 2 shows the ribbon (panel A) and surface representations (panel B) of the MmeI molecular model. Mapping of the sequence conservation in the MmeI family onto the protein structure revealed concentrations of conserved residues in the predicted catalytic pockets of the nuclease and MTase domains, as well as in the predicted DNA-binding region of the TRD. In particular, the model illustrates the predicted active sites of the nuclease and MTase domains and the predicted DNA-binding residues of the TRD.
FIG. 2.

Theoretical model of the MmeI structure. Coordinates are available for download from the FTP server (ftp://genesilico.pl/iamb/models/RM.MmeI/). (A) Ribbon representation, colored according to the predicted accuracy (agreement with the native structure estimated for individual residues using MetaMQAP), from blue (highly confident, predicted error of ∼1 Å), to yellow (predicted medium accuracy, expected differences between the model and the native structure of up to ∼5 Å), to red (predicted low accuracy, error difficult to estimate). Residues predicted to be important for DNA binding and/or catalysis are shown in the space-filled representation in red and are labeled. (B) MmeI model in the surface representation, colored according to sequence conservation in the MmeI family, from deep blue (invariant), to light blue (conserved), to yellow/red (highly variable).
According to the PROQ method, evaluation of individual domains revealed the following scores: 4.336 (“extremely good”) for the central MTase domain (residues 158 to 630), 2.254 (“fairly good”) for the PD-(D/E)XK domain (residues 1 to 157), and 2.241 (“fairly good”) for the C-terminal TRD (residues 631 to 919). These results reflect the correlation between the protein model quality and the similarity to the predicted structural templates. PROQ scores suggest that the predicted structure of the MTase catalytic core is of very high accuracy, while the peripheral domains are likely to have a correct overall structure but may exhibit local errors (e.g., misthreading of amino acids along the backbone due to imperfect alignment to the templates) and therefore should be independently evaluated (see below). In any case, from our experience with modeling of REases, the MmeI model presented in this work is most likely sufficiently robust to guide functional predictions.
The surprising prediction that a type IIS/C/G MmeI RM enzyme appears to comprise an incomplete assortment of domains otherwise found in type I enzymes deserves special attention and, above all, experimental confirmation (in particular with respect to the uncertain structural predictions of the N-terminal nuclease and C-terminal TRD). Therefore, we carried out site-directed mutagenesis of regions that were challenging to predict. In our experiments we were focused on the putative MmeI enzyme nuclease active site (Asp70, Glu80, and Lys82) and selected residues in the putative TRD (Ser754, Arg757, Asn773, Ser807, and Arg810) to test their functional importance. All the MmeI variant proteins were produced in E. coli ER2683, and their activities in vitro and in vivo were studied.
Assay for restriction activity of wt MmeI and the MmeI D70A, E80A, and K82A mutated variants.
In order to test the restriction activities of wt and MmeI variant proteins with Ala substitutions in the putative D70-X9-EXK82 catalytic/Mg2+-binding motif, plasmids carrying mutated versions of the mmeIRM gene were introduced into the E. coli ER1992 strain. This strain carries a dinD1::LacZ+ fusion and is deprived of methylation-dependent restriction systems. Induction of the SOS response is a result of DNA damage by REase due to an apparent lack of sufficient protection of genomic DNA by the cognate MTase. The SOS-induced cells form blue colonies on LA plates supplemented with X-Gal (18). This was the case for bacteria carrying pTB (wt MmeI R+ M+). In contrast, bacteria producing MmeI variants with alanine substitutions in the predicted nuclease active site (MmeI D70A, E80A, and K82A) formed only white colonies, indicating a lack of SOS response induction (apparently due to the absence of the MmeI REase activity). Furthermore, in the case of E. coli ER1992(pTB) cells carrying the wt mmeIRM gene we have observed filamentation, a feature characteristic of SOS-induced cells (data not shown). The same was not noticed for E. coli ER1992 cells producing restriction-deficient MmeI variants (MmeI D70A, E80A, and K82A).
In the next step, E. coli ER2683(pTB) (wt MmeI R+ M+) or the same bacteria harboring plasmids with mutated versions of the mmeIRM gene (MmeI D70A, E80A, and K82A) were examined for in vivo restriction of λvir phage. The substantial relative restriction was observed only with bacteria producing wt MmeI (Table 1). In the case of bacteria producing MmeI D70A, E80A, and K82A variants, the obtained EOP values were 1 order of magnitude higher than EOP values obtained for the wt MmeI (Table 1). This indicates that each of the alanine substitutions in the catalytic motif D70-X9-EXK82 affects restriction activity in vivo.
TABLE 1.
Enzymatic activities of wt MmeI and catalytic site mutated variantsa
| E. coli strain | EOP | Activity in cell lysateb | Induction of SOS response in E. coli ER1992c | Sp act
|
|
|---|---|---|---|---|---|
| Cleavaged | Methylationd | ||||
| ER2683 | 1 | ||||
| ER2683 (pMmeIwt) | 0.0342 | + | + | 250,000 | 200,000 |
| ER2683(pD70A) | 0.569 | − | − | − | ND |
| ER2683(pE80A) | 0.621 | − | − | − | 50,000 |
| ER2683(pK82A) | 0.521 | − | − | − | 33,000 |
All determinations were repeated three times. Efficiency of plaquing was determined as the phage titer on the host under investigation divided by the phage titer on a nonrestricting host (only E. coli ER2683).
−, no activity; +, detection of restriction activity.
E. coli strain ER1992 was transformed with plasmids carrying wt MmeI and mutant genes. +, appearance of dark blue colonies and the presence of cell filamentation in the microscopic view (magnification, ×1,000) indicated induction of the SOS response due to the presence of MmeI cleavage activity; −, white colonies on agar plates with X-Gal and no filamentation of tested bacteria indicated lack of SOS response induction.
Specific activities (in U/mg of protein) were determined as the average of three independent measurements. −, no activity; ND, not determined. Units were defined as described in Materials and Methods.
Endonucleolytic activity of wt MmeI and its restriction-deficient variants was also assayed in in vitro experiments. MmeI D70A, E80A, and K82A variants did not show any DNA endonucleolytic activity under standard buffer conditions, at concentrations of 6 μg/ml and 9 μg/ml. On the other hand cleavage of λ DNA by wt MmeI was observed at a concentration of 0.012 μg/ml (data not shown).
Since MmeI restriction activity strongly depends on the presence of AdoMet (40), we wanted to check whether alanine substitutions of Asp70, Glu80, or Lys82 could shift the balance between the REase and MTase activities of MmeI toward DNA methylation. Thus, the REase activity of the MmeI D70A, E80A, and K82A variants was tested in the presence of the AdoMet nonhydrolyzable analog sinefungin (SIN), a potent inhibitor of MTases. We found previously that SIN stimulates the REase activity of wt MmeI (61). However, we did not observe any endonucleolytic activity of MmeI variants when AdoMet was replaced by SIN (data not shown). This provides additional evidence that Asp70, Glu80, and Lys82 residues in the predicted PD-(D/E)XK domain are essential for the endonucleolytic activity of MmeI.
Binding of wt MmeI and the MmeI D70A, E80A, and K82A mutated variants to DNA.
An electrophoretic mobility shift assay was used to assess binding of wt MmeI and its variants to DNA fragments containing a single recognition site. We wanted to exclude the possibility that the observed restriction deficiency of the MmeI D70A, E80A, and K82A variants is due to compromised DNA binding rather than a direct defect in the MmeI endonuclease active site. In this experiment two DNA fragments were used: a 32P-labeled 146-bp NheI-BamHI DNA fragment from pBR322 containing a single MmeI site (5′-TCCGAC-3′) and a 27-bp synthetic oligonucleotide without the MmeI recognition site, used as competitor. Binding of wt MmeI or the D70A, E80A, and K82A MmeI variants to the DNA was determined (Table 2) as the concentration of the enzyme required to obtain the C50, determined from densitometric measurement of autoradiograms (Fig. 3A to E). The presumed catalytic mutants of MmeI showed no significant difference in DNA binding when compared to the wt enzyme (Table 2). These results showed that the analyzed restriction-deficient variants of MmeI can bind DNA in a specific manner.
TABLE 2.
Concentrations of wt MmeI and MmeI mutated variants required for 50% DNA binding
| Mutant type | MmeI variant | C50 (nM) |
|---|---|---|
| Wild type | wt | 1.1 |
| Catalytic site | D70A | 1.1 |
| Catalytic site | E80A | 0.7 |
| Catalytic site | K82A | 0.5 |
| TRD | S754A | 1.4 |
| TRD | N773A | 0.2 |
| TRD | R757A | DNA binding impaired |
| TRD | S807A | 0.02 |
| TRD | R810A | Lack of DNA binding |
FIG. 3.

Binding of wt MmeI and its mutated variants to specific DNA sequence. The 146-bp DNA fragment from pBR322 containing a single MmeI recognition site was used as a specific DNA. The nonspecific DNA was a synthetic double-stranded 27-bp oligonucleotide (1 pmol). The binding mixtures contained 10 mM Tris-HCl, pH 7.7, 20% glycerol, 50 mM KCl, 1 mM DTT, 0.1 mg/ml bovine serum albumin. A constant amount of 64 fmol of [32P]DNA fragment carrying the MmeI site (a 146-bp DNA fragment derived from pBR322 [32]) and an increasing amount of wt MmeI (0 to 3 nM) or its mutated variants were added as indicated on the x axis. (A) DNA shifts obtained in the presence of increasing concentrations of wt MmeI (from left to right, indicated by a triangle above the picture). I, free DNA; II, MmeI-DNA complexes; III, gel wells. The obtained bands were visualized by autoradiography, and intensities of the bands were processed digitally. (B to H) Graphical representations of binding of wt MmeI and MmeI mutated variants to a 146-bp DNA fragment carrying the MmeI site. The binding of MmeI to the DNA fragment carrying the MmeI site was determined as the concentration of MmeI required for recruitment of 50% of the DNA into a DNA-protein complex(C50). Curve fitting was done with the use of Sigma Plot 2000 v. 6.0.
Methylation activity of wt MmeI and MmeI restriction-deficient variants.
To study the influence of the MmeI endonucleolytic moiety on the MTase activity, we tested the methylation activity of the MmeI restriction-deficient variants (D70A, E80A, and K82A) in vitro and in vivo. Methylation activity of aforementioned enzymes was measured by using λ DNA protection assays (see Materials and Methods). We found that the specific activities of two MmeI variants, E80A and K82A, were four- and sevenfold lower, respectively, than the wt MmeI enzyme activity (Table 1).
Probing the function of selected residues in the MmeI target recognition domain.
Based on the preliminary model of the C-terminal domain of MmeI, we identified a set of amino acids suspected to play a role in DNA recognition. The importance of five residues (Ser754, Arg757, Asn773, Ser807, and Arg810) was evaluated with the use of MmeI variants with alanine substitutions. In the first experiment we tested the induction of the SOS response upon introduction of plasmids carrying mutated versions of the mmeIRM gene into E. coli ER1992. As a result we found that the MmeI variants R757A and R810A did not induce the SOS response in E. coli cells (white colonies on LA plates supplemented with X-Gal). This suggested a restriction-deficient phenotype. On the other hand, variants S754A and S807A induced the SOS response (dark blue colonies), proving high restriction activity with these enzymes. In the case of the MmeI N773A variant, induction of the SOS response was only partial (pale blue colonies), indicating a weak endonucleolytic activity.
In vitro endonuclease activities of MmeI variants S754A, N773A, S807A, R757A, and R810A were tested by digesting λ DNA with an increasing amount of enzyme. We found that 40 ng of wt MmeI was sufficient for complete digestion of 1 μg of λ DNA. The MmeI N773A, R757A, and R810A variants showed a lack of endonucleolytic activity upon λ DNA with any amount of enzyme added (4 to 40 ng) (Fig. 4, lanes 6 to 14). The result was surprising in the case of MmeI N773A, which partially induced the SOS response in E. coli ER1992, suggesting restriction activity, but at a low level. It seems that the endonucleolytic activity of this variant is extremely low. Unlike the Arg and Asn alanine substitutions, both Ser substitutions (MmeI S754A and S807A) showed restriction activity in vitro (Fig. 4, lanes 15 to 20). The MmeI S754A variant (40 ng) was unable to completely digest the same amount of DNA (1.0 μg) as the wt MmeI (40 ng), indicating low endonucleolytic activity. Surprisingly, the MmeI S807A variant cleaved the λ DNA even more effectively than the wt MmeI. Only 0.12 ng of the MmeI S807A variant was needed to digest the DNA to the same extent as 40 ng of the wt MmeI under the same reaction conditions (Fig. 4, lane 5 versus 20). The MmeI S807A variant also showed high affinity for DNA containing the MmeI site.
FIG. 4.

Restriction activity of MmeI TRD variants in vitro. Restriction activity of the enzymes was measured by digesting 1 μg of λ DNA in a 30-μl volume of standard reaction buffer supplemented with 0.04 mM AdoMet. The reaction mixtures were incubated at 37°C for 1 h and resolved on a 1% agarose gel. The amount of the enzyme used in a particular reaction mixture is indicated above each lane. M, molecular weight marker (Gene Ruler, 1-kb DNA ladder; Fermentas); λ, undigested λ DNA.
Binding of MmeI mutated variants in the target recognition domain to DNA.
An electrophoretic mobility shift assay was carried out to see whether the MmeI TRD variants were able to bind specific DNA sequence. The same procedure was used as for MmeI restriction-deficient variants. Among five TRD variants tested, MmeI S754A, N773A, and S807A formed complexes with the 146-bp DNA fragment (Fig. 3F to H). However, the highest affinity was shown by the highly active MmeI S807A (Fig. 3H; Table 2). Under the same conditions, two MmeI variants with substituted Arg residues did not exhibit specific DNA-binding activity (R810A), or binding to DNA was seriously impaired (R757A) (data not shown).
Arginine residues play an important role in the MmeI-DNA interaction.
The arginine residues can be engaged in hydrogen bonding with nucleotides within the recognition sites (54). There are 43 codons for arginine in the mmeIRM gene. Two Arg MmeI variants, namely R757A and R810A, did not show any endonucleolytic activity either in vitro (lack of λ DNA cleavage under optimal reaction conditions) (Fig. 4, lanes 6 to 11) or in vivo (lack of SOS response induction in E. coli ER1992). In order to examine the effect of Arg residues in DNA recognition we chemically modified these residues with 2,3-butanedione. This modifying agent reacts with arginine to yield a dihydroxyimidazoline derivative (13). We have found that 2,3-butanedione at a concentration of 0.4 mM completely abolishes MmeI endonucleolytic activity (Fig. 5, curve c). However, preincubation of the MmeI protein with DNA containing MmeI specific sequence (pUC18 was used as a substrate) preserves 60% of the enzyme activity, confirming the importance of Arg residues in the process of target recognition (Fig. 5, curve b). The activity of MmeI measured without the chemical modification constituted 100% activity (Fig. 5, curve a).
FIG. 5.
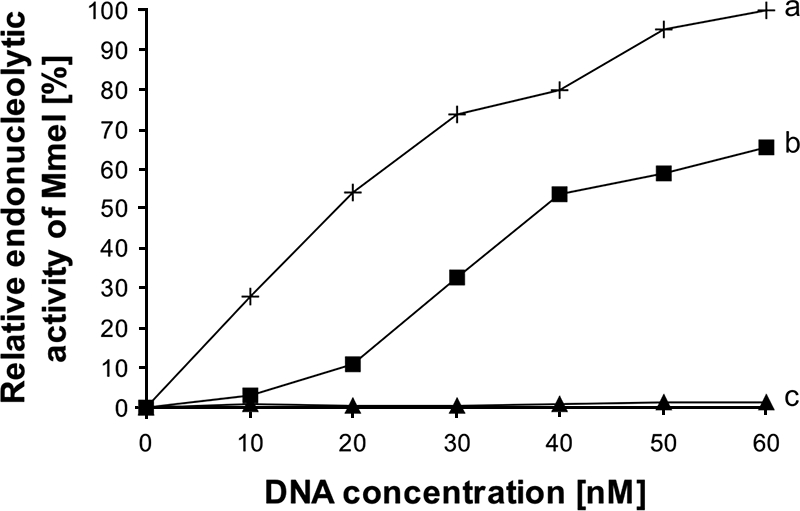
Protection of MmeI enzyme by substrate (pUC18) from chemical modification by 2,3-butanedione. Endonucleolytic activity of MmeI was measured using pUC18 DNA as a substrate. Three experiments were performed. Plot a (crosses) shows the activity of MmeI measured without modification with 2,3-butanedione (positive control); plot b (squares) shows the results of the substrate protection experiment, in which MmeI was preincubated with the substrate DNA (pUC18) before it was modified with 2,3-butanedione. Plot c (triangles) shows the activity of the enzyme in the presence of the 2,3-butanedione (without prior substrate protection).
DISCUSSION
Structure of a bifunctional RM protein.
Numerous crystal structures have been solved for type II REases composed only of the nuclease domain (type IIP and type IIF enzymes) or of the nuclease domain fused to a single DNA-binding domain (type IIE and type IIS enzymes), and various comparative analyses have been made to elucidate their evolutionary and functional relationships (7, 8, 41, 47). On the other hand, so far no structural or evolutionary studies have been reported for bifunctional restriction-modification enzymes that possess nuclease and MTase domains fused together in a single polypeptide; for example, type IIC REBASE analysis groups together as many as 538 sequences of these proteins, which together represent 38% of all type II REase sequences (50). To our knowledge, the only structural analysis concerns crystallization of the Eco57I enzyme (59), but so far its structure has not been solved. Here, we carried out a comprehensive sequence analysis of MmeI, a representative of type IIC (and also a member of types IIS and IIG), to identify its structural domains. Interestingly, in the process of the structure prediction, we found that the type II enzyme MmeI comprises domains characteristic of type I enzymes, including the N-terminal domain related to a variant of the PD-(D/E)XK domain previously identified in the type I HsdR subunit, the central region related to the HsdM subunit (comprising two structural domains), and the C-terminal region related to a TRD from the HsdS subunit (Fig. 1B). However, whereas the similarity of the central domain to the HsdM domain is evident and detectable even with PSI-BLAST searches (1), the sequence relationships of N and C termini to HsdR and HsdS are very remote and not detectable using standard tools for sequence analysis. Thus, we tested our structural predictions for the N-terminal and C-terminal domains by site-directed mutagenesis of amino acids implicated in catalysis of DNA cleavage or DNA recognition. Our computational model is of too low accuracy to provide precise estimation of contacts and distances between the individual functional groups of amino acid residues and the DNA; however, the predicted “residue-level” accuracy is sufficient to guide site-directed mutagenesis. We found that residues Asp70, Glu80, and Lys82 in the predicted PD-(D/E)XK domain are indeed indispensable for the nuclease activity of MmeI, while residues Arg757 and Arg810 in the predicted TRD are important for DNA binding. The residue Asn773 may be also somehow involved in interactions with DNA, as its substitution leads to increased DNA binding.
DNA binding and sequence recognition.
As far as protein-DNA interactions are concerned Arg residues are known to show some preference in guanine recognition (33). In R.EcoRI, Arg145 and Arg200 are engaged in direct sequence-specific interactions with EcoRI recognition sequence (23). Conservative substitutions of these arginines with lysine resulted in decreased affinity for DNA binding, whereas specificity of DNA recognition was saved (19). Similar observations were made for Asn141, which forms three hydrogen bonds with two adenines of R.EcoRI sequence (51). In extensive work presented by the Pingoud group, two Arg-rich regions present in several restrictions enzymes recognizing CC-GG sequences are responsible for backbone contacts and base recognition (46). On the other hand R.MboI, which recognizes the GATC sequence, lacks the region most likely to be involved in base recognition of CC-GG sequences (46, 59). Genetic experiments concerning MmeI variants Arg810 and Arg757 confirmed the structure prediction presented with the MmeI molecular model, although it is not possible at this moment to precisely determine the character of both Arg residues in the MmeI-DNA interaction. We can only claim that the Arg810 mutation, which resulted in complete loss of restriction activity as well as DNA binding, is crucial for DNA binding/cleavage by MmeI. Results obtained for the Arg757 mutant (DNA binding impaired but not totally abolished) indicate less significant participation in the MmeI-DNA interaction (probably protein-DNA complex stabilization but not specific binding). To probe the roles of both Arg residues in sequence discrimination, a more conservative substitution should be performed (for example, Arg to Lys). As the results concerning Arg mutants of MmeI quite clearly show their significance in DNA binding, the interpretation of the N773A substitution seems to be more difficult. Asparagine is the amino acid found to interact most readily with adenine and also with the phosphate backbone via hydrogen bond formation. Substitution of this amino acid with alanine reduces the hydrogen bonding potential about fourfold (33). Anyway, such a reduction still resulted in efficient DNA binding by the N773A MmeI variant, but without the ability to cleave the DNA in vitro. However, cleavage of the DNA in vivo as assayed by induction of the SOS response in E. coli ER1992 proved that this MmeI variant retains trace amounts of restriction activity, indicating that the nuclease domain of MmeI N773A remains unaffected. Such results do not exclude participation of Asn773 in DNA binding by MmeI, although its role seems to be different from that predicted. We hypothesize that Asn773 may participate in intra-/intersubunit communication. Experiments with R.EcoRV heterodimers carrying a single amino acid substitution (N188Q) in the DNA-binding site in one subunit proved that signals originating from specific DNA contact with one subunit are transferred to the catalytic centers of both subunits. Such a result demonstrates that Asn188 in R.EcoRV takes part in transition-state stabilization that does not affect DNA binding in the ground state (58). In R.EcoRI there was a structural element identified called the “cross-talk ring” which is responsible for communication between enzyme subunits. Glu144 plays the key role in the cross-talk ring by interacting with a similar element of the second subunit. The cross-talk ring couples not only recognition and catalysis in each monomer of EcoRI but also base recognition elements in each of the two half-sites to each other (31). There are no data on how recognition is coupled to catalysis in the case of enzymes with a domain architecture similar to MmeI. In the R.FokI crystal structure without DNA, the cleavage domain is sequestered in a piggyback fashion to the recognition domain. The interactions involve Gln420 and Arg422 of the cleavage domain, which form solvent-mediated hydrogen bonds with the side chain of Lys225 and the main chain carbonyl of Glu220 of the recognition domain. The latter two are responsible for base-specific contact in the R.FokI-DNA crystal structure (64).
Relation to type I RM systems.
Based on our predictions and supporting experimental data, we hypothesize that MmeI and other type IIC REases with similar domain architectures are more closely related to type I RM systems than to other type II REases. The molecular model of MmeI presented in this work corresponds to just one-half of the type I enzyme structure (42), suggesting that head-to-head dimerization may be required for its function. This inference agrees with the strong preference of MmeI for using oppositely oriented sites as substrates (e.g., the ability to damage the fully unmodified E. coli genome containing 1,201 MmeI sites, 111 in the forward and 1,090 in the reverse orientation), compared to single or tandemly repeated sites (an inability to cleave a newly replicated genome with one unmethylated strand, in which all unmodified MmeI sites are in the same orientation).
Thus, we hypothesize that MmeI and most likely many other type IIC REases may dimerize (at least temporarily) upon DNA binding to cleave a pair of their target sites. DNA recognition by two molecules of a type IIC enzyme, each comprising one TRD, may involve a similar mechanism, such as the recognition of a bipartite site by type I enzymes and type IIB enzymes (e.g., BcgI) that possess a separate subunit with two TRDs. It is intriguing that type I and type IIC enzymes share a helical domain that in type I enzymes is found in the N terminus of the HsdM subunit. In EcoKI this domain has been implicated in communication of the status of the methylatable base (methylated or not) between both HsdM subunits in the multisubunit restriction enzyme (HsdS1HsdM2HsdR2) (22), which is essential for the “decision” of this complicated molecular machine whether to methylate one base (if the other is already methylated) or to initiate cleavage (if both bases are unmethylated). Therefore, it is tempting to speculate that the helical “type I-like” domain mediates interactions between molecules of type IIC enzymes in a similar manner to interactions between HsdM subunits in type I enzymes.
Conclusions.
Based on structural predictions and detection of homology between MmeI and type I enzymes, we propose a low-resolution structural and functional model of MmeI (Fig. 2) that can be generalized to other type IIC enzymes. In this model, type IIC enzymes can be regarded as simpler variants of type I enzymes that do not require the DNA translocase module to trigger DNA cleavage. However, at this point it is not possible to decide whether type I RM systems evolved from type II systems by recruitment of a translocase domain and partition of nuclease, MTase, and DNA-binding modules into distinct subunits or if type IIC enzymes are a streamlined variant of type I enzymes that underwent fusion of subunits and lost the unnecessary domains. We hope our model will prompt further experiments for type IIC enzymes that will help to refine it and increase its resolution. If our hypothesis of close homology and mechanistic similarity between type IIC and type I enzymes holds true, it may provide a starting point for explaining the origin and evolution of complex, multidomain restriction enzymes.
Acknowledgments
This paper is dedicated to the memory of Anna J. Podhajska.
We thank Richard Morgan (New England Biolabs) for useful discussions and for sharing materials (plasmid pTB) and unpublished data on MmeI. We are grateful to Iwona Mruk, Monika Radlinska, and Krzysztof Skowronek for critical reading of the manuscript. Thanks are addressed also to Ania Gwizdek-Wisniewska for the help with preparation of figures.
This work was supported by grants 2 P04B 007 28 and 2 P04B 013 30 from the Ministry of Science and Higher Education (Warsaw, Poland). A.O.-K. and J.M.B. were supported by a 6FP grant from the European Union (MRTN-CT-2005-019566).
Footnotes
Published ahead of print on 7 November 2008.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aravind, L., K. S. Makarova, and E. V. Koonin. 2000. Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 28:3417-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Backman, K., M. Ptashne, and W. Gilbert. 1976. Construction of plasmids carrying the cI gene of bacteriophage lambda. Proc. Natl. Acad. Sci. USA 73:4174-4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourniquel, A. A., and T. A. Bickle. 2002. Complex restriction enzymes: NTP-driven molecular motors. Biochimie 84:1047-1059. [DOI] [PubMed] [Google Scholar]
- 5.Boyd, A. C., I. G. Charles, J. W. Keyte, and W. J. Brammar. 1986. Isolation and computer-aided characterization of MmeI, a type II restriction endonuclease from Methylophilus methylotrophus. Nucleic Acids Res. 14:5255-5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bujnicki, J. M. 1999. Comparison of protein structures reveals monophyletic origin of the AdoMet-dependent methyltransferase family and mechanistic convergence rather than recent differentiation of N4-cytosine and N6-adenine DNA methylation. In Silico Biol. 1:175-182. [PubMed] [Google Scholar]
- 7.Bujnicki, J. M. 2000. Phylogeny of the restriction endonuclease-like superfamily inferred from comparison of protein structures. J. Mol. Evol. 50:39-44. [DOI] [PubMed] [Google Scholar]
- 8.Bujnicki, J. M. 2003. Crystallographic and bioinformatic studies on restriction endonucleases: inference of evolutionary relationships in the “midnight zone” of homology. Curr. Protein Pept. Sci. 4:327-337. [DOI] [PubMed] [Google Scholar]
- 9.Bujnicki, J. M., and M. Radlinska. 1999. Molecular evolution of DNA-(cytosine-N4) methyltransferases: evidence for their polyphyletic origin. Nucleic Acids Res. 27:4501-4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bujnicki, J. M., and L. Rychlewski. 2001. Grouping together highly diverged PD-(D/E)XK nucleases and identification of novel superfamily members using structure-guided alignment of sequence profiles. J. Mol. Microbiol. Biotechnol. 3:69-72. [PubMed] [Google Scholar]
- 11.Case, D. A., T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, K. M. Merz, B. Wang, D. A. Pearlman, M. Crowley, S. Brozell, V. Tsui, H. Gohlke, J. Mongan, V. Hornak, G. Cui, P. Beroza, C. Schafmeister, J. W. Caldwell, W. S. Ross, and P. A. Kollman. 2004. AMBER 8. University of California, San Francisco.
- 12.Chmiel, A. A., J. M. Bujnicki, and K. J. Skowronek. 2005. A homology model of restriction endonuclease SfiI in complex with DNA. BMC Struct. Biol. 5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daemen, F. J., and J. F. Riordan. 1974. Essential arginyl residues in Escherichia coli alkaline phosphatase. Biochemistry 13:2865-2871. [DOI] [PubMed] [Google Scholar]
- 14.Davies, G. P., I. Martin, S. S. Sturrock, A. Cronshaw, N. E. Murray, and D. T. Dryden. 1999. On the structure and operation of type I DNA restriction enzymes. J. Mol. Biol. 290:565-579. [DOI] [PubMed] [Google Scholar]
- 15.Dunn, J. J., S. R. McCorkle, L. A. Praissman, G. Hind, D. Van Der Lelie, W. F. Bahou, D. V. Gnatenko, and M. K. Krause. 2002. Genomic signature tags (GSTs): a system for profiling genomic DNA. Genome Res. 12:1756-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emmersen, J., A. M. Heidenblut, A. L. Høgh, S. A. Hahn, K. G. Welinder, and K. L. Nielsen. 2007. Discarding duplicate ditags in long SAGE analysis may introduce significant error. BMC Bioinformatics 8:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fomenkov, A., J. P. Xiao, D. Dila, E. Raleigh, and S. Y. Xu. 1994. The ‘endo-blue method’ for direct cloning of restriction endonuclease genes in E. coli. Nucleic Acids Res. 22:2399-2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fritz, A., W. Kuster, and J. Alves. 1998. Asn141 is essential for DNA recognition by EcoRI restriction endonuclease. FEBS Lett. 438:66-70. [DOI] [PubMed] [Google Scholar]
- 20.Grazulis, S., E. Manakova, M. Roessle, M. Bochtler, G. Tamulaitiene, R. Huber, and V. Siksnys. 2005. Structure of the metal-independent restriction enzyme BfiI reveals fusion of a specific DNA-binding domain with a nonspecific nuclease. Proc. Natl. Acad. Sci. USA 102:15797-15802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaroszewski, L., L. Rychlewski, Z. Li, W. Li, and A. Godzik. 2005. FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res. 33:W284-W288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelleher, J. E., A. S. Daniel, and N. E. Murray. 1991. Mutations that confer de novo activity upon a maintenance methyltransferase. J. Mol. Biol. 221:431-440. [DOI] [PubMed] [Google Scholar]
- 23.Kim, Y. C., J. C. Grable, R. Love, P. J. Greene, and J. M. Rosenberg. 1990. Refinement of EcoRI endonuclease crystal structure: a revised protein chain tracing. Science 249:1307-1309. [DOI] [PubMed] [Google Scholar]
- 24.Kneale, G. G. 1994. A symmetrical model for the domain structure of type I DNA methyltransferases. J. Mol. Biol. 243:1-5. [DOI] [PubMed] [Google Scholar]
- 25.Kosinski, J., M. Feder, and J. M. Bujnicki. 2005. The PD-(D/E)XK superfamily revisited: identification of new members among proteins involved in DNA metabolism and functional predictions for domains of (hitherto) unknown function. BMC Bioinformatics 6:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosinski, J., I. A. Cymerman, M. Feder, M. A. Kurowski, J. M. Sasin, and J. M. Bujnicki. 2003. A “FRankenstein's monster” approach to comparative modeling: merging the finest fragments of Fold-Recognition models and iterative model refinement aided by 3D structure evaluation. Proteins 53(Suppl. 6):369-379. [DOI] [PubMed] [Google Scholar]
- 27.Kosinski, J., M. J. Gajda, I. A. Cymerman, M. A. Kurowski, M. Pawlowski, M. Boniecki, A. Obarska, G. Papaj, P. Sroczynska-Obuchowicz, K. L. Tkaczuk, P. Sniezynska, J. M. Sasin, A. Augustyn, J. M. Bujnicki, and M. Feder. 2005. FRankenstein becomes a cyborg: the automatic recombination and realignment of fold recognition models in CASP6. Proteins 61(Suppl. 7):106-113. [DOI] [PubMed] [Google Scholar]
- 28.Kosinski, J., E. Kubareva, and J. M. Bujnicki. 2007. A model of restriction endonuclease MvaI in complex with DNA: a template for interpretation of experimental data and a guide for specificity engineering. Proteins 68:324-336. [DOI] [PubMed] [Google Scholar]
- 29.Kozbial, P. Z., and A. R. Mushegian. 2005. Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurowski, M. A., and J. M. Bujnicki. 2003. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 31:3305-3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurpiewski, M. R., L. E. Engler, L. A. Wozniak, A. Kobylanska, M. Koziolkiewicz, W. J. Stec, and L. Jen-Jacobson. 2004. Mechanisms of coupling between DNA recognition specificity and catalysis in EcoRI endonuclease. Structure 12:1775-1788. [DOI] [PubMed] [Google Scholar]
- 32.Lundstrom, J., L. Rychlewski, J. M. Bujnicki, and A. Elofsson. 2001. Pcons: a neural-network-based consensus predictor that improves fold recognition. Protein Sci. 10:2354-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luscombe, N. M., R. A. Laskowski, and J. M. Thornton. 2001. Amino acid-base interactions: a three dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 29:2860-2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malone, T., R. M. Blumenthal, and X. Cheng. 1995. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 253:618-632. [DOI] [PubMed] [Google Scholar]
- 35.Mantovani, J., N. Holic, K. Martinez, O. Danos, and J. Perea. 2006. A high throughput method for genome-wide analysis of retroviral integration. Nucleic Acids Res. 34:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGuffin, L. J., and D. T. Jones. 2003. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics 19:874-881. [DOI] [PubMed] [Google Scholar]
- 37.Meisel, A., T. A. Bickle, D. H. Kruger, and C. Schroeder. 1992. Type III restriction enzymes need two inversely oriented recognition sites for DNA cleavage. Nature 355:467-469. [DOI] [PubMed] [Google Scholar]
- 38.Miyazono, K., M. Watanabe, J. Kosinski, K. Ishikawa, M. Kamo, T. Sawasaki, K. Nagata, J. M. Bujnicki, Y. Endo, M. Tanokura, and I. Kobayashi. 2007. Novel protein fold discovered in the PabI family of restriction enzymes. Nucleic Acids Res. 35:1908-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan, R. D., T. Bhatia, T. Davis, and L. Lovasco. 2004. Recombinant type II restriction endonucleases, MmeI and related endonucleases and methods for producing the same. Patent WO 2004007670.
- 40.Nakonieczna, J., J. W. Zmijewski, B. Banecki, and A. J. Podhajska. 2007. Binding of MmeI restriction-modification enzyme to its specific recognition sequence is stimulated by S-adenosyl-L-methionine. Mol. Biotechnol. 37:127-135. [DOI] [PubMed] [Google Scholar]
- 41.Niv, M. Y., D. R. Ripoll, J. A. Vila, A. Liwo, E. S. Vanamee, A. K. Aggarwal, H. Weinstein, and H. A. Scheraga. 2007. Topology of type II REases revisited; structural classes and the common conserved core. Nucleic Acids Res. 35:2227-2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obarska, A., A. Blundell, M. Feder, S. Vejsadova, E. Sisakova, M. Weiserova, J. M. Bujnicki, and K. Firman. 2006. Structural model for the multisubunit type IC restriction-modification DNA methyltransferase M. EcoR124I in complex with DNA. Nucleic Acids Res. 34:1992-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orlowski, J., and J. M. Bujnicki. 2008. Structural and evolutionary classification of type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res. 36:3552-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pawlowski, M., M. J. Gajda, R. Matlak, and J. M. Bujnicki. 2008. MetaMQAP: a meta-server for the quality assessment of protein models BMC Bioinformatics 9:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pingoud, A., M. Fuxreiter, V. Pingoud, and W. Wende. 2005. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 62:685-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pingoud, V., C. Conzelmann, S. Kinzebach, A. Sudina, V. Metelev, E. Kubareva, J. M. Bujnicki, R. Lurz, G. Luder, S. Y. Xu, and A. Pingoud. 2003. PspGI, a type II restriction endonuclease from the extreme thermophile Pyrococcus sp.: structural and functional studies to investigate an evolutionary relationship with several mesophilic restriction enzymes. J. Mol. Biol. 329:913-929. [DOI] [PubMed] [Google Scholar]
- 47.Pingoud, V., A. Sudina, H. Geyer, J. M. Bujnicki, R. Lurz, G. Luder, R. Morgan, E. Kubareva, and A. Pingoud. 2005. Specificity changes in the evolution of type II restriction endonucleases: a biochemical and bioinformatic analysis of restriction enzymes that recognize unrelated sequences. J. Biol. Chem. 280:4289-4298. [DOI] [PubMed] [Google Scholar]
- 48.Pósfai, J., A. S. Bhagwat, and R. J. Roberts. 1988. Sequence motifs specific for cytosine methyltransferases. Gene 74:261-265. [DOI] [PubMed] [Google Scholar]
- 49.Roberts, R. J., M. Belfort, T. Bestor, A. S. Bhagwat, T. A. Bickle, J. Bitinaite, R. M. Blumenthal, S. Degtyarev, D. T. Dryden, K. Dybvig, K. Firman, E. S. Gromova, R. I. Gumport, S. E. Halford, S. Hattman, J. Heitman, D. P. Hornby, A. Janulaitis, A. Jeltsch, J. Josephsen, A. Kiss, T. R. Klaenhammer, I. Kobayashi, H. Kong, D. H. Kruger, S. Lacks, M. G. Marinus, M. Miyahara, R. D. Morgan, N. E. Murray, V. Nagaraja, A. Piekarowicz, A. Pingoud, E. Raleigh, D. N. Rao, N. Reich, V. E. Repin, E. U. Selker, P. C. Shaw, D. C. Stein, B. L. Stoddard, W. Szybalski, T. A. Trautner, J. L. Van Etten, J. M. Vitor, G. G. Wilson, and S. Y. Xu. 2003. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 31:1805-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roberts, R. J., T. Vincze, J. Posfai, and D. Macelis. 2007. REBASE—enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 35:D269-D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenberg, J. M. 1991. Structure and function of restriction endonucleases. Curr. Opin. Struct. Biol. 1:104-113. [DOI] [PubMed] [Google Scholar]
- 52.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 53.Sasin, J. M., and J. M. Bujnicki. 2004. COLORADO3D, a web server for the visual analysis of protein structures. Nucleic Acids Res. 32:W586-W589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seeman, N. C., J. M. Rosenberg, and A. Rich. 1976. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl. Acad. Sci. USA 73:804-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiraki, T., S. Kondo, S. Katayama, K. Waki, T. Kasukawa, H. Kawaji, R. Kodzius, A. Watahiki, M. Nakamura, T. Arakawa, S. Fukuda, D. Sasaki, A. J. Podhajska, M. Harbers, J. Kawai, P. Carninci, and Y. Hayashizaki. 2003. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 100:15776-15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simons, K. T., C. Kooperberg, E. Huang, and D. Baker. 1997. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268:209-225. [DOI] [PubMed] [Google Scholar]
- 57.Soding, J. 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951-960. [DOI] [PubMed] [Google Scholar]
- 58.Stahl, F., W. Wende, A. Jeltsch, and A. Pingoud. 1996. Introduction of asymmetry in the naturally symetric restriction endonuclease EcoRV to investigate intersubunit communication in the homodimeric protein. Proc. Natl. Acad. Sci. USA 93:6175-6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamulaitiene, G., S. Grazulis, A. Janulaitis, R. Janowski, G. Bujacz, and M. Jaskolski. 2004. Crystallization and preliminary crystallographic studies of a bifunctional restriction endonuclease Eco57I. Biochim. Biophys. Acta 1698:251-254. [DOI] [PubMed] [Google Scholar]
- 60.Tucholski, J., P. M. Skowron, and A. J. Podhajska. 1995. MmeI, a class-IIS restriction endonuclease: purification and characterization. Gene 157:87-92. [DOI] [PubMed] [Google Scholar]
- 61.Tucholski, J., J. W. Zmijewski, and A. J. Podhajska. 1998. Two intertwined methylation activities of the MmeI restriction-modification class-IIS system from Methylophilus methylotrophus. Gene 223:293-302. [DOI] [PubMed] [Google Scholar]
- 62.Velculescu, V. E., L. Zhang, B. Vogelstein, and K. W. Kinzler. 1995. Serial analysis of gene expression. Science 270:484-487. [DOI] [PubMed] [Google Scholar]
- 63.Venclovas, C., A. Timinskas, and V. Siksnys. 1994. Five-stranded beta-sheet sandwiched with two alpha-helices: a structural link between restriction endonucleases EcoRI and EcoRV. Proteins 20:279-282. [DOI] [PubMed] [Google Scholar]
- 64.Wah, D. A., J. Bitinaite, I. Schildkraut, and A. K. Aggarwal. 1998. Structure of FokI has implications for DNA cleavage. Proc. Natl. Acad. Sci. USA 95:10564-10569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wallner, B., and A. Elofsson. 2003. Can correct protein models be identified? Protein Sci. 12:1073-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wallner, B., and A. Elofsson. 2006. Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Sci. 15:900-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Winkler, F. K., D. W. Banner, C. Oefner, D. Tsernoglou, R. S. Brown, S. P. Heathman, R. K. Bryan, P. D. Martin, K. Petratos, and K. S. Wilson. 1993. The crystal structure of EcoRV endonuclease and of its complexes with cognate and noncognate DNA fragments. EMBO J. 12:1781-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]