

Abstract
In this study, the binding of F components of the staphylococcal bicomponent leukotoxins Panton-Valentine leucocidin (LukF-PV) and gamma-hemolysin (HlgB) on polymorphonuclear neutrophils (PMNs), monocytes, and lymphocytes was determined using labeled mutants and flow cytometry. Leukotoxin activity was evaluated by measuring Ca2+ entry or pore formation using spectrofluorometry or flow cytometry. Although HlgB had no affinity for cells in the absence of an S component, LukF-PV had high affinity for PMNs (dissociation constant [Kd], 6.2 ± 1.9 nM; n = 8), monocytes (Kd, 2.8 ± 0.8 nM; n = 7), and lymphocytes (Kd, 1.2 ± 0.2 nM; n = 7). Specific binding of HlgB was observed only after addition of LukS-PV on PMNs (Kd, 1.1 ± 0.2 nM; n = 4) and monocytes (Kd, 0.84 ± 0.31 nM; n = 4) or after addition of HlgC on PMNs, monocytes, and lymphocytes. Addition of LukS-PV or HlgC induced a second specific binding of LukF-PV on PMNs. HlgB and LukD competed only with LukF-PV molecules bound after addition of LukS-PV. LukF-PV and LukD competed with HlgB in the presence of LukS-PV on PMNs and monocytes. Use of antibodies and comparisons between binding and activity time courses showed that the LukF-PV molecules that bound to target cells before addition of LukS-PV were the only LukF-PV molecules responsible for Ca2+ entry and pore formation. In contrast, the active HlgB molecules were the HlgB molecules bound after addition of LukS-PV. In conclusion, LukF-PV must be linked to LukS-PV and to a binding site of the membrane to have toxin activity.
Staphylococcus aureus secretes water-soluble protein monomers which assemble to form well-characterized two-component pore-forming leukotoxins that define a toxin subfamily (17). Each leukotoxin is formed by a class S protein (LukS-PV, HlgA, HlgC, or LukE) that is associated with a class F protein (LukF-PV, HlgB, or LukD). The Panton-Valentine leucocidin (PVL) (16), composed of LukS-PV and LukF-PV, is secreted by strains isolated from humans suffering from abscesses, furuncles (6, 8), and necrotizing pneumonia (11, 12). PVL has been shown to contribute to the severity of acute hematogenous osteomyelitis caused by community-acquired methicillin-resistant S. aureus in children (2). Moreover, reports of hospital-acquired and community-acquired methicillin-resistant S. aureus possessing PVL genes are increasing (3). The LukE-LukD-secreting S. aureus strains have been shown to be associated with postantibiotic diarrhea (10). However, gamma-hemolysin (HlgA-HlgB and HlgC-HlgB) is produced by all human clinical S. aureus strains.
The main target cells of staphylococcal leukotoxins are human polymorphonuclear cells (PMNs), monocytes, and lymphocytes (13). However, PVL is not toxic for lymphocytes, and gamma-hemolysin is hemolytic. After binding to the membrane, leukotoxins assemble as a ring-shaped prepore (15) consisting of heterologous octamers with a molar ratio of 1:1 (14). They induce an increase in the intracellular Ca2+ level by opening Ca2+ channels (19, 1), and the deployment of stems forms beta-barrels that create transmembrane pores in target cells (7, 1). Previous perfusion studies of PMNs showed that the initial binding of S components was a prerequisite for the binding of F components to obtain toxic activity on PMNs (4). Conversely, Yokota and Kamio (22) showed that the binding of the F component LukF (HlgB) on human erythrocytes was a prerequisite for the subsequent binding of Hlg2 (HlgA). Later, Gauduchon et al. (9) determined a dissociation constant (Kd) of 0.07 nM for LukS-PV binding on PMNs and showed that HlgC competed with LukS-PV for binding but HlgA and LukE did not compete. However, no data are available yet for the binding of F components.
The present study was performed to analyze the binding of the F components of leukotoxins, particularly the binding of LukF-PV compared with that of HlgB, two F components which exhibit 71% identity. LukF-PV* and HlgB* leukotoxins with cysteine mutations were labeled with fluorescein to follow their binding to human leukocytes by flow cytometry. This analysis demonstrated that LukF-PV, in contrast to HlgB, binds to a specific membrane site of target cells before its association with an S component for opening of a Ca2+ channel and integration in a transmembrane pore.
MATERIALS AND METHODS
Bacterial strains and vectors.
Escherichia coli XL1 Blue cells [recA1 endA1 gvrA96 thi1 supE44 relA1 lac (F′ proAB lacIqZΔM15 Tn10) (Tetr)] (Stratagene, Amsterdam, The Netherlands) were used as recipient cells after site-directed mutagenesis of recombinant plasmids. E. coli BL21 [F− ompT hsdS(rB− mB−) gal] was used for overexpression of pGEX-6P-1 glutathione S-transferase-fused leukotoxins as recommended by the manufacturer (Pharmacia, Uppsala, Sweden) (1).
Cloning and expression of leukotoxins in E. coli and purification.
LukS-PV, HlgC, HlgA, LukF-PV, HlgB, and the functional mutant proteins LukF-PV S27C and HlgB S27C were constructed using a QuikChange mutagenesis kit (Stratagene) and dedicated oligonucleotides as described previously (1). Proteins were purified by affinity chromatography on glutathione-Sepharose 4B, followed by cation-exchange fast-performance chromatography (1, 21), after removal of the glutathione S-transferase tag with Precision protease (Amersham-Pharmacia), and the homogeneity was checked by radial gel immunoprecipitation and sodium dodecyl sulfate-polyacrylamide gel electrophoresis before the proteins were stored at −80°C until they were used.
Fluorescein labeling.
Mutant proteins LukF-PV S27C and HlgB S27C were labeled with fluorescein to obtain LukF-PV* and HlgB*. A 10 μM LukF-PV S27C or HlgB S27C solution with a fivefold excess of fluorescein 5-maleimide (Molecular Probes, Eugene, OR) was incubated for 30 min at room temperature in 50 mM sodium phosphate, 0.15 M NaCl, 1 mM Na2-EDTA (pH 7.0). The coupling reaction was stopped by addition of 10 mM β-mercaptoethanol. The mixture was then desalted, and the coupling yield for LukF-PV* and HlgB* was determined by determining the ratio of the concentration of fluorescein (ɛ490 nm = 81,900 cm−1 mol−1) to the concentration of the protein determined by Bradford titration (Bio-Rad, Ivry sur Seine, France) (1). Only leukotoxins with a coupling yield that was more than 0.95 but less than 1 were used in this study.
PMN preparation.
PMNs were prepared from buffy coats obtained from healthy donors of either sex (Etablissement Français du Sang, Strasbourg, France) as described previously (7). Briefly, 40 ml of a 1/3 (vol/vol) dilution of blood cells in 0.9% (wt/vol) NaCl was layered on 12 ml of J Prep (Techgen International, Voisins le Bretonneux, France). After centrifugation at 800 × g for 20 min, the pellet was suspended in 30 ml of 0.9% NaCl, added to 10 ml of 6% (wt/vol) dextran, and sedimented for 30 min. Thirty milliliters of the supernatant was centrifuged for 10 min at 800 × g. The pellet was suspended in HEPES buffer (140 mM NaCl, 5 mM KCl, 10 mM glucose, 0.1 mM EGTA, 10 mM HEPES, 3 mM Tris; pH 7.3), and the contaminating erythrocytes were removed by hypotonic lysis for 45 s and subsequent washing in HEPES buffer. The concentration of the final suspension was adjusted to 6 × 106 PMNs/ml, and 0.1% (wt/vol) bovine serum albumin was added to prevent nonspecific leukotoxin adherence to tube walls.
Spectrofluorometric determinations.
Variations in intracellular free Ca2+ levels were determined by recording the variations in the fluorescence of Fura2- or Fluo3-containing PMNs as previously described (7, 9). Briefly, 107 cells/ml were incubated in HEPES buffer containing 1.1 mM CaCl2 and 4 μM Fura2-AM or 2 μM Fluo3-AM (Molecular Probes, Eugene, OR) for 45 min at room temperature. Subsequently, the PMNs were washed twice and then suspended a second time (6 × 106 cells/ml) in HEPES buffer. One milliliter of PMNs was added to 1 ml HEPES buffer in a 4-ml quartz thermostat-equipped cuvette (with a 1-cm light path) at 37°C and homogenized by continuous stirring. Variations in fluorescence intensity were recorded with a dual-excitation spectrofluorometer (Deltascan; PTI, United States) operated in a ratio mode for Fura-2 at excitation wavelengths of 340 and 380 nm (slit widths, 4 nm) and an emission wavelength of 510 nm (slit width, 4 nm). For Fluo3 recording, the wavelengths were 488 and 530 nm.
Flow cytometry determinations.
Flow cytometry data were obtained using a FACSort cytometer (Becton Dickinson, Le Pont de Claix, France) equipped with a 15-mW argon laser tuned to 488 nm. PMNs were classically distinguished by forward and side light scatter, and then their fluorescence was recorded. Pore formation was determined by recording variations in the fluorescence intensity of ethidium penetrating PMNs via pores as described previously (13). Variations in intracellular Ca2+ levels were determined by using PMNs loaded with Fluo3-AM as described above. Fluorescein and Fluo3 fluorescence intensities were recorded in the FL1 channel (emission wavelength, 530 nm), and ethidium fluorescence intensity was recorded in the FL3 channel (emission wavelength, >650 nm). The FACSort cytometer was set in such a way that calibrated fluorescent microbeads (Immuno-Brite; Coulter Corporation, Hialeah, FL) displayed the same fluorescence intensity for each experiment. Thus, mean fluorescein fluorescence intensity was expressed in standardized fluorescence units.
Determination of binding kinetics.
Maximal binding of leukotoxins (Bm) and their apparent Kd were calculated from saturation curves obtained by cytometric measurement of fluorescence intensity (F) (expressed in standardized fluorescence units) in PMNs incubated with different concentrations of fluorescent leukotoxins ([Toxin*]) (expressed in nM). Fitting of saturation curves was performed by nonlinear regression analysis (SigmaPlot; SSI, San Jose, CA) using the following equation:
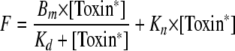 |
where Kn is the nonspecific coefficient.
Fifty percent inhibitory concentrations (IC50s) were determined by using the one-site competition model from the “ligand” software (SigmaPlot).
Antileukotoxin antibodies.
Anti-LukF-PV and anti-HlgB polyclonal antibodies were obtained from rabbits and were purified by using affinity columns as previously described (8). These antibodies cross-react with each other, but they do not react with leukotoxin S components.
RESULTS
LukF-PV and HlgB binding constants.
Until now, the binding of F components was thought to occur after the initial S component binding (4). In order to study this hypothesis in greater detail, suspensions of PMNs were incubated with different concentrations of LukF-PV* and HlgB* with and without a saturating concentration (2 nM) of LukS-PV in HEPES buffer (9). The fluorescence intensity of cells was recorded under these conditions over time by flow cytometry until we obtained a plateau value. The plateau values were then plotted against F component concentrations, which revealed that binding of LukF-PV* (Fig. 1A) and HlgB* (Fig. 1B) is concentration dependent for both leukotoxins. However, the binding of LukF-PV* was found to be saturable (Fig. 1A) (Kd, 6.2 nM ± 1.9; n = 8) and therefore mediated by a site on the membrane, but the binding of HlgB* was proportional to its concentration (Fig. 1B) (slope, 6.05 ± 0.13; n = 4) and consequently not specific. After 10 min of incubation of PMNs with different concentrations of LukF-PV*, 2 nM LukS-PV was added. This resulted in increases in PMN fluorescence intensity, which revealed that there was a second specific binding of LukF-PV* after a time lag (an example is shown in Fig. 5). The differences between the binding values obtained for the LukF-PV* concentrations with and without LukS-PV were calculated and plotted against the LukF-PV* concentrations (Fig. 1A). The LukS-PV-dependent binding of LukF-PV* was specifically concentration dependent (Kd, 10.7 ± 5 nM; n = 8) and limited by the number of LukS-PV molecules bound to PMNs (about 200,000 molecules per PMN) (8). The equilibrium constants obtained are summarized in Table 1. Similar experiments performed with and without 2 nM HlgC resulted in comparable determinations for kinetic constants of LukF-PV* binding (Table 1). When 2 nM LukS-PV was added after 10 min of incubation of PMNs with HlgB*, the specific binding of HlgB* increased considerably (Fig. 1B) (Kd, 1.1 ± 0.2 nM; n = 4). Similar results were obtained after addition of 2 nM HlgC (Table 2).
FIG. 1.

Determination by flow cytometry of the binding of LukF-PV* and HlgB* labeled with fluorescein before and after addition of LukS-PV on human PMNs from the same donor. ▪, binding after 10 min of incubation for different concentrations of LukF-PV* (A) and HlgB* (B) with PMNs; •, additional binding 10 min after addition of 2 nM LukS-PV. Equilibrium constants calculated by nonlinear regression are shown in Tables 1 and 2. The slope of HlgB* binding obtained in the absence of LukS-PV is 6.05 ± 0.13 (mean ± standard deviation; n = 4). SFUF, standardized fluorescent units for LukF-PV*; SFUB, standardized fluorescent units for HlgB*.
FIG. 5.

Flow cytometry comparison of binding time courses (• and ▪) for LukF-PV* (A) and HlgB* (B) and increases in the intracellular Ca2+ level (○ and □) before and after addition of LukS-PV on PMNs. The LukF-PV* concentrations were 1 (○ and •) and 5 nM (□ and ▪), and the HlgB* concentrations were 0.3 (○ and •) and 1.2 nM (□ and ▪). LukS-PV (2 nM) was added 10 min after LukF-PV* was added (A) and 5 min after HlgB* was added (B), when binding was observed. The PMNs (5 × 104 cells/ml) used for both determinations were from the same donor and were loaded with Fluo3. The LukF-PV and HlgB concentrations used were lower than and close to the apparent Kds. The experiment was carried out four times, and the same results were obtained each time. Therefore, the results of only one experiment are shown. SFUF, standardized fluorescent units for LukF-PV*; SFUB, standardized fluorescent units for HlgB*; a.u., arbitrary units.
TABLE 1.
Equilibrium constants determined by flow cytometry for LukF-PV* binding in the presence or in the absence of LukS-PV or HlgC on human PMNs, monocytes, and lymphocytesa
| Cells | LukF*-PV
|
LukF*-PV plus LukS-PV
|
LukF*-PV
|
LukF*-PV plus HlgC
|
||||
|---|---|---|---|---|---|---|---|---|
| Kd (nM) | Maximal binding (SFUF) | Kd (nM) | Maximal binding (SFUF) | Kd (nM) | Maximal binding (SFUF) | Kd (nM) | Maximal binding (SFUF) | |
| PMNs | 6.2 ± 1.9b | 50 ± 7b | 10.7 ± 5.0b | 186 ± 46b | 4.8 ± 1.2c | 38 ± 4c | 2.9 ± 0.7c | 122 ± 11c |
| Monocytes | 2.8 ± 0.8d | 231 ± 38d | NDe | ND | ND | ND | ND | ND |
| Lymphocytes | 1.2 ± 0.2d | 120 ± 8d | NBf | NB | 1.2 ± 0.3c | 120 ± 6c | 4.9 ± 1.2c | 46 ± 4c |
All data were obtained as described in the legend to Fig. 1. The HlgC concentrations used were 2 nM for PMNs and 50 nM for lymphocytes. No data were obtained for LukF-PV* binding on monocytes in the presence of LukS-PV or HlgC due to quenched fluorescence. The data are means ± standard deviations. SFUF, standardized fluorescent units for LukF-PV*.
n = 8.
n = 3.
n = 7.
ND, not determined.
NB, no LukS-PV binding.
TABLE 2.
Equilibrium constants determined by flow cytometry for HlgB* binding in the presence of LukS-PV or HlgC on human PMNs, monocytes, and lymphocytesa
| Cells | HlgB* plus LukS-PV
|
HlgB* plus HlgC
|
||
|---|---|---|---|---|
| Kd (nM) | Maximal binding (SFUB) | Kd (nM) | Maximal binding (SFUB) | |
| PMNs | 1.1 ± 0.2b | 484 ± 47b | 0.37 ± 0.11b | 474 ± 53b |
| Monocytes | 0.84 ± 0.31b | 143 ± 22b | 0.11 ± 0.02c | 200 ± 10c |
| Lymphocytes | NBd | NB | 3.6 ± 1.0b | 132 ± 12b |
All data were obtained as described in the legend to Fig. 1. The HlgC concentrations used were 2 nM for PMNs and monocytes and 50 nM for lymphocytes. The data are means ± standard deviations. SFUB, standardized fluorescent units for HlgB*.
n = 4.
n =3.
NB, no LukS-PV binding.
This type of experiment was also performed with human monocytes and lymphocytes. The binding of LukF-PV* was shown to be specific on monocytes and lymphocytes, and there was higher affinity on lymphocytes (Table 1). However, the binding of HlgB* was as nonspecific as it was on PMNs (data non shown). The total number of binding sites for LukF-PV* was higher on both monocytes and lymphocytes, particularly monocytes, than on PMNs.
The binding of LukF-PV* on monocytes after addition of LukS-PV or HlgC could not be determined by flow cytometry due to the unexplained quenching of fluorescence in LukF-PV* bound to these cells. No additional binding of LukF-PV* was observed on lymphocytes after addition of LukS-PV, since no receptors for LukS-PV were observed on these cells (9). However, addition of a high concentration (50 nM) of HlgC induced secondary specific binding of LukF-PV* on lymphocytes (Table 1).
Another analysis of the apparent Kd of LukF-PV binding was carried out using the increase in the intracellular Ca2+ level as a criterion. After incubation of Fluo3-containing PMNs with different LukF-PV concentrations for 10 min, addition of 2 nM LukS-PV resulted in variations in fluorescence intensity, which were recorded by spectrofluorometry (Fig. 2A). The maximum slope obtained for each increase in fluorescence intensity was determined using PTI software and then was plotted (Fig. 2B) as percentage of the maximum value obtained in the experiment. The apparent Kd calculated (2.2 ± 0.4 nM; n = 5) was the same order as the values obtained directly by LukF-PV* binding (Table 1). The same experiment done with LukF-PV* resulted in calculation of an apparent Kd of 2.3 ± 0.6 nM (n = 3).
FIG. 2.

Spectrofluorometric determination of the Kd of LukF-PV binding on PMNs by recording the time course of the increase in the intracellular Ca2+ level after addition of LukS-PV. (A) Fluorescence intensity for PMNs loaded with Fluo3 after 10 min of incubation with different LukF-PV concentrations and then addition of 2 nM LukS-PV. Only some of the concentrations used in one of five experiments are shown for clarity. arb. units, arbitrary units. (B) Plot of the higher slope values calculated for each LukF-PV concentration by nonlinear regression by the PTI software. The slopes were expressed as percentages of the maximal slope (100%) calculated for each experiment by nonlinear regression (single rectangular hyperbola plus nonspecific component) using the Sigma plot software (Systat Software Inc., San Jose, CA).
Binding competition between LukF-PV* or HlgB* and F components of leukotoxins.
The specificity of the binding of LukF-PV was investigated further by examining binding competition between LukF-PV* and the F components LukF-PV, HlgB, and LukD (Fig. 3). Examination of the competition between 5 nM LukF-PV* and LukF-PV resulted in different IC50s depending on the cells tested (IC50 for PMNs, 19.4 ± 1.0 nM [n = 6] [Fig. 3A]; IC50 for monocytes, 14.2 ± 1.0 nM [n = 4] [Fig. 3B]; IC50 for lymphocytes, 6.8 ± 1.1 nM [n = 5] [Fig. 3C]). Thus, a molar ratio of 1:1 for the IC50 of LukF-PV competing with LukF-PV* was obtained only with lymphocytes. However, in any one cell type, neither HlgB nor LukD competed with LukF-PV* in the concentration range assayed. Nevertheless, in PMNs the increase in binding of LukF-PV* after the addition of LukS-PV decreased as the concentrations of HlgB and LukD increased, until the fluorescence intensity value reached a value corresponding to the value for binding of LukF-PV* alone (Fig. 3A). In PMNs, HlgB (IC50, 3.4 ± 1.3 nM; n = 3) exhibited greater affinity for LukS-PV than LukF-PV (IC50, 19.61.0 nM; n = 5) or LukD (IC50, 17.5 ± 1.0 nM; n = 2). In monocytes precise competition values could not be determined due to quenched fluorescence, and in lymphocytes addition of LukS-PV had no effect since it does not bind to these cells (9). The opposite experiment was performed by examining the competition between HlgB* fixed in the presence of LukS-PV on PMNs (Fig. 4A) and monocytes (Fig. 4B) and HlgB, LukF-PV, or LukD. The competition between 2 nM HlgB* and HlgB was adequate on PMNs (Fig. 4A) (IC50, 2.4 ± 0.6 nM; n = 5) and on monocytes (Fig. 4B) (IC50, 1.5 ± 0.2 nM; n = 3). LukF-PV and LukD competed with HlgB* for binding on both types of cells. The IC50 of LukF-PV was higher on PMNs (Fig. 4A) (46 ± 1 nM; n = 4) than on monocytes (Fig. 4B) (9.2 ± 1.5 nM; n = 3), but it was the same for LukD on both types of cells (20.4 ± 1.1 and 20.6 ± 2.7 nM, respectively; n = 3).
FIG. 3.

Flow cytometry determination of the competition between LukF-PV* and F components (LukF-PV, HlgB, and LukD) for binding in human PMNs (A), monocytes (B), and lymphocytes (C) in the presence and in the absence of LukS-PV. LukF-PV* at a concentration of 5 nM and different concentrations of F components were simultaneously added 10 min after 2 nM LukS-PV was added. The binding of 5 nM LukF-PV* was defined as 100%. The leukocyte concentration was 5 × 104 cells/ml, and the incubation time was 15 min. The IC50s were as follows (means ± standard deviations): (A) 19.2 ± 2.2 nM for LukF-PV and 18.2 ± 4.2 nM for LukS-PV-LukF-PV (n = 6); (B) 14.1 ± 1.6 nM for LukF-PV (n = 4); (C) 7.1 ± 1.6 nM for LukF-PV (n = 4).
FIG. 4.

Flow cytometry determination of the competition between HlgB* and F components (HlgB, LukF-PV, and LukD) for binding in human PMNs (A) and monocytes (B) in the presence of LukS-PV. HlgB* at a concentration of 2 nM and different concentrations of F components were simultaneously added 10 min after 2 nM LukS-PV was added. The binding of 2 nM HlgB* was defined as 100%. The incubation time was 15 min. The IC50s were as follows (means ± standard deviations): (A) 2.3 ± 0.3 nM for HlgB (n = 5), 38.8 ± 1.6 nM for LukF-PV (n = 3), and 26.0 ± 1.6 nM for LukD (n = 4); (B) 1.5 ± 0.5 nM for HlgB (n = 3) and 9.5 ± 1.2 nM for LukF-PV (n = 3), and 20.4 ± 1.2 nM for LukD (n = 3).
Activity of LukF-PV molecules.
This study showed that LukF-PV can bind to two specific sites, one of which is on the membrane and one of which is the previously fixed LukS-PV molecule, although HlgB binds only to LukS-PV. The aim of this study was to determine if the active LukF-PV molecules were the LukF-PV molecules bound to a membrane site, the LukF-PV molecules bound directly to LukS-PV, or both. To answer this question, the binding time courses for both labeled F components were compared with the time courses for the first event induced by leukotoxins (i.e., the increase in the intracellular Ca2+ level after opening of Ca2+ channels) (19). Thus, the binding of LukF-PV* and HlgB* and the variations in fluorescence intensity of Fluo3-containing PMNs were recorded for 10 min and subsequently after addition of LukS-PV or HlgC to PMNs from the same donor using flow cytometry. Figure 5 shows results obtained for LukF-PV* and HlgB* and addition of LukS-PV. When 2 nM LukS-PV was added 10 min after 1 nM LukF-PV* was added, there was a significant increase in the fluorescence intensity of Fluo3 and there was no increase in LukF-PV* binding (Fig. 5A). Furthermore, when 2 nM LukS-PV was added 10 min after 5 nM LukF-PV* was added, the increase in the fluorescence intensity of Fluo3 clearly preceded the increase in LukF-PV* binding induced by addition of LukS-PV (Fig. 5A). However, when LukS-PV was added after 0.3 or 1.2 nM HlgB* was added, the binding of HlgB* preceded or was concomitant with the increase in Fluo3 fluorescence intensity (Fig. 5B). Similar results were obtained when LukS-PV was added before HlgB* was added (data not shown). Thus, the entry of Ca2+ observed in PMNs was linked to the association of LukS-PV with the LukF-PV* molecules already bound to the membrane or to the HlgB* molecules subsequently bound to LukS-PV after its addition. Similar results were obtained after HlgC addition under the same conditions (data not shown).
Further proof of the different fixation styles of the F components was obtained when we analyzed the effect of anti-LukF-PV and anti-HlgB, both of which are able to completely inhibit leukotoxin-induced increases in intracellular Ca2+ levels. However, no nonspecific interactions of antibodies with PMNs were observed if no other element was added (data not shown).
In these experiments, PMNs containing Fura-2 were incubated for 10 min with LukF-PV or HlgB before LukS-PV addition, which resulted in an increase in Fura-2 fluorescence intensity (Fig. 6A and B, lines a). Figure 6A shows that when anti-LukF-PV was added 10 s before (line b) or after (line c) LukS-PV, there was no inhibition of the leukotoxin activity. Partial inhibition of the increase in the intracellular Ca2+ level was observed when anti-LukF-PV was added 1 min before LukS-PV was added (line d). There was complete inhibition when anti-LukF-PV was added at least 5 min before LukS-PV was added (line e) or 10 s before associated LukS-PV-LukF-PV was added (line f). Conversely, anti-HlgB inhibited the increase in the intracellular Ca2+ level when it was added 10 s before HlgB was added (Fig. 6B, line b) or 5 min (line c) or 10 s (line d) before LukS-PV was added. Identical results were obtained when anti-HlgB was used instead of anti-LukF-PV or when anti-LukF-PV was used instead of anti-HlgB. Consequently, we concluded that the increases in intracellular Ca2+ levels were due only to LukS-PV associated with the LukF-PV molecules directly bound to a cell membrane site.
FIG. 6.

Spectrofluorometric determination of anti-LukF-PV (A) and anti-HlgB (B) antibody influence on the increase in the intracellular Ca2+ level induced in the presence of LukF-PV and HlgB following addition of LukS-PV in Fluo3-containing human PMNs. (A) PMNs were incubated for 10 min with 5 nM LukF-PV before addition of 2 nM LukS-PV (lines a, b, c, d, and e), and LukF-PV and LukS-PV were also added simultaneously (line f). Anti-LukF-PV was added as follows: line a, no addition; line b, 10 s before addition of LukS-PV; line c, 10 s after addition of LukS-PV; line d, 5 min before addition of LukS-PV; line e, 10 s before addition of LukF-PV; line f, 10 s before addition of LukF-PV-LukS-PV. (B) PMNs were incubated for 10 min with 2 nM HlgB before addition of 2 nM LukS-PV (lines a, b, c, and d). Anti-HlgB was added as follows: line a, no addition; line b, 10 s before addition of LukS-PV; line c, 10 s before addition of HlgB; line d, 5 min before addition of LukS-PV. The experiment was done four times, and the same results were obtained each time. arb. units, arbitrary units.
The effects of both types of LukF-PV binding on pore formation were also examined. It has been shown previously that the extent of pore formation is dependent on the concentration of LukF-PV (7). Consequently, after 10 min of incubation of PMNs with a nonsaturating concentration (5 nM) of LukF-PV*, different LukS-PV concentrations were used to obtain an increase in the number of LukF-PV* molecules directly bound to LukS-PV (Fig. 7A). Pore formation was measured under the same conditions with PMNs from the same donors by using ethidium fluorescence (Fig. 7B). As there was no correlation between the secondary binding of LukF-PV* on LukS-PV and pore formation (which did not change when the LukS-PV concentration was changed), we presumed that the LukF-PV molecules fixed after the addition of LukS-PV were devoid of activity.
FIG. 7.

Comparison by flow cytometry of LukF-PV* binding induced by LukS-PV and pore formation obtained under the same conditions. (A) Binding of LukF-PV* after addition of different concentrations of LukS-PV to PMNs after 10 min of incubation with 5 nM LukF-PV*. (B) Pore formation measured by ethidium fluorescence after addition of LukS-PV under the conditions described above for panel A and expressed as percentages of the maximal fluorescence intensity. The maximal fluorescence intensity (100%), corresponding to the maximal possible entry of ethidium into PMNs, was calculated by nonlinear regression of a sigmoidal model (SigmaPlot). The experiment was carried out three times, and the same results were obtained each time. The results of one experiment are shown.
The ability of LukF-PV molecules fixed on lymphocytes to be active was tested by comparing the effects of addition of LukS-PV or HlgA on Ca2+ entry and pore formation in lymphocytes and monocytes. As shown in Fig. 8, these associations were fully active in monocytes. In lymphocytes, however, LukF-PV-LukS-PV was not active, as previously shown, but LukF-PV-HlgA was fully active, although to a lower extent than it was on monocytes, demonstrating the toxicity of LukF-PV bound to lymphocytes.
FIG. 8.

Flow cytometry determination of the time course of the increase in the intracellular Ca2+ level and pore formation induced by LukF-PV in the presence of LukS-PV or HlgA in human monocytes and lymphocytes. The intracellular Ca2+ level was determined by Fluo3 fluorescence, and pore formation was evaluated using ethidium fluorescence with 5 × 103 mixed monocytes and lymphocytes. Cells were incubated with 5 nM LukF-PV for 10 min before addition of 2 nM LukS-PV or 5 nM HlgA at time zero. arb. units, arbitrary units.
DISCUSSION
The binding of leukotoxin F components was studied using mutated proteins in which a serine was replaced by a cysteine so that they could be labeled by a fluorescein maleimide moiety. However, LukF-PV did not seem to compete at a molar ratio of 1:1 with LukF-PV* in PMNs and monocytes for unknown reasons. Modification of the affinity by labeling could have been responsible for the possible allosteric problem, but this problem was not observed in lymphocytes. However, the discrepancy could have been due to conformity differences in the binding sites depending on the cells used. Furthermore, the Kd of the nonlabeled and labeled leukotoxins measured by using the increase in the intracellular Ca2+ level and the Kd of the labeled leukotoxin measured by using binding were the same order. Consequently, the labeling did not modify the activity of the leukotoxin, and LukF-PV* was considered suitable for binding studies and determination of apparent kinetic constants. Since the specific fluorescence intensities of LukF-PV* and HlgB* were not known, the maximal binding values obtained for these two components could not be compared; rather, the maximal binding values obtained for different cell types for each component were compared.
This study showed that LukF-PV binds to a specific site on the cell membrane, in contrast to HlgB, for which the initial binding of an S component is a prerequisite for binding. Several findings support this conclusion. (i) When applied alone to target cells, LukF-PV presented a saturable binding component, but HlgB did not. (ii) The binding of LukF-PV was very specific since in the absence of LukS-PV neither HlgB nor LukD competed with LukF-PV* in the concentration range tested. However, they competed for binding to the LukS-PV(HlgC)-receptor complex. Moreover, LukD competed with 2 nM HlgB* on PMNs and monocytes with similar IC50s (20.4 and 20.6, respectively), but LukF-PV had fivefold-higher affinity for the LukS-PV-receptor complex on PMNs than on monocytes (IC50, 45.7 and 9.2, respectively). This suggests that there is variable complexity among leukotoxin receptors with different target cells. (iii) The molecules that bound to target cells in the absence of LukS-PV were the molecules active with Ca2+ channels, while HlgB molecules required the presence of previously fixed LukS-PV to bind and be active. (iv) The LukF-PV molecules bound after addition of LukS-PV did not play any role in pore formation. (v) Antibodies which inhibited the activity of LukF-PV and HlgB if they were added 10 s before these components were added were unable to inhibit the activity of LukF-PV molecules already bound if they were added 10 s before or after LukS-PV was added.
The quenching of LukF-PV* fluorescence after addition of LukS-PV or HlgC in monocytes suggests that the fluorescein moiety was hidden by a membrane component after octamerization or by LukS-PV (HlgC) itself. However, the S component seems not to be responsible since this effect was not observed in PMNs, but the possibility that the LukS-PV (HlgC) receptor in monocytes is involved cannot be eliminated.
It is important to note that saturable binding of LukF-PV on lymphocytes has also been observed, although LukS-PV, the natural S component of PVL, does not bind to lymphocytes at any concentration. However, although HlgC competes with LukS-PV on PMNs and monocytes (9), a high concentration of HlgC (50 nM) was able to induce the binding of HlgB to lymphocytes. Thus, in contrast to LukS-PV, HlgC was able to bind to lymphocytes.
LukF-PV could bind to the LukS-PV-receptor complex on PMNs and monocytes, but LukS-PV could not bind to the LukF-PV-receptor complex on lymphocytes. However, LukF-PV was active on lymphocytes, since after addition of HlgA, which binds to these cells, both an increase in the intracellular Ca2+ level and pore formation were induced.
In a previous study (9), the calculated ratio of the LukS-PV receptors of PMNs to monocytes was 2.4. This proportion was verified in the present study since the maximal binding ratios for HlgB* binding to LukS-PV and HlgC in PMNs and monocytes were 3.3 and 2.4, respectively. In contrast, the ratio of LukF-PV binding sites of PMNs to LukF-PV binding sites of monocytes was 0.22.
Early perfusion experiments with PMNs (4) showed that the binding of LukS-PV was maintained after cells were rinsed, but the binding of LukF-PV was not maintained. Thus, in contrast to the binding of LukS-PV, the binding of LukF-PV is reversible, although it is saturable. This binding is required for activity since the LukF-PV molecules that bound after the addition of LukS-PV were not associated with pore formation. Recently, a new concept has been proposed to explain the binding of alpha-toxin, another member of the family of staphylococcal pore-forming toxins which forms homotypic heptamers. Besides low-affinity binding sites, the role of high-affinity receptors for this toxin would be played by clusters of phosphocholine head groups in sphingomyelin-cholesterol-rich microdomains (20) and consequently would not be protein mediated. The present study did not result in any final conclusions concerning the receptors of bicomponent leukotoxins. However, these leukotoxins differ from alpha-toxin in important ways. They have few target cells and several receptors (9). Furthermore, the receptors of LukS-PV which appear in terminally differentiated PMNs (13) are downregulated by protein kinase C (9). Moreover, LukS-PV and LukF-PV have only high-affinity receptors, which have been shown to be irreversible for LukS-PV but reversible for LukF-PV, and form heterotypic octamers (14).
In conclusion, LukF-PV binds specifically to the membrane of PMNs, monocytes, and lymphocytes, and association of this molecule with its binding site is a prerequisite for its toxic activity. In contrast, the presence of an S component is required for the binding of HlgB. We suggest (Fig. 9) that after binding to its fixation site, LukS-PV is associated (i) with LukF-PV (bound to its receptor) to induce opening of nonspecific Ca2+ channels before formation of pores (1, 18) through the membrane (Fig. 9, panel I), (ii) with LukF-PV in an inactive form (Fig. 9, panel II), and (iii) with HlgB to induce the opening of Ca2+ channels before pore formation (1, 18) (Fig. 9, panel III). However, although both the LukS-PV(HlgC)-LukF-PV and LukS-PV(HlgC)-HlgB associations induce activation of target cells by increasing the intracellular Ca2+ level, the binding of LukF-PV to a specific membrane component could produce intracellular reactions of the target cells responsible for the epidemiology of PVL+ strains. These reactions could inhibit the defenses of target cells or activate cell products, resulting in environmental conditions favorable for the growth of bacteria. For instance, it has been shown that LukF-PV associated with LukS-PV could, at low concentrations, induce an oxidative burst in the extracellular medium, but no such effect has been observed for HlgB (5).
FIG. 9.

Schematic diagram of the association of PVL components and HlgB. (Panel I) LukS-PV (S) and LukF-PV (F) bind to their membrane receptors (RS and RF, respectively) and associate to open a Ca2+ channel (C) and form a pore (P) through the membrane (M). (Panel II) LukF-PV binds to the LukS-PV-LukS-PV membrane receptor complex, forming an inactive association. (Panel III) HlgB binds to the LukS-PV-LukS-PV membrane receptor complex to open a calcium channel and form a pore through the membrane.
Acknowledgments
We are very grateful to Daniel Keller for expert toxin purification and labeling and to Joanna Lignot for English language improvement.
This work was supported by grant EA-3432 from the Direction de la Recherche et des Etudes Doctorales.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 6 October 2008.
REFERENCES
- 1.Baba Moussa, L., S. Werner, D. A. Colin, L. Mourey, J. D. Pédelacq, J. P. Samama, A. Sanni, H. Monteil, and G. Prévost. 1999. Discoupling the Ca2+-activation from the pore-forming function of the bi-component Panton-Valentine leucocidin in human PMNs. FEBS Lett. 461280-286. [DOI] [PubMed] [Google Scholar]
- 2.Bocchini, C. E., K. G. Hulten, E. O. Mason, Jr., B. E. Gonzalez, W. A. Hammerman, and S. L. Kaplan. 2006. Panton-Valentine leukocidin genes are associated with enhanced inflammatory response and local disease in acute hematogenous Staphylococcus aureus ostomyelitis in children. Pediatrics 117433-440. [DOI] [PubMed] [Google Scholar]
- 3.Boyle-Vavra, S., and R. S. Daum. 2007. Community-acquired methicillin-resistant Staphylococcus aureus: the role of Panton-Valentine leukocidin. Lab. Investig. 873-9. [DOI] [PubMed] [Google Scholar]
- 4.Colin, D. A., I. Mazurier, S. Sire, and V. Finck-Barbançon. 1994. Interaction of the two components of leukocidin from Staphylococcus aureus with human polymorphonuclear leukocyte membranes: sequential binding and subsequent activation. Infect. Immun. 623184-3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colin, D. A., and H. Monteil. 2003. Control of the oxidative burst of human neutrophils by staphylococcal leukotoxins. Infect. Immun. 713724-3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cribier, B., G. Prévost, P. Couppié, V. Finck-Barbançon, E. Grosshans, and Y. Piémont. 1992. Staphylococcus aureus leukocidin: a new virulence factor in cutaneous infections? An epidemiological and experimental study. Dermatology 185175-180. [DOI] [PubMed] [Google Scholar]
- 7.Finck-Barbançon, V., G. Duportail, O. Meunier, and D. A. Colin. 1993. Pore formation by a two component leukocidin from Staphylococcus aureus within the membrane of human polymorphonuclear leukocytes. Biochim. Biophys. Acta 1182275-282. [DOI] [PubMed] [Google Scholar]
- 8.Finck-Barbançon, V., G. Prévost, and Y. Piémont. 1991. Improved purification of leukocidin from Staphylococcus aureus and toxin distribution among hospital strains. Res. Microbiol. 14275-85. [DOI] [PubMed] [Google Scholar]
- 9.Gauduchon, V., S. Werner, G. Prévost, H. Monteil, and D. A. Colin. 2001. Flow cytometric determination of Panton-Valentine leucocidin S component binding. Infect. Immun. 692390-2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gravet, A., D. A. Colin, D. Keller, R. Girardot, H. Monteil, and G. Prévost. 1998. Characterization of a novel structural member, LukE-LukD, of the bi-component staphylococcal leucotoxin family. FEBS Lett. 436202-208. [DOI] [PubMed] [Google Scholar]
- 11.Labandeira-Rey, M., F. Couzon, S. Boisset, E. L. Brown, M. Bes, Y. Benito, E. M. Barbu, V. Vazquez, M. Höök, J. Etienne, F. Vandenesch, and M. G. Bowden. 2007. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science 3151130-1133. [DOI] [PubMed] [Google Scholar]
- 12.Lina, G. Y., Piémont, F. Godail-Gamot, M. Bes, M.-O. Peter, V. Gauduchon, F. Vandenesch, and J. Etienne. 1999. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin. Infect. Dis. 291128-1132. [DOI] [PubMed] [Google Scholar]
- 13.Meunier, O., A. Falkenrodt, H. Monteil, and D. A. Colin. 1995. Application of flow cytometry in toxinology: pathophysiology of human polymorphonuclear leucocytes damaged by a pore-forming toxin from Staphylococcus aureus. Cytometry 21241-247. [DOI] [PubMed] [Google Scholar]
- 14.Miles, G., L. Movileanu, and H. Bayley. 2002. Subunit composition of a bicomponent toxin: staphylococcal leukocidin forms an octameric transmembrane pore. Protein Sci. 11894-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen, V. T., H. Higuchi, and Y. Kamio. 2002. Controlling pore assembly of staphylococcal gamma-haemolysin by low temperature and by disulphide bond formation in double-cysteine LukF mutants. Mol. Microbiol. 451485-1498. [DOI] [PubMed] [Google Scholar]
- 16.Panton, P. N., and F. C. O. Valentine. 1932. Staphylococcal toxins. Lancet ii506-508. [Google Scholar]
- 17.Prévost, G., B. Cribier, P. Couppié, P. Petiau, G. Supersac, V. Finck-Barbançon, H. Monteil, and Y. Piémont. 1995. Panton-Valentine leucocidin and gamma-hemolysin from Staphylococcus aureus ATCC 49775 are encoded by distinct genetic loci and have different biological activities. Infect. Immun. 634121-4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prévost, G., T. Bouhakam, Y. Piémont, and H. Monteil. 1995. Characterization of a synergohymenotropic toxin produced by Staphylococcus intermedius. FEBS Lett. 376135-140. [DOI] [PubMed] [Google Scholar]
- 19.Staali, L., H. Monteil, and D. A. Colin. 1998. The staphylococcal pore-forming leukotoxins open Ca2+ channels in the membrane of human polymorphonuclear neutrophils. J. Membr. Biol. 162209-216. [DOI] [PubMed] [Google Scholar]
- 20.Valeva, A., N. Hellman, I. Walev, D. Strand, M. Plate, F. Boukhallouk, A. Brack, K. Hanada, H. Decker, and S. Bhakdi. 2006. Evidence that clustered phosphocholine head groups serve as sites for binding and assembly of an oligomeric protein pore. J. Biol. Chem. 28126014-26021. [DOI] [PubMed] [Google Scholar]
- 21.Werner, S., D. A. Colin, M. Coraiola, G. Menestrina, H. Monteil, and G. Prévost. 2002. Retrieving biological activity from LukF-PV mutants combined with different S components implies compatibility between the stem domains of these staphylococcal bicomponent leucotoxins. Infect. Immun. 701310-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yokota, K., and Y. Kamio. 2000. Tyrosine72 residue at the bottom of rim domain in LukF crucial for the sequential binding of the staphylococcal gamma-hemolysin to human erythrocytes. Biosci. Biotechnol. Biochem. 642744-2747. [DOI] [PubMed] [Google Scholar]