

Abstract
Klebsiella pneumoniae is a leading cause of both community-acquired and nosocomial gram-negative bacterial pneumonia. A significant clinical complication of Klebsiella pulmonary infections is peripheral blood dissemination, resulting in a systemic infection concurrent with the localized pulmonary infection. We report here on the critical importance of β2-microglobulin expression during murine K. pneumoniae bacteremia. β2-Microglobulin knockout mice displayed significantly increased mortality upon intravenous inoculation that correlated with increased bacterial burden in the blood, liver, and spleen. As β2-microglobulin knockout mice lack both CD8+ T cells and invariant NK T cells, mouse models specifically deficient in either cell population were examined to see if this would account for the increased mortality noted in β2-microglobulin knockout mice. Surprisingly, neither CD8 T-cell-deficient (TAP-1 knockout; in vivo anti-CD8 antibody treatment) nor invariant NK (iNK) T-cell-deficient (CD1d knockout, Jα281 knockout) mice were more susceptible to K. pneumoniae bacteremia. Combined, these studies clearly indicate the importance of a β2-microglobulin-dependent but CD8 T-cell- and iNK T-cell-independent mechanism critical for survival during K. pneumoniae bacteremia.
Klebsiella pneumoniae is a leading cause of nosocomial and community-acquired gram-negative bacterial pneumonia, resulting in a severe pyogenic infection with high mortality rates in the absence of therapeutic intervention (19). A significant clinical complication of Klebsiella pneumonia is dissemination of bacteria from within the pulmonary airspace into the bloodstream, resulting in bacteremia concurrent with the localized pulmonary infection (21). Inability to clear blood-borne bacteria can lead to a state of overwhelming bacteremia, which can culminate in multiple organ dysfunction syndrome and increased mortality.
We have previously reported on the differential murine host response to localized pulmonary versus systemic K. pneumoniae infection. Mice lacking γδ T cells had an impaired ability to resolve disseminated bacterial infections subsequent to the initial pulmonary infection. Interestingly, γδ T-cell knockout (KO) mice displayed increased peripheral blood dissemination while pulmonary bacterial clearance was unimpaired (14). To address the importance of gamma interferon (IFN-γ) in localized pulmonary versus disseminated blood-borne Klebsiella infection, IFN-γ KO mice were intratracheally or intravenously inoculated with K. pneumoniae. These studies indicated that IFN-γ is a critical mediator for the resolution of localized, pulmonary gram-negative pneumonia, whereas resolution of systemic, blood-borne gram-negative bacterial infections is independent of IFN-γ secretion (16). In contrast, in our murine model of Klebsiella bacteremia, mice receiving anti-tumor necrosis factor alpha (anti-TNF-α) treatment displayed increased mortality that correlated with impaired bacterial clearance (13, 15). These studies, however, did not address the cellular source of these cytokines induced during Klebsiella bacteremia.
Mice deficient in β2-microglobulin (β2-m)-dependent lymphocytes have been utilized to examine the relative contributions of these cells in several models of systemic infections (28). Increased susceptibility to intravenous Mycobacterium tuberculosis infection or lipopolysaccharide-induced lethal shock has been reported in β2-m KO mice compared to their wild-type counterparts (4, 7). In contrast, β2-m KO mice have been shown to be resistant to lethal polymicrobial sepsis based on studies using the cecal ligation and puncture model (25).
To assess the relative contribution of β2-m-dependent lymphocytes during gram-negative blood-borne infection, we utilized a murine model of K. pneumoniae bacteremia. We report here on the critical dependence of β2-m expression for survival following intravenous infection with K. pneumoniae. Unexpectedly, mice rendered specifically deficient in CD8 T cells (TAP-1 KO, in vivo anti-CD8 antibody treatment) or invariant NK (iNK) T cells (CD1d KO, Jα281 KO) were no more susceptible than wild-type infected animals. Combined, these studies clearly indicate the importance of a β2-m-dependent but CD8 T-cell- and iNK T-cell-independent mechanism critical for survival during K. pneumoniae bacteremia.
MATERIALS AND METHODS
Animals.
C57BL/6J wild-type mice, β2-m KO (B6.129P2-B2 mtm1Unc/J) mice, TAP-1 KO (B6.129S2-Tap1tm1Arp/J) mice, and CD1d KO (C.129S2-Cd1tm1Gru/J) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Jα281 KO mice on the C57BL/6 background were obtained from the RIKEN Research Center for Allergy and Immunology (Yokohama, Japan) by way of Luc Van Kaer (Vanderbilt University School of Medicine, Nashville, TN). CD1d KO mice on the C57BL/6 background were obtained from Luc Van Kaer and from Chyung-Ru Wang (University of Chicago, Chicago, IL). Animals were housed under specific-pathogen-free conditions within the animal care facility at the University of Michigan until the day of sacrifice. All experimental animal procedures were approved by the University Committee on Use and Care of Animals at the University of Michigan.
Klebsiella pneumoniae inoculation.
K. pneumoniae strain 43816 serotype O1:K2 (ATCC, Rockville, MD) was grown in tryptic soy broth (Difco, Detroit, MI) overnight at 37°C. The bacterial concentration was determined by measuring the amount of absorbance at 600 nm and compared to a predetermined standard curve. Bacteria were then diluted to the desired concentration for inoculation. For intravenous infection, mice were warmed under a heat lamp for an appropriate time to allow vasodilation of the tail vein. Bacteria, diluted in pyrogen-free saline, were injected in a 0.5-ml volume through a 27-gauge needle. For all experiments, an aliquot of the inoculated K. pneumoniae suspension was serially diluted onto blood agar plates to determine the actual dose of injected bacteria.
For survival studies, mice intravenously inoculated with bacteria were monitored twice daily (morning and late afternoon) for signs of illness. Animals appearing moribund (as outlined in the University Committee on Use and Care of Animals policy for end-stage illness and humane endpoints) were euthanized to prevent any unnecessary suffering.
Whole liver or spleen homogenization for CFU analyses.
At designated time points, mice were euthanized by inhalation of CO2. The liver was perfused with 2 to 3 ml phosphate-buffered saline-5 mM EDTA and removed for analyses as previously described (13, 15, 16). Briefly, liver or spleen was homogenized using a tissue homogenizer (Biospec Products, Bartlesville, OK) in 1 ml phosphate-buffered saline. For organ CFU determinations, a small aliquot of tissue homogenate was serially diluted and plated on blood agar plates and incubated at 37°C, and colonies were counted.
Peripheral blood CFU analyses.
For determination of peripheral blood bacterial numbers, mice were euthanized and heparinized blood was collected by cardiac puncture at the indicated time points. Serial dilutions were plated onto blood agar plates and incubated at 37°C, and colonies were counted.
Isolation and RT-PCR amplification of liver mRNA.
Liver (two lobes) was harvested at the indicated time points, immediately snap-frozen in liquid nitrogen, and then stored at −70°C for further analyses. Total cellular RNA from frozen tissue was isolated by homogenizing in 3 ml TRIzol reagent (Gibco BRL, Gaithersburg, MD) following the TRIzol protocol. Total RNA was determined by spectrometric analysis at a 260-nm wavelength. Expression of mRNA was determined by reverse transcription-PCR (RT-PCR) using the Access RT-PCR system kit from Promega (Madison, WI) following the manufacturer's protocol. The following primer pairs (all primers are shown 5′ to 3′) were used for specific mRNA amplification: mTNF-α sense, CCT GTA GCC CAC GTC GTA GC; mTNF-α antisense, AGC AAT GAC TCC AAA GTA GAC C; mKC sense, TGA GCT GCG CTG TCA GTG CCT; mKC antisense, AGA AGC CAG CGT TCA CCA GA; mMCP-1 sense, CTC ACC TGC TGC TAC TCA TTC; mMCP-1 antisense, GCT TGA GGT GGT TGT GGA AAA; mIP-10 sense, ATC ATC CCT GCG AGC CTA TC; mIP-10 antisense, GAA CTG ACG AGC CTG AGC TA; mβ-actin sense, CTT CTA CAA TGA GCT GCG TGT G; mβ-actin antisense, GAT TCC ATA CCC AAG AAG GAA GG. cDNA products were detected on a 2% agarose gel containing ethidium bromide, and bands were visualized and photographed using UV transillumination.
In vivo CD8 T-cell depletion.
Mice were injected intraperitoneally with 200 μg anti-CD8α monoclonal antibody (clone YTS169) 5 days prior to intravenous inoculation with K. pneumoniae. In vivo depletion of CD8+ T cells was confirmed by flow cytometry and resulted in >95% depletion of cells for a minimum of 7 to 10 days.
Plasma AST analyses.
Plasma levels of aspartate aminotransferase (AST), as an indication of hepatic cellular injury, were determined on peripheral blood samples collected at various time points following K. pneumoniae inoculation. AST activities from plasma samples were quantitated by the Clinical Chemistry Laboratory at the University of Michigan Medical Center using an automated spectrophotometric assay.
Statistical analyses.
Statistical significance was determined using the unpaired, two-tailed Student t test or the nonparametric Mann-Whitney test, analysis of variance for multiple group using the Student-Newman-Keuls post test, and the Fisher's exact test. Calculations were performed using InStat 3 for Macintosh (GraphPad Software, San Diego, CA). Statistical analyses of survival curves were performed by the log rank test using the Prism 3 for Macintosh software program (GraphPad Software).
RESULTS
Increased mortality of β2-microglobulin-deficient mice during K. pneumoniae bacteremia.
To determine whether mice deficient in β2-m expression had altered antibacterial host defenses during blood-borne K. pneumoniae infection, β2-m KO mice and their C57BL/6 wild-type littermates were intravenously inoculated with 5 × 104 CFU of K. pneumoniae. Survival following infection was then observed over the course of 10 days. As we have previously reported (13, 15, 16), this dose of K. pneumoniae results in a mortality rate of 50% in wild-type animals, with the majority of death occurring within 2 to 4 days of infection. Interestingly, β2-m KO mice were significantly more susceptible to K. pneumoniae infection, with 90% of the infected animals succumbing by day 5 postinfection (Fig. 1). No further mortality was observed in either group past day 10 of infection.
FIG. 1.
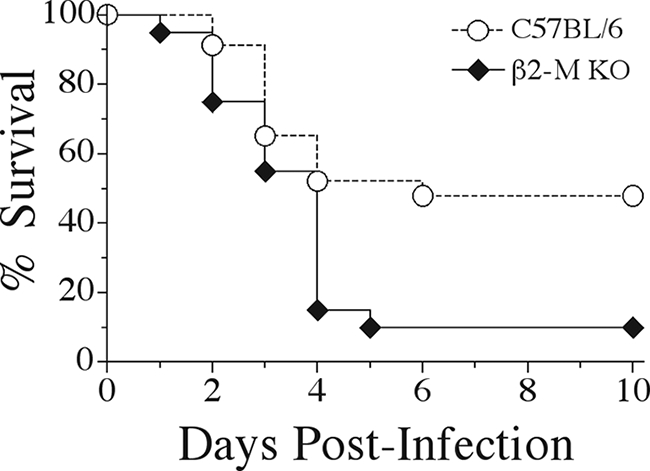
Increased mortality of β2-microglobulin-deficient mice during K. pneumoniae bacteremia. C57BL/6 wild-type and β2-m KO mice were intravenously inoculated with 5 × 104 CFU of K. pneumoniae, and survival was observed for 10 days postinfection. Mortality differences were statistically significant (P < 0.01) as determined by a log rank test. Survival curves were generated from three independent experiments using a total of 20 β2-m KO and 23 C57BL/6 mice.
Impaired early bacterial clearance in β2-microglobulin-deficient mice following K. pneumoniae inoculation.
Mice lacking β2-m were clearly more susceptible to blood-borne Klebsiella infection. Impaired bacterial clearance may, in part, account for the dramatic increase in mortality noted in β2-m KO mice. To determine the kinetics of bacterial clearance, β2-m KO mice and their wild-type littermates were intravenously inoculated and bacterial burdens were determined in blood, liver, and spleen tissues. Since differences in survival were noted by day 2 postinoculation, we examined bacterial clearance at time points prior to observed mortality. Within 12 h of infection, β2-m KO mice displayed a 10-fold increase in the number of blood-associated bacteria (Fig. 2A). This impaired blood clearance was more dramatic by 24 h, with a greater-than-100-fold increase in blood-borne bacteria present in β2-m KO mice. Additionally, only 40% of wild-type mice exhibited blood-associated bacteria, while 90% of the inoculated β2-m KO mice were highly bacteremic at 24 h postinfection. Similar data were obtained when examining bacterial burdens in both liver and spleen tissues from β2-m KO and wild-type infected animals. At both 12 and 24 h postinfection, mice lacking β2-m contained significantly more bacteria in liver and spleen, with differences approaching 100-fold in both tissues by 24 h of infection (Fig. 2B and C).
FIG. 2.

Increased bacterial burden and liver injury in β2-microglobulin-deficient mice during K. pneumoniae bacteremia. Mice were intravenously inoculated with K. pneumoniae and euthanized at the indicated time points following infection. Bacterial burdens from blood (A), liver (B), and spleen (C), along with plasma AST levels (D), were determined as described in Materials and Methods. Bacterial numbers for liver and spleen are for the entire organ, while blood bacterial numbers are per ml of blood. Data are displayed as means (with standard errors of the means) of the log10 of bacterial CFU from one (12 h) or three (24 h) independent experiments, with asterisks indicating that P was <0.005. At 24 h postinfection, the frequency of animals containing blood-borne bacteria is indicated above each bar; this difference was statistically significant (P < 0.001).
Liver injury in β2-microglobulin-deficient mice and wild-type mice following intravenous infection.
The significantly impaired clearance of bacteria from the blood, liver, and spleens of β2-m KO mice likely contributes to the increased mortality observed in these animals. To determine if excessive liver injury may also contribute to increased mortality, release of the hepatocyte-associated enzyme AST into peripheral blood was examined (Fig. 2D). At 12 h postinfection, β2-m KO and wild-type infected mice both displayed a similar 30-fold increase in AST levels that were not significantly different from each other. AST levels were lower by 24 h of infection for both animal groups; however, β2-m KO mice displayed statistically higher levels compared to wild-type infected mice. This increased AST activity was possibly due to the approximately 50-fold increase in K. pneumoniae bacteria in the livers of β2-m KO mice at 24 h of infection. Liver histology confirmed the presence of focal hepatocyte cellular injury at 12 and 24 h postinfection. However, no significant differences were noted between wild-type and β2-m KO mice (data not shown). These data suggest that elevated liver injury may possibly contribute to the increased mortality in β2-m KO mice; however, it is unlikely that increased liver injury alone would account for the increased mortality noted in these mice.
Rapid but unaltered production of liver-associated proinflammatory cytokines and chemokines in β2-microglobulin-deficient mice during K. pneumoniae bacteremia.
We have previously reported the rapid induction of liver-associated proinflammatory cytokines and chemokines following induction of K. pneumoniae bacteremia in C57BL/6 mice. Within 6 h of infection, significant induction of hepatic TNF-α, IFN-γ, interleukin-12, monocyte chemoattractant protein 1 (MCP-1), and macrophage inflammatory protein 2 (MIP-2) was observed (13, 15, 16). As β2-m KO mice display increased mortality within 2 days postinfection, we examined at time points prior to animal mortality whether dysregulated cytokine and/or chemokine induction contributed to increased mortality in these animals. Wild-type and β2-m KO mice were inoculated, and at 1, 3, and 6 h after infection hepatic cytokine and chemokine induction was determined by RT-PCR. Proinflammatory cytokine/chemokine induction occurred rapidly following bacterial inoculation in wild-type mice; within 1 h TNF-α, KC, MCP-1, and IFN-inducible protein 10 (IP-10) mRNA levels were readily detected and remained elevated through 6 h postinfection. Interestingly, mRNA induction kinetics in β2-m KO mice were indistinguishable from wild-type mice (Fig. 3). Message levels remained elevated through 24 h postinfection in both wild-type and β2-m KO mice (data not shown). Induction kinetics of IFN-γ, MIP-2, MIP-1α, and monokine-induced IFN-γ were also indistinguishable in both animals groups. Additionally, no differences were observed in splenic cytokine induction between β2-m KO and wild-type mice (data not shown). These data strongly suggest that dysregulated induction of cytokines and chemokines following bacterial infection does not contribute toward the significantly increased rate of mortality seen in β2-m KO mice during K. pneumoniae bacteremia.
FIG. 3.
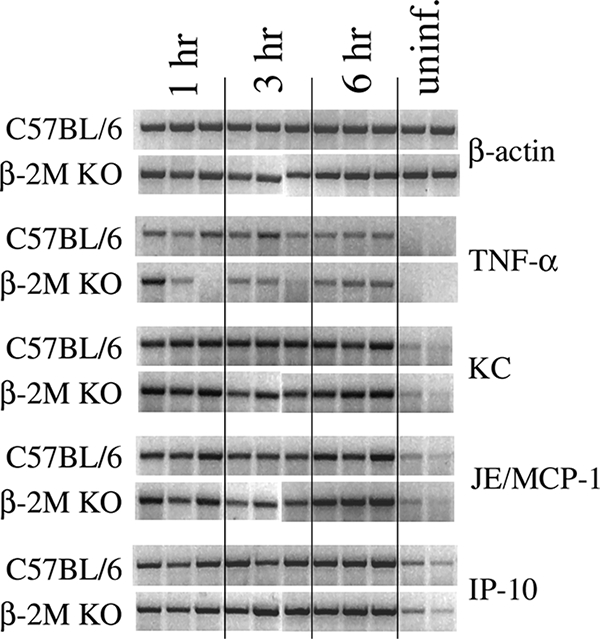
Induction of hepatic proinflammatory cytokines and chemokines during K. pneumoniae bacteremia. Mice were intravenously inoculated with K. pneumoniae and euthanized, and liver RNA was isolated at the indicated time points following infection. Cytokine and chemokine mRNA induction levels were determined as described in Materials and Methods. Three C57BL/6 and three β2-m KO mice were analyzed at each time point, with each lane representing an individual animal.
CD8 T-cell-deficient mice display unaltered susceptibility to K. pneumoniae bacteremia.
Mice deficient in β2-m expression lack surface major histocompatibility complex (MHC) class I antigens and thus lack mature CD8+ T cells (30). We wished to determine whether the specific absence of these cells could account for the increased mortality seen in β2-m KO mice following intravenous K. pneumoniae infection. To address this question we utilized mice genetically deficient in TAP-1. These mice are unable to transport cytosolic peptides into the endoplasmic reticulum for loading into MHC class I molecules and therefore lack both class I expression and CD8+ T cells (11, 12). As can be seen in Fig. 4, TAP-1 KO mice were not more susceptible to K. pneumoniae bacteremia, displaying identical survival rates as C57BL/6 wild-type control mice. To confirm these data, we examined the susceptibility of C57BL/6 wild-type mice depleted of CD8+ T cells by administration of an in vivo depleting monoclonal antibody. These CD8 T-cell-depleted mice were no more susceptible than nondepleted animals following bacterial inoculation (data not shown), in agreement with data generated with TAP-1 KO mice.
FIG. 4.
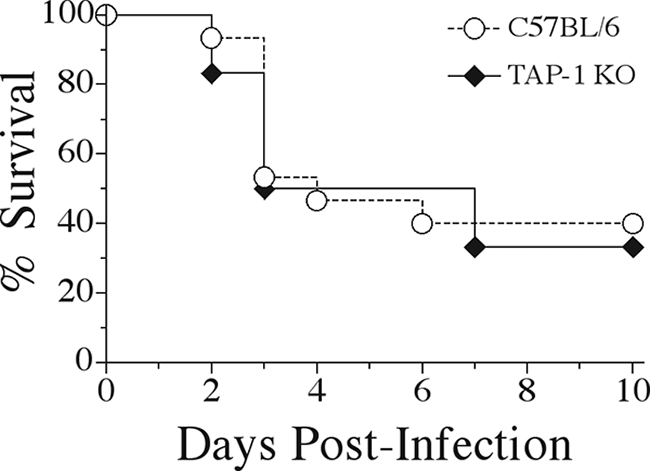
CD8-deficient TAP-1 KO mice display unaltered survival during K. pneumoniae bacteremia. C57BL/6 wild-type and TAP-1 KO mice were intravenously inoculated with 5 × 104 CFU of K. pneumoniae, and survival was observed for 10 days postinfection. Survival curves were generated from two independent experiments using a total of 15 C57BL/6 and 12 TAP-1 KO mice.
These survival data indicated that the absence of mature CD8+ T cells had no detrimental impact during K. pneumoniae bacteremia. To further confirm this observation, we examined bacterial clearance in TAP-1 KO mice or CD8-depleted mice and compared this to control mice 24 h after infection. Neither CD8-deficient mouse model displayed altered bacterial clearance in blood, liver, or spleen tissues when compared to wild-type mice (Table 1). This is in sharp contrast to the data obtained from β2-m KO mice, in which bacterial burden was significantly increased 24 h postinfection (Fig. 2). Additionally, no differences were observed in the levels of plasma AST in CD8-depleted versus wild-type infected animals (data not shown).
TABLE 1.
Bacterial clearance in CD8 T-cell-deficient mice 24 h post-K. pneumoniae intravenous inoculation
| Expt no. and animal group |
K. pneumoniae CFU (log10)a in:
|
||
|---|---|---|---|
| Blood | Liver | Spleen | |
| Expt 1 | |||
| C57BL/6 | 2.48 (0.26) | 6.45 (0.19) | 3.83 (0.38) |
| TAP-1 KO | 2.27 (0.27) | 5.76 (0.21) | 4.15 (0.88) |
| Expt 2 | |||
| C57BL/6 | 0.52 (0.52) | 5.04 (0.65) | 2.13 (0.44) |
| Anti-CD8 treatment | 1.35 (0.5) | 5.19 (0.59) | 2.58 (0.10) |
Bacterial numbers for liver and spleen are for the entire organ, while blood bacterial numbers are per ml of blood. CFU data are displayed as means (with standard errors of means in parentheses) of the log10 bacterial CFU and were generated from 7 to 10 animals per experimental group.
Invariant NK T-cell-deficient mice display unaltered susceptibility to K. pneumoniae bacteremia.
In addition to lacking MHC class I expression, β2-m KO mice also lack expression of CD1d and are therefore deficient in iNK T cells (2, 28). Data generated from CD8 T-cell-deficient mice support the hypothesis that β2-m KO mice may be more susceptible to K. pneumoniae bacteremia due to the lack of iNK T cells rather than from the absence of CD8 T cells. If this were correct, one could postulate that mice specifically deficient in iNK T cells display a similar pattern of susceptibility to that seen in β2-m KO mice. To directly test this hypothesis, CD1d KO mice specifically lacking iNK T cells were examined for susceptibility to K. pneumoniae infection. To our surprise, CD1d KO mice displayed survival rates identical to their CD57BL/6 littermates (Fig. 5). By definition, iNK T cells express a T-cell receptor utilizing the invariant Vα14-Jα281 chain paired with a restricted subset of Vβ chains. Mice deficient in Jα281 have been generated and have been shown to largely parallel CD1d KO mice. When intravenously inoculated, Jα281 KO mice were no more susceptible than the control infected animals, in agreement with the survival data obtained with CD1d KO mice (data not shown).
FIG. 5.
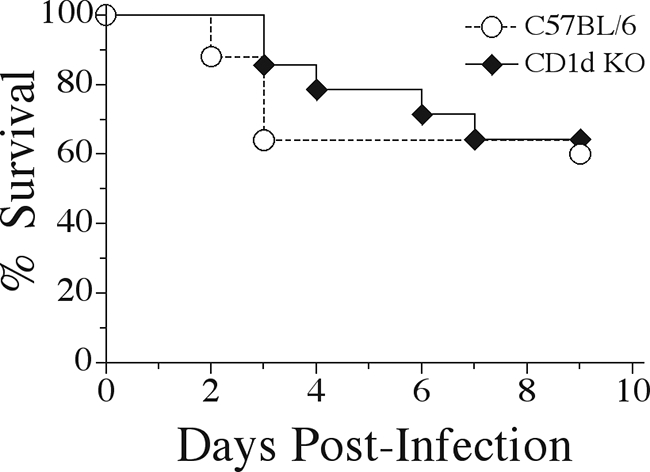
Unaltered survival of CD1d-deficient mice during K. pneumoniae bacteremia. CD1d KO mice and C57BL/6 wild-type mice were intravenously inoculated with K. pneumoniae and monitored for survival over the course of 9 days. Survival curves were generated from three independent experiments using a total of 14 CD1d KO and 24 C57BL/6 mice.
When bacterial clearance in CD1d KO mice was analyzed 24 h postinfection, no differences were observed in blood, liver, and spleen tissues (Table 2). To determine if the lack of increased susceptibility of CD1d KO mice was independent of the mouse genetic background, BALB/c CD1d KO mice were infected and bacterial clearance determined 24 h after inoculation. As was seen on the C57BL/6 background, CD1d KO mice on the BALB/c background contained the same numbers of bacteria in blood, liver, and spleen (data not shown). Again, these results were in sharp contrast to the significant impairment of bacterial clearance noted in β2-m KO mice 24 h postinfection.
TABLE 2.
Bacterial clearance in CD1d-deficient mice 24 h post-K. pneumoniae intravenous inoculation
| Animal group |
K. pneumoniae CFU (log 10)a in:
|
||
|---|---|---|---|
| Blood | Liver | Spleen | |
| C57BL/6 | 0.92 (0.38) | 3.59 (0.33) | 3.22 (0.50) |
| CD1d KO | 0.51 (0.34) | 3.07 (0.49) | 3.01 (0.53) |
Bacterial numbers for liver and spleen are for the entire organ, while blood bacterial numbers are per ml of blood. CFU data are displayed as means (with standard errors of the means in parentheses) of the log10 bacterial CFU and were generated from two independent experiments for a total of 8 to 10 animals per group.
DISCUSSION
Klebsiella pneumoniae is a leading cause of gram-negative bacterial infections. Antibacterial host responses during acute Klebsiella pneumonia have been well-characterized (17, 27). However, a significant clinical complication of pulmonary K. pneumoniae infection is peripheral blood dissemination, resulting in bacteremia concurrent with the localized pulmonary infection (21). Inability to clear blood-borne bacteria can lead to a state of overwhelming bacteremia, which can culminate in multiple organ dysfunction syndrome and increased mortality. We have previously reported on the requirements for the proinflammatory cytokines TNF-α and IFN-γ during Klebsiella bacteremia; however, these studies did not address the cellular source of these and other requisite cytokines (13, 15, 16). Here we report on the critical importance of β2-m expression for survival during blood-borne Klebsiella infection. Mice lacking β2-m display significantly increased mortality to intravenous K. pneumoniae infection that correlates with impaired bacterial clearance from blood, liver, and spleen tissues.
Mice deficient in β2-m expression lack both CD8+ T cells and iNK T cells due to the absence of surface MHC class I and CD1d, respectively (2, 11, 12, 28, 30). Therefore, the increased susceptibility to Klebsiella bacteremia could result from the absence of either one or both of these lymphocyte populations. To determine if the specific absence of CD8+ T cells would recapitulate the data from β2-m KO mice, we utilized two mouse models of CD8 T-cell deficiency. Infection of TAP-1 KO mice resulted in animal survival and bacterial clearance essentially identical to their wild-type counterparts. To confirm these observations that antibacterial defenses during Klebsiella bacteremia are CD8 T-cell independent, we infected mice acutely depleted of CD8+ T cells by the in vivo administration of a depleting anti-CD8 monoclonal antibody. As was observed with TAP-1 KO mice, CD8-depleted animals displayed unaltered survival and bacterial clearance. These data indicate that the absence of CD8 T cells alone does not explain the increased susceptibility of β2-m KO mice. We then addressed the importance of iNK T cells during K. pneumoniae infection by using two mouse models of iNK T-cell deficiency. Surprisingly, neither CD1d-deficient nor Jα281-deficient mice were more susceptible to infection compared to wild-type control animals. Combined, these data indicate that the absence of either CD8 T cells or iNK T cells does not explain the increased susceptibility seen in β2-m KO mice. In a pilot study with limited numbers of animals, we examined the effect on bacterial clearance in anti-CD8-treated CD1d-KO mice. This preliminary experiment suggested that CD8 T-cell-depleted CD1d-KO mice displayed unimpaired bacterial clearance compared to CD1d-KO mice. Combined, these data suggest that β2-m KO mice are more susceptible to Klebsiella bacteremia due to the lack of a β2-m-dependent protein separate from MHC class I and CD1d.
Recently, it has been reported that the human iron overload disease hereditary hemochromatosis results from the absence of the novel β2-m-dependent, MHC class I-like molecule, HFE (5, 18). Mice deficient in β2-m expression have been shown to recapitulate the parenchymal iron overload seen in hereditary hemochromatosis patients (20, 22). Generation of HFE KO mice has confirmed the linkage between β2-m, HFE, and the iron overload phenotype (1, 33). Iron is an essential element for bacterial growth; however, bioavailability of ferric iron is extremely low in mammalian hosts (24). Since β2-m KO mice have excessive iron levels, it is possible that intravenously inoculated K. pneumoniae bacteria have a competitive proliferation advantage in these mice that results in increased bacterial growth and subsequent mortality. A recent report examining the susceptibility of β2-m KO mice to pulmonary Mycobacterium tuberculosis infection supported this possibility (23). Previously it had been shown that β2-m KO mice displayed impaired M. tuberculosis clearance from lung, liver, and spleen and that this impaired clearance was worse than that seen in CD8-deficient, MHC class I-KO, or CD1d-KO mice (3, 26). The authors speculated that excessive iron in β2-m KO mice might impair host immune responses and/or enhance M. tuberculosis growth; therefore, they depleted extracellular iron by administration of the iron chelators lactoferrin or deferoxamine. Iron chelation lowered bacterial numbers down to the levels seen in MHC class I KO mice (23). These results were intriguing, as they closely paralleled our observations using β2-m KO, class I KO, and CD1d KO mice in our Klebsiella bacteremia model, in that β2-m KO mice displayed heightened susceptibility compared to either class I KO or CD1d KO mice. In a pilot study, we treated β2-m KO mice with deferoxamine prior to infection and monitored the animals for survival. However, iron chelation had no survival benefit in β2-m KO mice. This would suggest that the increased susceptibility of β2-m KO mice to Klebsiella blood-borne infections is not due to excessive iron in these animals and therefore is likely to be independent of the expression of HFE.
β2-m is also utilized by other cell surface molecules in addition to MHC class I, CD1d, and HFE. One such candidate is the neonatal Fc receptor (FcRn) (6, 8, 9). The FcRn is responsible for transport of maternal immunoglobulin G (IgG) across the placenta and neonatal intestinal epithelium (31). Additionally, in adult mice it functions to protect plasma IgG from catabolism (10). Two recent reports also suggested a role for FcRn during bacterial infections. FcRn was shown to be required for the bidirectional transport of IgG antibody into the intestinal lumen, allowing retrieval of luminal antigens which were then transported back through intestinal epithelial cells for presentation to dendritic cells. Interestingly, mice deficient in FcRn expression displayed enhanced susceptibility to Citrobacter rodentium infection (32). Selective expression of FcRn on the intestinal epithelium led to reduced susceptibility but only in the presence of circulating pathogen-specific IgG. Recently, FcRn expression in murine neutrophil granules has been shown to relocate to phagolysosomes following phagocytosis of IgG-opsonized bacteria. Neutrophils from β2-m or FcRn KO mice displayed impaired phagocytosis of IgG-opsonized bacteria (29). Of relevance for our model, both of these studies required the presence of bacteria-specific IgG antibodies. It is unclear whether FcRn plays an active role in our model, as it is unlikely that preexisting anti-K. pneumoniae IgG antibodies would be present in our experimental animals. However, this has not been formally addressed.
In summary, we report here on the critical importance of β2-microglobulin expression during murine K. pneumoniae bacteremia. β2-m KO mice display significantly increased mortality upon intravenous inoculation that correlates with increased bacterial burdens in blood, liver, and spleen. Surprisingly, neither CD8 T-cell-deficient (TAP-1 KO; in vivo anti-CD8 antibody treatment) nor iNK T-cell-deficient mice (CD1d KO, Jα281 KO) were more susceptible to K. pneumoniae blood-borne infection. Furthermore, preliminary data do not support a role for the β2-m-dependent, MHC class I-like molecule HFE, whose absence is responsible for the human iron overload disease hereditary hemochromatosis. Further studies will be required to determine the specific β2-m-dependent but CD8 T-cell-independent and iNK T-cell-independent mechanisms critical for survival during K. pneumoniae bacteremia.
Acknowledgments
This work was supported in part by grants AI49448 (T.A.M.) from the National Institutes of Health and a Career Investigator Award from the American Lung Association (T.A.M.).
We are indebted to Luc Van Kaer and Chyung-Ru Wang for their willingness to provide CD1d and Jα281 KO mice and Gary Huffnagle for critical review of the manuscript.
Editor: R. P. Morrison
Footnotes
Published ahead of print on 3 November 2008.
REFERENCES
- 1.Bahram, S., S. Gilfillan, L. C. Kuhn, R. Moret, J. B. Schulze, A. Lebeau, and K. Schumann. 1999. Experimental hemochromatosis due to MHC class I HFE deficiency: immune status and iron metabolism. Proc. Natl. Acad. Sci. USA 9613312-13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berzins, S. P., M. J. Smyth, and D. I. Godfrey. 2005. Working with NKT cells—pitfalls and practicalities. Curr. Opin. Immunol. 17448-454. [DOI] [PubMed] [Google Scholar]
- 3.D'Souza, C. D., A. M. Cooper, A. A. Frank, S. Ehlers, J. Turner, A. Bendelac, and I. M. Orme. 2000. A novel nonclassic β2-microglobulin-restricted mechanism influencing early lymphocyte accumulation and subsequent resistance to tuberculosis in the lung. Am. J. Respir. Cell Mol. Biol. 23188-193. [DOI] [PubMed] [Google Scholar]
- 4.Emoto, M., M. Miyamoto, I. Yoshizawa, Y. Emoto, U. E. Schaible, E. Kita, and S. H. Kaufmann. 2002. Critical role of NK cells rather than V alpha 14+ NKT cells in lipopolysaccharide-induced lethal shock in mice. J. Immunol. 1691426-1432. [DOI] [PubMed] [Google Scholar]
- 5.Enns, C. A. 2001. Pumping iron: the strange partnership of the hemochromatosis protein, a class I MHC homolog, with the transferrin receptor. Traffic 2167-174. [DOI] [PubMed] [Google Scholar]
- 6.Firan, M., R. Bawdon, C. Radu, R. J. Ober, D. Eaken, F. Antohe, V. Ghetie, and E. S. Ward. 2001. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int. Immunol. 13993-1002. [DOI] [PubMed] [Google Scholar]
- 7.Flynn, J. L., M. M. Goldstein, K. J. Triebold, B. Koller, and B. R. Bloom. 1992. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 8912013-12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghetie, V., and E. S. Ward. 2000. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu. Rev. Immunol. 18739-766. [DOI] [PubMed] [Google Scholar]
- 9.Israel, E. J., D. F. Wilsker, K. C. Hayes, D. Schoenfeld, and N. E. Simister. 1996. Increased clearance of IgG in mice that lack beta 2-microglobulin: possible protective role of FcRn. Immunology 89573-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Junghans, R. P., and C. L. Anderson. 1996. The protection receptor for IgG catabolism is the β2-microglobulin-containing neonatal intestinal transport receptor. Proc. Natl. Acad. Sci. USA 935512-5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lankat-Buttgereit, B., and R. Tampe. 1999. The transporter associated with antigen processing TAP: structure and function. FEBS Lett. 464108-112. [DOI] [PubMed] [Google Scholar]
- 12.Lankat-Buttgereit, B., and R. Tampe. 2002. The transporter associated with antigen processing: function and implications in human diseases. Physiol. Rev. 82187-204. [DOI] [PubMed] [Google Scholar]
- 13.Moore, T. A., H. Y. Lau, A. L. Cogen, C. L. Monteleon, and T. J. Standiford. 2003. Anti-tumor necrosis factor-alpha therapy during murine Klebsiella pneumoniae bacteremia: increased mortality in the absence of liver injury. Shock 20309-315. [DOI] [PubMed] [Google Scholar]
- 14.Moore, T. A., B. B. Moore, M. W. Newstead, and T. J. Standiford. 2000. Gamma delta-T cells are critical for survival and early proinflammatory cytokine gene expression during murine Klebsiella pneumonia. J. Immunol. 1652643-2650. [DOI] [PubMed] [Google Scholar]
- 15.Moore, T. A., M. L. Perry, A. G. Getsoian, C. L. Monteleon, A. L. Cogen, and T. J. Standiford. 2003. Increased mortality and dysregulated cytokine production in tumor necrosis factor receptor 1-deficient mice following systemic Klebsiella pneumoniae infection. Infect. Immun. 714891-4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore, T. A., M. L. Perry, A. G. Getsoian, M. W. Newstead, and T. J. Standiford. 2002. Divergent role of gamma interferon in a murine model of pulmonary versus systemic Klebsiella pneumoniae infection. Infect. Immun. 706310-6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore, T. A., and T. J. Standiford. 2001. Cytokine immunotherapy during bacterial pneumonia: from benchtop to bedside. Semin. Respir. Infect. 1627-37. [DOI] [PubMed] [Google Scholar]
- 18.Parkkila, S., O. Niemela, R. S. Britton, R. E. Fleming, A. Waheed, B. R. Bacon, and W. S. Sly. 2001. Molecular aspects of iron absorption and HFE expression. Gastroenterology 1211489-1496. [DOI] [PubMed] [Google Scholar]
- 19.Podschun, R., and U. Ullmann. 1998. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11589-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothenberg, B. E., and J. R. Voland. 1996. β2 knockout mice develop parenchymal iron overload: a putative role for class I genes of the major histocompatibility complex in iron metabolism. Proc. Natl. Acad. Sci. USA 931529-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahm, D. F., M. K. Marsilio, and G. Piazza. 1999. Antimicrobial resistance in key bloodstream bacterial isolates: electronic surveillance with the Surveillance Network Database—USA. Clin. Infect. Dis. 29259-263. [DOI] [PubMed] [Google Scholar]
- 22.Santos, M., M. W. Schilham, L. H. Rademakers, J. J. Marx, M. de Sousa, and H. Clevers. 1996. Defective iron homeostasis in beta 2-microglobulin knockout mice recapitulates hereditary hemochromatosis in man. J. Exp. Med. 1841975-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaible, U. E., H. L. Collins, F. Priem, and S. H. Kaufmann. 2002. Correction of the iron overload defect in beta-2-microglobulin knockout mice by lactoferrin abolishes their increased susceptibility to tuberculosis. J. Exp. Med. 1961507-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaible, U. E., and S. H. Kaufmann. 2004. Iron and microbial infection. Nat. Rev. 2946-953. [DOI] [PubMed] [Google Scholar]
- 25.Sherwood, E. R., C. Y. Lin, W. Tao, C. A. Hartmann, J. E. Dujon, A. J. French, and T. K. Varma. 2003. Beta 2 microglobulin knockout mice are resistant to lethal intraabdominal sepsis. Am. J. Respir. Crit. Care Med. 1671641-1649. [DOI] [PubMed] [Google Scholar]
- 26.Sousa, A. O., R. J. Mazzaccaro, R. G. Russell, F. K. Lee, O. C. Turner, S. Hong, L. Van Kaer, and B. R. Bloom. 2000. Relative contributions of distinct MHC class I-dependent cell populations in protection to tuberculosis infection in mice. Proc. Natl. Acad. Sci. USA 974204-4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Standiford, T. J., W. C. Tsai, B. Mehrad, and T. A. Moore. 2000. Cytokines as targets of immunotherapy in bacterial pneumonia. J. Lab. Clin. Med. 135129-138. [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi, M., M. Harada, S. Kojo, T. Nakayama, and H. Wakao. 2003. The regulatory role of Vα14 NKT cells in innate and acquired immune response. Annu. Rev. Immunol. 21483-513. [DOI] [PubMed] [Google Scholar]
- 29.Vidarsson, G., A. M. Stemerding, N. M. Stapleton, S. E. Spliethoff, H. Janssen, F. E. Rebers, M. de Haas, and J. G. van de Winkel. 2006. FcRn: an IgG receptor on phagocytes with a novel role in phagocytosis. Blood 1083573-3579. [DOI] [PubMed] [Google Scholar]
- 30.Williams, A., C. A. Peh, and T. Elliott. 2002. The cell biology of MHC class I antigen presentation. Tissue Antigens 593-17. [DOI] [PubMed] [Google Scholar]
- 31.Yoshida, M., S. M. Claypool, J. S. Wagner, E. Mizoguchi, A. Mizoguchi, D. C. Roopenian, W. I. Lencer, and R. S. Blumberg. 2004. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 20769-783. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida, M., K. Kobayashi, T. T. Kuo, L. Bry, J. N. Glickman, S. M. Claypool, A. Kaser, T. Nagaishi, D. E. Higgins, E. Mizoguchi, Y. Wakatsuki, D. C. Roopenian, A. Mizoguchi, W. I. Lencer, and R. S. Blumberg. 2006. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Investig. 1162142-2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou, X. Y., S. Tomatsu, R. E. Fleming, S. Parkkila, A. Waheed, J. Jiang, Y. Fei, E. M. Brunt, D. A. Ruddy, C. E. Prass, R. C. Schatzman, R. O'Neill, R. S. Britton, B. R. Bacon, and W. S. Sly. 1998. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA 952492-2497. [DOI] [PMC free article] [PubMed] [Google Scholar]