

Abstract
The bipartite anthrax lethal toxin (LeTx) consisting of protective antigen (PA) and lethal factor (LF) is a major virulence factor contributing to death from systemic Bacillus anthracis infection. The current vaccine elicits antibodies directed primarily to PA; however, in experimental settings serologic responses to LF can neutralize LeTx and contribute to protection against infection. The goals of the present study were to identify sequential B-cell epitopes of LF and to determine the capacity of these determinants to bind neutralizing antibodies. Sera of recombinant LF-immunized A/J mice exhibited high titers of immunoglobulin G anti-LF reactivity that neutralized LeTx in vitro 78 days after the final booster immunization and protected the mice from in vivo challenge with 3 50% lethal doses of LeTx. These sera bound multiple discontinuous epitopes, and there were major clusters of reactivity on native LF. Strikingly, all three neutralizing, LF-specific monoclonal antibodies tested bound specific peptide sequences that coincided with sequential epitopes identified in polyclonal antisera from recombinant LF-immunized mice. This study confirms that LF induces high-titer protective antibodies in vitro and in vivo. Moreover, the binding of short LF peptides by LF-specific neutralizing monoclonal antibodies suggests that generation of protective antibodies by peptide vaccination may be feasible for this antigen. This study paves the way for a more effective anthrax vaccine by identifying discontinuous peptide epitopes of LF.
The use of anthrax spores as a bioterror weapon in 2001 highlighted the importance of obtaining an improved understanding of Bacillus anthracis disease pathogenesis and developing improved vaccination strategies. B. anthracis is a gram-positive, rod-shaped, spore-forming bacterium that is commonly found in soil and is the causative agent of anthrax infection (47). Humans can become infected by the cutaneous, gastrointestinal, and inhalation routes of exposure to spores. Inhalational anthrax, if left untreated, usually leads to fatal systemic disease with a mortality approaching 100% (48).
The virulence of B. anthracis is attributable to two major virulence factors: a poly-d-glutamic acid capsule and a secreted tripartite protein complex toxin (12). The tripartite protein complex consists of lethal factor (LF), edema factor (EF), and protective antigen (PA). Individually, none of the three components is toxic, but the combination of PA and LF forms lethal toxin (LeTx) and the combination of PA and EF forms edema toxin (11). The 83-kDa PA component is responsible for binding to and toxin entry into host cells (6) by two different receptors expressed ubiquitously on many cell types (8, 38). Surface proteases, such as furin, cleave PA into 20- and 63-kDa components, allowing the 63-kDa protein to heptamerize and bind to three molecules of LF, EF, or both in a competitive manner (32). This complex is internalized and, upon endosome acidification, forms a pore that delivers the individual toxin components into the cytosol (30, 31, 40). Although substantial work has been done to understand the biology and pathogenic mechanisms of this infectious agent, considerable gaps in our understanding of the immune response to B. anthracis remain.
LF is a 90-kDa, zinc-dependent metalloprotease that specifically cleaves and inactivates mitogen-activated protein kinase kinases, leading to the inhibition of one or more signaling pathways (14, 44, 45). LF has been shown to induce vascular collapse and subsequent hypoxia-mediated toxicity in mice (2). It is also known to subvert host immunity by inducing apoptosis in macrophages and dendritic cells and is ultimately lethal to mice (2). Studies have revealed that even when aggressive, early antibiotic therapy eradicates the bacterial load within 72 h, these toxins are still present at concentrations sufficient to cause death (13, 19). Since death can result even when the bacteria have been cleared from the system, vaccine development is essential to prevent or stop infection at an early stage.
Vaccine development has been a key part of protection against anthrax infection for many years. In 1881, Pasteur used a vaccine that was attenuated by growing vegetative B. anthracis organisms at raised temperatures to protect livestock against anthrax (39). After over 50 years of broad use, this vaccine was replaced by a more stable live attenuated spore vaccine in the 1930s, when Sterne isolated a B. anthracis variant that produces the toxins but lacks the capsule. The Sterne strain vaccine has been safe and effective for domestic animals, but there are still concerns about the safety of using a live vaccine for humans (39). Currently, the only human vaccine available in the United States, anthrax vaccine absorbed (AVA), contains mainly PA as the protective component, as shown by the general lack of antibodies to LF and EF constituents in vaccinees (3, 5, 43). AVA is an aluminum hydroxide-adsorbed, formalin-treated culture supernatant of a toxigenic, nonencapsulated, nonproteolytic B. anthracis strain derived from the Sterne strain (36). The existing vaccine has many disadvantages, including a requirement for containment facilities for production, batch-to-batch variation of the protective bacterial components, a limited duration of protection that requires subcutaneous injections at 0, 2, and 4 weeks and 6, 12, and 18 months and then subsequent yearly boosters for continued immunity, and transient reactogenicity in some vaccinees (17, 18, 47).
While it is clear that PA is the primary protective component in the currently licensed vaccine, studies in which mice were immunized with mutant strains of B. anthracis that did not produce individual toxin components revealed the significant individual contributions of antibodies to LF and EF to immunoprotection (35). Several studies have shown that passive transfer of neutralizing polyclonal or monoclonal antibodies (MAb) to LF can protect experimental animals from challenge with anthrax toxin or bacteria (1, 9, 23, 27, 41, 51). Moreover, the protective efficacy of neutralizing antibodies was greatly enhanced by using a combination of PA- and LF-neutralizing antibodies (9). Although a recombinant PA vaccine is currently in clinical trials, this vaccine does not elicit antibodies to LF and EF (4). The fine specificities of LF-neutralizing MAbs have not been delineated to our knowledge (1, 27, 41, 51).
The purposes of this study were to identify sequential B-cell epitopes within LF and to further identify subsets of these epitopes that may be candidates for induction of neutralizing antibodies by peptide vaccination. Although some B-cell epitopes are conformational in nature, others are comprised of a single continuous stretch of amino acids. The latter category of sequential epitopes is useful in rational design of peptide vaccines, and these epitopes have been demonstrated to elicit neutralizing or protective antibodies to viruses (15, 21, 25, 42), bacteria (10), and parasites (22). Addition of key LF humoral epitopes to a recombinant PA vaccine is expected to increase the titers of LeTx-neutralizing antibodies in the sera of vaccinees and thus provide enhanced efficacy. We hypothesized that neutralizing antibodies to LF would recognize discrete, sequential peptide sequences of LF and that these antibodies would be capable of neutralizing LeTx activity. We found that immunization of A/J mice with recombinant LF (rLF) generated high antibody titers capable of neutralizing toxin in vitro and in vivo. Furthermore, neutralizing LF-specific MAbs 10G3, 9A11, and LF8 (27, 41, 51) bound specific peptide sequences that overlapped or coincided with epitopes bound by neutralizing polyclonal sera of mice immunized with rLF.
MATERIALS AND METHODS
Mice.
Six-week-old A/J strain female mice were purchased from Jackson Laboratories (Bar Harbor, ME). The mice were housed in the Oklahoma Medical Research Foundation Chapman Animal Facility. All mouse experiments were approved by the Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee.
Immunization and blood sampling.
For antibody studies, experimental mice were immunized subcutaneously with rLF protein (100 μg) emulsified 1:1 in complete Freund's adjuvant (Difco, Lawrence, KS) on day 0 using a previously described immunization schedule (16). Mice were boosted three times with 50 μg of the immunizing protein emulsified 1:1 in incomplete Freund's adjuvant on days 10, 24, and 38. Control mice were similarly immunized and boosted with adjuvant alone. Retroorbital bleeding of anesthetized mice was performed on days 14, 28, 42, and 116.
Production of rLF protein.
rLF protein was produced as an amino-terminal His6-tagged protein (37, 46). Cultures of BL-21/pET15b (containing LF cDNA from B. anthracis) were grown in Luria broth containing ampicillin (50 μg/ml) to an optical density at 600 nm (OD600) of 0.6 to 0.8, and protein expression was induced by addition of 1 mM isopropyl β-d-thiogalactoside (IPTG) and incubation overnight at 16°C. Cells were then pelleted and disrupted with a French press. The lysed cells were then centrifuged, and the supernatant was passed over a Ni2+-charged column equilibrated with a binding buffer (Novagen, Gibbstown, NJ). The bound fusion protein was removed with 0.5 M imidazole according to the manufacturer's instructions (Novagen, Gibbstown, NJ). The eluted protein was then dialyzed against 20 mM Tris-HCl (pH 7.5). The protein concentration was determined by a standard Bradford assay (Bio-Rad, Hercules, CA) and stored at 4°C on ice. Purified LF (20 μg) was electrophoresed in a 12.5% sodium dodecyl sulfate-polyacrylamide gel and visualized by Coomassie blue staining to ensure the purity of the sample.
Assessment of LeTx activity.
RAW 264.7 mouse macrophages were grown in 96-well plates (1 × 105 cells per well) in 100 μl of Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum, 2 mM Glutamax, 2 mM HEPES, and 50 μg/ml gentamicin at 37°C in a 5% CO2-95% air atmosphere. To assess the functionality of the protein, LeTx was added at a PA-to-LF ratio of 3:1 (75 ng/well PA and 25 ng/well LF; final well volume, 100 μl) and serially diluted 1:2 to assess the point at which toxin activity decreased. Cells treated with medium alone served as negative controls. An additional set of controls involved treatment of cells with LF alone and with PA alone at each concentration. All experiments were done in duplicate. Cells were incubated for 2 h at 37°C, followed by addition of WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] (Dojindo Molecular Technologies, Inc., Gaithersburg, MD) at a concentration equivalent to 0.01% of the total volume of the wells. OD450 measurements for wells that were subjected to different treatments were analyzed to assess whether LeTx significantly affected RAW 264.7 cell viability.
Standard enzyme-linked immunosorbent assay (ELISA).
Ninety-six-well microtiter plates (Corning CoStar, Lowell, MA) were coated with 2.5 μg of antigen/ml in carbonate coating buffer (0.125 μg of antigen/well; pH 9.6) overnight at 4°C. The plates were then washed four times with 0.05% Tween 20 in phosphate-buffered saline (PBS) and blocked with 0.1% bovine serum albumin-0.2% NaN3 in PBS for 1 h at room temperature. The plates were washed again, and appropriately diluted samples were added and incubated for 2 h at room temperature. After washing, alkaline phosphatase-labeled goat anti-mouse immunoglobulin G (IgG) (Sigma, Saint Louis, MO) diluted 1:5,000 was added. After 2 h of incubation at room temperature, ρ-nitrophenyl phosphate (Sigma, St. Louis, MO) in substrate buffer (0.167 M NaHCO3, 0.012 M Na2CO3, 0.001 M MgCl2, 0.02% NaN3) was added to the plates, and the OD410 and OD490 of the plates were determined.
In vitro LeTx neutralization assay.
RAW 264.7 mouse macrophages were grown in Dulbecco's modified Eagle medium containing 4 mM l-glutamine (ATCC) supplemented with 10% fetal calf serum at 37°C in a 5% CO2-95% air atmosphere. To determine toxin neutralization, 1 × 105 cells were plated in each well of a 96-well flat-bottom tissue culture plate and cultured overnight at 37°C with 5% CO2. The next day, serum samples were diluted to obtain specific concentrations in culture medium and incubated for 1 h at room temperature with LeTx (composed of PA and LF at a 3:1 ratio [75 ng/well PA and 25 ng/well LF; final well volume, 100 μl]). Following incubation, the medium was removed from the cultured cells, and 100 μl of the serum-toxin mixture was added to each well. Cells treated with medium alone served as negative controls. An additional set of controls involved treatment of cells with LF alone, with PA alone, and with LeTx alone. All experiments were carried out in duplicate. Following addition of the serum-toxin mixture, the cells were incubated for 2 h at 37°C with 5% CO2. After 2 h, 10 μl of WST-8 (CCK-8; Dojindo Molecular Technologies, Inc., Gaithersburg, MD) was added to each well and incubated for an additional 3 h at 37°C with 5% CO2. The OD450 was determined using a Dynex MRX II microplate reader (Dynex Technologies, Chantilly, VA). The percentage of viability was calculated based on the absorbance for the control wells containing only cells (medium alone).
In vivo LeTx challenge.
rLF and PA were produced as described above. rLF-immunized mice and mice treated with adjuvant alone were challenged with 3 A/J 50% lethal doses (LD50) of LeTx (300 μg PA plus 125 μg LF; experimentally determined). Mice were then monitored for 7 days, and mortality was recorded. Survival curves were generated and percentages of survival were determined by using GraphPad Prism (GraphPad Software Inc., La Jolla, CA).
Solid-phase ELISAs.
To determine the fine specificity of the anti-LF antibodies, a solid-phase peptide ELISA was used. Peptides that were 10 amino acids long, overlapped by 8 amino acids, and covered the entire length of the LF protein (GenBank accession number AAM26117) were covalently synthesized on polyethylene pins using a 96-well format as previously described (28, 29). Washing and incubation were carried out in sealed plastic containers. Other assay steps were performed by lowering the pins into microtiter plate wells. First, the pins were blocked with 3% low-fat milk in PBS for 1 h at room temperature. The pins were then incubated in 1:200 dilutions of sera in 3% milk-PBS with 0.05% Tween for 2 h at room temperature, washed with PBS-Tween, and incubated with a 1:20,000 dilution of horseradish peroxidase-labeled anti-human IgG conjugate (Kirkegaard & Perry Laboratories [KPL], Gaithersburg, MD) at 4°C overnight. Following washing, the pins were incubated with SureBlue Reserve TMB microwell peroxidase substrate (KPL) for 5 min at room temperature, and the reaction was terminated by addition of TMB stop solution (KPL). The OD450 was determined by using a Dynex MRX II microplate reader (Dynex Technologies). The results for each plate were then standardized by comparison with the results for positive control pins. The same control pins were used for all plates and were developed to a specific optical density with a known concentration of a standard control serum. After completion of an assay, the pins were sonicated for 2 h in sonication buffer (40 g sodium dodecyl sulfate, 4 ml β-mercaptoethanol, and 62.4g sodium phosphate in 4 liters) to remove the antibodies, conjugate, and substrate. After sonication, the pins were washed twice in hot water and boiled in methanol for 2 min. The pins were then allowed to air dry for 15 min and were stored with desiccant or used for another assay. An epitope was defined as two or more consecutive solid-phase ELISA pins exhibiting an OD450 greater than or equal to the average OD450 plus 3 standard deviations (SD) for “adjuvant-only” sera on all decapeptides.
Screening of LF MAbs.
LF-specific MAbs were obtained from various sources and used to determine if a neutralizing MAb could bind linear peptides. MAb 10G3 (commercial name, MAb 291) was an antibody that was provided by the Biodefense and Emerging Infections Research Resources Repository (BEI, Manassas, VA). MAb 10G3 was an antibody produced and characterized by Little et al. (27), MAb LF 9A11 was an antibody produced and characterized by Staats et al. (41), and MAb LF8 was an antibody produced and characterized by Zhao et al. (51). All antibodies were screened as described above for rLF-specific polyclonal antisera.
Assay for in vitro peptide inhibition of neutralization.
Peptides (10-mers) (see Fig. 4B) bound by MAbs were synthesized as linear peptides by using standard Fmoc chemistry and were purified by high-performance liquid chromatography to obtain ≥95% purity (GenScript, Piscataway, NJ). Peptide inhibition of MAb-mediated toxin neutralization was achieved by titrating various concentrations of peptides into tubes containing the corresponding MAbs at fixed concentrations. The peptide-antibody mixtures were incubated for 30 min at room temperature. After incubation, LeTx (at the concentration used in the in vitro neutralization assay described above) was added to each tube and incubated for 30 min at room temperature. The concentration of each MAb was chosen so that it was the last 2× dilution exhibiting maximal neutralization capacity with a starting MAb concentration of 5 μg/ml. Following incubation, the medium was removed from the cultured cells, and 100 μl of the peptide-antibody-toxin mixture was added to each well. Cells treated with medium alone served as negative controls. All experiments were carried out in duplicate. Following addition of the peptide-antibody-toxin mixture, the cells were incubated for 2 h at 37°C with 5% CO2. After 2 h, 10 μl of WST-8 (CCK-8; Dojindo Molecular Technologies, Inc., Gaithersburg, MD) was added to each well and incubated for an additional 3 h at 37°C with 5% CO2. The OD450 was determined using a Dynex MRX II microplate reader (Dynex Technologies, Chantilly, VA). The percentage of viability was calculated based on the absorbance of the control wells containing a mixture of irrelevant control peptide, antibody, and toxin.
FIG. 4.

Neutralizing LF-specific MAbs bind sequential B-cell epitopes of LF. (A) Anti-LF MAbs 10G3, 9A11, and LF8 (27, 41, 51) neutralize anthrax LeTx in vitro. (B) Epitope mapping of LF-specific, LeTx-neutralizing MAbs 10G3, 9A11, and LF8. The sequences of bound decapeptides, the numbers of SD above the background level, and the corresponding epitopes shown in Table 1 are indicated. The colored letters correspond to the colored letters in panel C. (C) Structural representation of five epitopes bound by neutralizing LF-specific MAbs. The colored letters correspond to the colored letters in panel B. Two residues of epitope c are not resolved in the LF crystal structure and thus are not shown. (D) Linear peptides corresponding to MAbs 10G3, 9A11, and LF8 mediate measurable reductions in neutralization in vitro (striped bars). Inhibition of neutralization is compared to the results for the same MAb mixed with toxin and irrelevant peptide from ovalbumin (OVA) (open bars). All experiments were carried out in duplicate. Since duplicate values were nearly identical, error bars are not shown. The concentrations of MAbs used for peptide inhibition (see Materials and Methods) were 1.25 μg/ml for MAb 9A11 and 5 μg/ml for MAbs 10G3 and LF8.
RESULTS
rLF immunization results in high titers of neutralizing anti-LF antibodies and protection from LeTx challenge.
Groups of A/J mice were immunized on day 0 with rLF in complete Freund's adjuvant and boosted with rLF in incomplete Freund's adjuvant on days 10, 24, and 38. Sera were collected 3 to 4 days after each boost and again at day 116 to determine the IgG anti-LF antibody levels by ELISA. The antibody titers were high by 2 weeks after the initial immunization and reached maximum levels (>1 × 106) by day 28 (Fig. 1A). The titers of anti-rLF IgG were relatively undiminished at day 116, nearly 80 days after the final booster immunization. Sera collected from mice immunized with adjuvant alone (control group) failed to bind LF antigen, as expected.
FIG. 1.

Immunization of A/J mice results in high-titer anti-LF antibodies that are neutralizing in vitro and protective in vivo. A/J mice were immunized with either rLF or adjuvant alone on days 10, 24, and 38 and bled on days 14, 28, 42, and 116. (A) IgG anti-LF antibody titers of pooled sera from A/J mice immunized with rLF or adjuvant alone were assessed at specified time points to quantitate the IgG anti-LF response. Antibody levels were measured by using a standard ELISA, OD410, and OD490. (B) Sera collected were subjected to an in vitro neutralization assay using a 3:1 ratio of PA to LF (LeTx). Sera exhibited near-maximal neutralization of toxin activity at day 116 (78 days after the final booster immunization). (C) A/J mice immunized with rLF are protected from an in vivo LeTx challenge using 3 times the A/J LD50 derived experimentally (300 μg PA plus 125 μg LF) for LeTx (P = 0.009).
To determine whether the antisera of rLF-immunized A/J mice contained LeTx-neutralizing activity, serum samples obtained at various time points were assessed for the capacity to neutralize LeTx in vitro in a RAW 264.7 mouse macrophage cell death assay. Various dilutions of antisera were preincubated with a LeTx concentration (3:1 molar ratio of PA to LF) previously determined to result in maximal cell death and then added to RAW 264.7 mouse macrophage cultures. Measurement of macrophage cell viability at 24 h revealed the presence of significant neutralizing antibodies at day 14 (75% viability at a 1:300 serum dilution) and maximal titers of neutralizing antibodies as late as day 116 (100% protection at a 1:900 dilution) (Fig. 1B). Sera from mice immunized with adjuvant alone exhibited no capacity to neutralize LeTx activity.
Groups of rLF-immunized mice and controls immunized using the same schedule but with PBS-adjuvant alone were then challenged in an in vivo setting with three times the experimentally derived A/J LD50 for LeTx (300 μg PA and 125 μg LF) (data not shown) to determine whether the anti-LF responses raised could protect mice from LeTx in vivo. As shown in Fig. 1C, seven of eight A/J mice immunized with rLF were protected from LeTx challenge, while only one of seven A/J controls immunized with adjuvant alone survived the challenge (P = 0.009). These findings reveal that rLF induces a robust immune response that results in neutralization of toxin and protection both in vitro and in vivo, further supporting previously described data (1, 9, 23, 27, 41, 51) indicating that LF can provide protection against a LeTx challenge.
Sequential mouse B-cell epitopes cluster in PA and substrate-binding domains of LF.
To analyze the fine specificity of the neutralizing antisera from rLF-immunized A/J mice, we subjected their sera to solid-phase peptide mapping utilizing a modified ELISA performed with a series of decapeptides that overlapped by eight amino acids and represented the whole protein. Reactive epitopes were defined by carrying out three independent immunization and mapping experiments. Figure 2 shows the mapping results for LF-immunized A/J mice for day 14, 28, and 42 bleeds compared to the day 42 results for mice immunized with adjuvant alone (control group). A total of 16 major antigenic regions throughout the molecule were identified. All 16 epitopes defined at day 42 were still present at day 116, although the intensities of epitopes 6, 10, 11, and 15 had increased by that time (data not shown). Mice immunized with adjuvant alone did not significantly bind any of the peptides. An antigenic epitope was defined as described in Materials and Methods. These epitopes were ranked in order of reproducibility and reactivity based on the results of three separate immunization and mapping experiments. The reproducibility and optical density (average ± standard error of the mean) are shown for each defined epitope in Table 1.
FIG. 2.

Fine specificity of the humoral anti-LF response following A/J immunization. IgG antibody binding to decapeptides of LF overlapping by eight amino acids was measured using a solid-phase ELISA. The data are the antibody responses in sera from rLF-adjuvant-immunized mice on days 14, 28, and 42 and in sera from mice immunized with adjuvant alone. The data are the averages of the results of three independent immunization and mapping experiments. Mapping within each experiment was performed using sera pooled from 18 to 20 mice. Epitopes were numbered in order of reproducibility and reactivity at day 42, as shown in Table 1. The epitope numbers correspond to the epitope numbers in Tables 1 and 2. The horizontal lines indicate extended antigenic regions of epitopes 3 and 9 as defined at day 42.
TABLE 1.
LF epitope reproducibility
| Epitope no. | Decapeptide no. | Amino acid no. | Epitope reproducibilitya | Optical density (avg ± SEM)b |
|---|---|---|---|---|
| 1 | 18-20 | 35-48 | 3/3 | 1.23 ± 0.517435 |
| 2 | 173-174 | 345-356 | 3/3 | 1.49 ± 0.310315 |
| 3 | 133-146 | 265-300 | 3/3 | 0.841 ± 0.19005 |
| 4 | 192-193 | 383-394 | 3/3 | 0.811 ± 0.25580 |
| 5 | 181-183 | 361-374 | 3/3 | 0.751 ± 0.22844 |
| 6 | 217-219 | 433-446 | 2/3 | 0.512 ± 0.13748 |
| 7 | 28-33 | 55-74 | 2/3 | 0.408 ± 0.14685 |
| 8 | 68-77 | 135-162 | 2/3 | 0.335 ± 0.12405 |
| 9 | 335-340 | 669-678 | 2/3 | 0.362 ± 0.0894 |
| 10 | 324-331 | 647-668 | 2/3 | 0.226 ± 0.1341 |
| 11 | 50-53 | 99-114 | 1/3 | 0.649 ± 0.36971 |
| 12 | 246-249 | 491-506 | 1/3 | 0.435 ± 0.25005 |
| 13 | 395-399 | 789-806 | 1/3 | 0.433 ± 0.31542 |
| 14 | 108-117 | 215-242 | 1/3 | 0.424 ± 0.32111 |
| 15 | 203-204 | 405-416 | 1/3 | 0.421 ± 0.22485 |
| 16 | 371-373 | 741-754 | 1/3 | 0.345 ± 0.29422 |
Epitope reproducibility is based on the number of times that a region was identified as an epitope in three independent immunization and mapping experiments. An epitope was defined as described in Materials and Methods.
Values obtained using the results of three independent epitope mapping studies.
The 16 identified epitopes shown in Fig. 2 are superimposed on the LF crystal structure in Fig. 3. Sequences, domain locations, and proposed functions of the epitopes are shown in Table 2. Interestingly, of the 16 epitopes identified by this method, the most reproducible epitopes tended to cluster in domains I (PA-binding domain) and III (substrate recognition domain, which has some residues that contact the mitogen-activated protein kinase kinase [or MEK] substrate) (Fig. 3 and Table 2). Specifically, five epitopes were in the PA-binding domain, one epitope overlapped the PA-binding domain and the VIP2-like domain, four epitopes were in the substrate recognition domain, two epitopes were in the VIP2-like domain, and four epitopes were in the zinc-binding domain (Table 2).
FIG. 3.
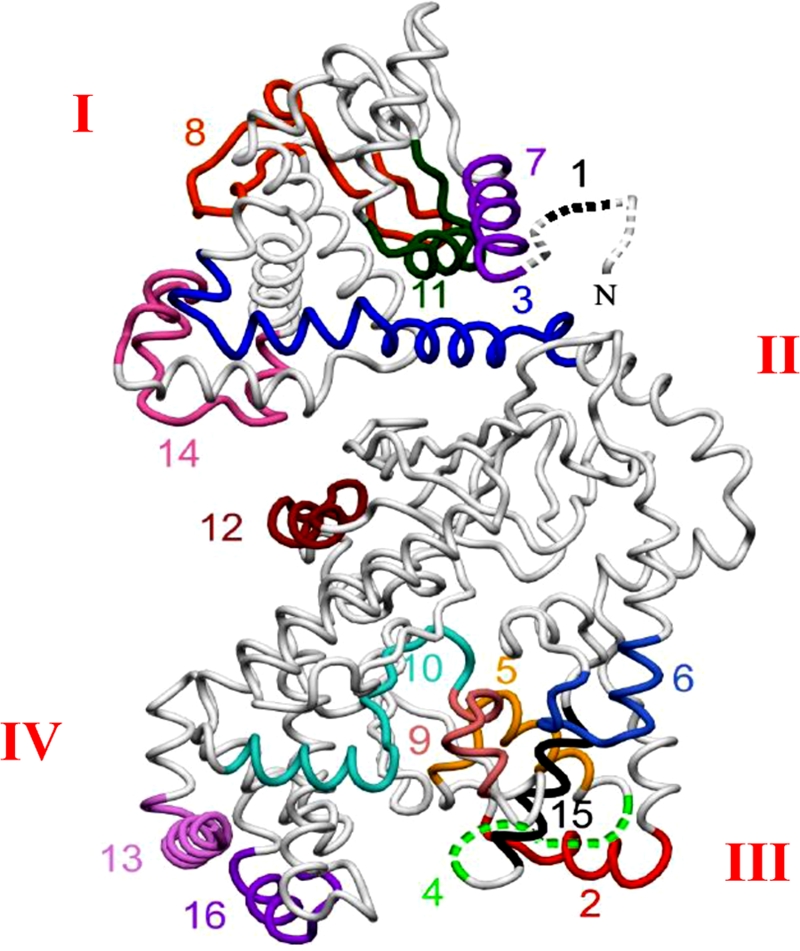
Sequential epitopes of LF. The 16 most reactive A/J epitopes are superimposed on the LF crystal structure (33) (Protein Data Bank accession code 1J7N [7]). Roman numerals indicate structural domains. The epitope numbers correspond to those shown in Fig. 2 and Tables 1 and 2.
TABLE 2.
LF most reactive epitopes
| Epitope no. | Sequence | Domain | Function | Secondary structure |
|---|---|---|---|---|
| 1 | GGHGDVGMHVKEKE | I | PA binding | Large ordered loop |
| 7 | KRKDEERNKTQEEHLKEIMK | I | PA binding | Alpha helix |
| 11 | VPSDVLEMYKAIGGKI | I | PA binding | Alpha helix |
| 8 | KKIKDIYGKDALLHEHYVYAKEGYEPVL | I | PA binding | Loop/beta |
| 14 | DSDGQDLLFTNQLKEHPTDFSVEFLEQN | I | PA binding | Alpha helix |
| 3 | VLQLYAPEAFNYMADKFNEQEINLSLEELKDQRMLAR | I/II | PA binding/VIP2-likea | Alpha helix |
| 2 | SQEEKELLKRIQ | III | Substrate recognition | Alpha helix |
| 5 | DFLSTEEKEFLKKL | III | Substrate recognition | Alpha helix |
| 4 | SEEEKELLNRIQ | III | Substrate recognition | Alpha helix |
| 15 | EKEFLKKLKLDI | III | Substrate recognition | Alpha helix |
| 6 | DSPSINLDVRKQYK | II | VIP2-likea | Alpha helix |
| 12 | TDNTKINRGIFNEFKK | II | VIP2-likea | Alpha helix |
| 10 | LIKKVTNYLVDGNGRFVYTDITLP | IV | Zinc metalloproteinase | Alpha/beta/loop |
| 9 | LPNIAEQYTHQDEIYEQVHS | IV | Zinc metalloproteinase | Loop/beta |
| 16 | VTNSKKFIDIFKEE | IV | Zinc metalloproteinase | Alpha helix |
| 13 | QKNAPKTFQFINDQIKFI | IV | Zinc metalloproteinase | Alpha helix |
This region has structural similarity to the catalytic domain of the Bacillus cereus toxin VIP2 (32).
Two of the five most reactive and reproducible epitopes bound antigenic regions in the PA-binding domain. The PA-binding domain has been shown to bind LF-neutralizing antibodies, directly inhibiting the binding of LF to PA to neutralize LeTx (9, 27, 34, 50). Three of the five most reactive and reproducible epitopes, which were identified in all three independent mapping studies and had average optical densities ranging from 0.75 to 0.8, were located in the substrate-binding region. This domain, identified as a 101-residue segment containing five tandem repeats, was previously shown to be required for LF activity based on insertion mutagenesis (33) and to bind neutralizing antibodies (26, 33, 51); however, the fine specificities of the MAbs were not reported previously.
LF-specific neutralizing MAbs bind epitopes identified in LF-immunized mice.
To determine whether sequential epitopes of LF identified in anti-LF antisera could bind neutralizing antibodies, we screened a panel of known LeTx-neutralizing MAbs with specificity for LF using solid-phase epitope mapping and in vitro neutralization assays (27, 41, 51). As shown in Fig. 4A, the neutralizing capacity of all three anti-LF MAbs tested (MAbs 10G3, 9A11, and LF8) was verified, although there was with some variability across the MAbs. MAb 9A11 had the greatest neutralization capacity (93.5% at concentrations as low as 0.313 μg/ml), while MAbs 10G3 (92% at a concentration of 5 μg/ml) and LF8 (80% at a concentration of 5 μg/ml) also exhibited high neutralizing activity.
To examine the fine specificity of these neutralizing LF MAbs, we performed a standard solid-phase peptide assay as described above. As shown in Fig. 4B, all MAbs bound linear epitopes of LF to various degrees. Furthermore, the decapeptides bound by these MAbs either coincided with or overlapped epitopes identified by using polyclonal anti-LF antisera (Fig. 2 and Table 1). MAb 10G3 bound an epitope that overlapped epitope 4, located in the substrate recognition domain, at a level 52 SD above the background level. The sequence (DIRDSLSEEE) overlapped epitope 4 (SEEEKELLNRIQ) by four amino acids. MAb 9A11 bound two different epitopes, an epitope that coincided with epitope 13 (domain IV) at a level 9 SD above the background level (INDQIKFIIN) and an epitope not identified by using polyclonal anti-LF antisera at a level 8 SD above the background level (VTNYLVDGNG). Lastly, MAb LF8 also bound two sequences that exhibit significant sequence similarity, one that strongly bound and coincided with epitope 2 (EEKELLKRIQ) at a level 379 SD above the background level and one that coincided with epitope 13 (TEEKEFLKKL) at a level 36 SD above the background level. These epitopes have the sequence EEKEXLK (R/K)(I/L), where X indicates a position where there is no amino acid similarity, and both occur in the substrate recognition domain. These data indicate that linear peptides of LF identified in our study bind neutralizing LF MAb.
To determine whether the peptide sequences of these epitopes had the capacity to inhibit neutralization of the corresponding MAbs, linear peptides corresponding to epitopes 2, 4, and 13 and the VTNYLVDGNG sequence were synthesized. These peptides were used in a modified in vitro neutralization assay to verify the binding specificity of MAbs and their ability to neutralize LeTx. As shown in Fig. 4D, all peptides mediated measurable reductions in MAb-mediated toxin neutralization activities in dose-dependent fashions. Two of these peptides (epitopes 2 and 4) essentially completely blocked neutralization under the conditions tested, whereas peptides corresponding to both epitopes recognized by MAb 9A11 exhibited measurable but less potent inhibition of MAb 9A11 neutralization capacity. Inhibition of neutralization was determined by comparing the percentages of viability of macrophages exposed to mixtures of MAb, toxin, and peptide to those of mixtures of MAb, toxin, and irrelevant control peptide from ovalbumin (ISQAVHAAHAEINEAGR) (control). The data obtained further supported the binding of linear peptides by LeTx-neutralizing antibodies. To our knowledge, this is the first time that linear peptides have been shown to inhibit neutralization of LeTx by these MAbs (27, 41, 51).
DISCUSSION
Vaccine development is a key part of protection against anthrax infection. Concerns regarding the transient reactogenicity of the current AVA vaccine, the burdensome immunization schedule, and the inefficiency of the AVA vaccine for eliciting antibodies to LF and EF suggest that this vaccine should be improved. As animal studies have demonstrated the importance of LF and EF in providing protection against anthrax infection and toxemia, we believe that induction of appropriate immune responses against these components would enhance the efficacy of the current vaccine. Moreover, construction of a vaccine from well-defined components may decrease the transient reactogenicity.
Synthetic peptides have the potential to provide safe, noninfective, well-defined, and stable vaccines that can focus antibody responses to neutralizing epitopes. For instance, peptide vaccination has been successfully used to neutralize or protect against viruses (15, 21, 25, 42), bacteria (10), and parasites (22). Based on data presented here, we propose peptide sequences of LF that are excellent candidates for a peptide-based vaccine for anthrax.
Using a solid-phase ELISA technique, we identified 16 antigenic regions, and some of the most reactive epitopes coincided with sequences bound by LeTx-neutralizing, LF-specific MAbs. Based on our data and previously published data (26, 51), the substrate-binding region (domain III) is extremely antigenic and, unpredictably, is an important target of the neutralizing antibody response. This may be due to the presence of a 101-residue segment comprising five tandem repeats (residues 249 to 349). Three of the five most reactive epitopes identified in this study (epitopes 2, 4, and 5) bind tandem repeats within the substrate recognition domain. The extent of cross-reactivity among polyclonal antibodies that bind to these epitopes is currently being investigated. Two of the tandem repeats coinciding with epitopes 2 and 5 bound a single neutralizing LF-specific MAb (MAb LF8), while a third tandem repeat overlapping epitope 4 was bound by LF-neutralizing MAb 10G3. Both of these MAbs were previously shown to inhibit binding of LF to PA; however, the specific peptide sequences bound by these MAbs were not reported. Here, we show that these neutralizing MAbs bind the substrate recognition domain of LF and therefore may alter the conformation of LF or sterically inhibit the capacity of LF to bind PA. Cross-reactivity among antibodies targeting the tandem repeats of the substrate recognition domain may increase the avidity of such antibodies for intact LF and enhance their neutralization capacity.
The most potent LF-neutralizing MAb tested (MAb 9A11) bound two different sequences within the zinc-binding domain, one of which corresponds to epitope 13, an epitope with moderate reactivity identified in one of our three mapping studies. Although the two sequences bound by this MAb exhibit no obvious similarity, examination of their location in the LF crystal structure revealed the possibility that several unresolved residues of epitope c (Fig. 4C), indicative of peptide flexibility, may contact epitope b to form one conformational epitope.
Antibodies directed to the PA-binding domain of LF are expected to interfere with the LF-PA interaction and thus be neutralizing. Other workers have demonstrated that mutations within the PA-binding domain of LF, which are located within two epitopes that we identified here, inhibit PA binding (24). Additionally, known LF-neutralizing antibodies have been shown to abrogate the binding of LF to PA and also cross-react with EF by binding to the PA-binding domain (9, 27, 34, 50). We defined five sequential B-cell epitopes in the PA-binding domain and one epitope that overlapped the PA-binding domain and the VIP2-like domain.
Although the rLF-immunized mice were not challenged with anthrax spores in the present study, protection of experimental animals, including A/J mice, from lethal anthrax spore challenge has been demonstrated in studies in which mice or rabbits were actively immunized with detoxified LF (either in plasmid [20] or protein [49] forms) or passively immunized with LF-specific MAbs (1). In these studies, mice were challenged with Sterne strain spores, whereas rabbits were challenged with Ames strain spores. The data strongly suggest that rLF-immunized animals are likely to be protected from challenge with lethal doses of anthrax spores.
In summary, we carefully mapped sequential B-cell epitopes of B. anthracis LF using polyclonal neutralizing antisera and known neutralizing MAbs specific for LF. Together, these studies highlight discrete peptides in the substrate recognition, zinc metalloproteinase, and PA-binding domains of LF that are important candidates for the generation of LeTx-neutralizing antibodies by peptide vaccination.
Acknowledgments
We thank the individuals who generously provided MAbs. MAb 10G3 was obtained from the NIH Biodefense and Emerging Infections Repository, NIAID, NIH (Bethesda, MD), and MAb 9A11 was obtained from the laboratory of Barton Haynes at Duke University Medical Center. Additionally, we thank Linda Ash and Clay Nelson for their technical assistance and Beverly Hurt for formatting figures and tables.
This work was supported by grant U19 AI062629 from the NIAID.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 3 November 2008.
REFERENCES
- 1.Albrecht, M. T., H. Li, E. D. Williamson, C. S. LeButt, H. C. Flick-Smith, C. P. Quinn, H. Westra, D. Galloway, A. Mateczun, S. Goldman, H. Groen, and L. W. Baillie. 2007. Human monoclonal antibodies against anthrax lethal factor and protective antigen act independently to protect against Bacillus anthracis infection and enhance endogenous immunity to anthrax. Infect. Immun. 755425-5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alileche, A., E. R. Serfass, S. M. Muehlbauer, S. A. Porcelli, and J. Brojatsch. 2005. Anthrax lethal toxin-mediated killing of human and murine dendritic cells impairs the adaptive immune response. PLoS Pathog. 1e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baillie, L., R. Hebdon, H. Flick-Smith, and D. Williamson. 2003. Characterisation of the immune response to the UK human anthrax vaccine. FEMS Immunol. Med. Microbiol. 3683-86. [DOI] [PubMed] [Google Scholar]
- 4.Baillie, L. W. 2005. Bacillus anthracis, a story of nature subverted by man. Lett. Appl. Microbiol. 41227-229. [DOI] [PubMed] [Google Scholar]
- 5.Baillie, L. W., K. Fowler, and P. C. Turnbull. 1999. Human immune responses to the UK human anthrax vaccine. J. Appl. Microbiol. 87306-308. [DOI] [PubMed] [Google Scholar]
- 6.Beauregard, K. E., R. J. Collier, and J. A. Swanson. 2000. Proteolytic activation of receptor-bound anthrax protective antigen on macrophages promotes its internalization. Cell. Microbiol. 2251-258. [DOI] [PubMed] [Google Scholar]
- 7.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradley, K. A., J. Mogridge, M. Mourez, R. J. Collier, and J. A. Young. 2001. Identification of the cellular receptor for anthrax toxin. Nature 414225-229. [DOI] [PubMed] [Google Scholar]
- 9.Brossier, F., M. Levy, A. Landier, P. Lafaye, and M. Mock. 2004. Functional analysis of Bacillus anthracis protective antigen by using neutralizing monoclonal antibodies. Infect. Immun. 726313-6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christodoulides, M., and J. E. Heckels. 1994. Immunization with a multiple antigen peptide containing defined B- and T-cell epitopes: production of bactericidal antibodies against group B Neisseria meningitidis. Microbiology 1402951-2960. [DOI] [PubMed] [Google Scholar]
- 11.Collier, R. J., and J. A. Young. 2003. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 1945-70. [DOI] [PubMed] [Google Scholar]
- 12.Crawford, M. A., C. V. Aylott, R. W. Bourdeau, and G. M. Bokoch. 2006. Bacillus anthracis toxins inhibit human neutrophil NADPH oxidase activity. J. Immunol. 1767557-7565. [DOI] [PubMed] [Google Scholar]
- 13.Dixon, T. C., M. Meselson, J. Guillemin, and P. C. Hanna. 1999. Anthrax. N. Engl. J. med. 341815-826. [DOI] [PubMed] [Google Scholar]
- 14.Duesbery, N. S., C. P. Webb, S. H. Leppla, V. M. Gordon, K. R. Klimpel, T. D. Copeland, N. G. Ahn, M. K. Oskarsson, K. Fukasawa, K. D. Paull, and G. F. Vande Woude. 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280734-737. [DOI] [PubMed] [Google Scholar]
- 15.El Kasmi, K. C., and C. P. Muller. 2001. New strategies for closing the gap of measles susceptibility in infants: towards vaccines compatible with current vaccination schedules. Vaccine 192238-2244. [DOI] [PubMed] [Google Scholar]
- 16.Farris, A. D., L. Brown, P. Reynolds, J. B. Harley, J. A. James, R. H. Scofield, J. McCluskey, and T. P. Gordon. 1999. Induction of autoimmunity by multivalent immunodominant and subdominant T cell determinants of La (SS-B). J. Immunol. 1623079-3087. [PubMed] [Google Scholar]
- 17.Flick-Smith, H. C., E. L. Waters, N. J. Walker, J. Miller, A. J. Stagg, M. Green, and E. D. Williamson. 2005. Mouse model characterisation for anthrax vaccine development: comparison of one inbred and one outbred mouse strain. Microb. Pathog. 3833-40. [DOI] [PubMed] [Google Scholar]
- 18.Friedlander, A. M., P. R. Pittman, and G. W. Parker. 1999. Anthrax vaccine: evidence for safety and efficacy against inhalational anthrax. JAMA 2822104-2106. [DOI] [PubMed] [Google Scholar]
- 19.Guarner, J., J. A. Jernigan, W. J. Shieh, K. Tatti, L. M. Flannagan, D. S. Stephens, T. Popovic, D. A. Ashford, B. A. Perkins, and S. R. Zaki. 2003. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am. J. Pathol. 163701-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hermanson, G., V. Whitlow, S. Parker, K. Tonsky, D. Rusalov, M. Ferrari, P. Lalor, M. Komai, R. Mere, M. Bell, K. Brenneman, A. Mateczun, T. Evans, D. Kaslow, D. Galloway, and P. Hobart. 2004. A cationic lipid-formulated plasmid DNA vaccine confers sustained antibody-mediated protection against aerosolized anthrax spores. Proc. Natl. Acad. Sci. USA 10113601-13606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hovanessian, A. G., J. P. Briand, E. A. Said, J. Svab, S. Ferris, H. Dali, S. Muller, C. Desgranges, and B. Krust. 2004. The caveolin-1 binding domain of HIV-1 glycoprotein gp41 is an efficient B cell epitope vaccine candidate against virus infection. Immunity 21617-627. [DOI] [PubMed] [Google Scholar]
- 22.Joshi, M. B., A. A. Gam, R. A. Boykins, S. Kumar, J. Sacci, S. L. Hoffman, H. L. Nakhasi, and R. T. Kenney. 2001. Immunogenicity of well-characterized synthetic Plasmodium falciparum multiple antigen peptide conjugates. Infect. Immun. 694884-4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobiler, D., Y. Gozes, H. Rosenberg, D. Marcus, S. Reuveny, and Z. Altboum. 2002. Efficiency of protection of guinea pigs against infection with Bacillus anthracis spores by passive immunization. Infect. Immun. 70544-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacy, D. B., M. Mourez, A. Fouassier, and R. J. Collier. 2002. Mapping the anthrax protective antigen binding site on the lethal and edema factors. J. Biol. Chem. 2773006-3010. [DOI] [PubMed] [Google Scholar]
- 25.Langeveld, J. P., J. Martinez-Torrecuadrada, R. S. Boshuizen, R. H. Meloen, and J. Ignacio Casal. 2001. Characterisation of a protective linear B cell epitope against feline parvoviruses. Vaccine 192352-2360. [DOI] [PubMed] [Google Scholar]
- 26.Lim, N. K., J. H. Kim, M. S. Oh, S. Lee, S. Y. Kim, K. S. Kim, H. J. Kang, H. J. Hong, and K. S. Inn. 2005. An anthrax lethal factor-neutralizing monoclonal antibody protects rats before and after challenge with anthrax toxin. Infect. Immun. 736547-6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Little, S. F., S. H. Leppla, and A. M. Friedlander. 1990. Production and characterization of monoclonal antibodies against the lethal factor component of Bacillus anthracis lethal toxin. Infect. Immun. 581606-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McClain, M. T., L. D. Heinlen, G. J. Dennis, J. Roebuck, J. B. Harley, and J. A. James. 2005. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat. Med. 1185-89. [DOI] [PubMed] [Google Scholar]
- 29.McClain, M. T., B. D. Poole, B. F. Bruner, K. M. Kaufman, J. B. Harley, and J. A. James. 2006. An altered immune response to Epstein-Barr nuclear antigen 1 in pediatric systemic lupus erythematosus. Arthritis Rheum. 54360-368. [DOI] [PubMed] [Google Scholar]
- 30.Milne, J. C., D. Furlong, P. C. Hanna, J. S. Wall, and R. J. Collier. 1994. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 26920607-20612. [PubMed] [Google Scholar]
- 31.Mogridge, J., K. Cunningham, D. B. Lacy, M. Mourez, and R. J. Collier. 2002. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc. Natl. Acad. Sci. USA 997045-7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mogridge, J., M. Mourez, and R. J. Collier. 2001. Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J. Bacteriol. 1832111-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pannifer, A. D., T. Y. Wong, R. Schwarzenbacher, M. Renatus, C. Petosa, J. Bienkowska, D. B. Lacy, R. J. Collier, S. Park, S. H. Leppla, P. Hanna, and R. C. Liddington. 2001. Crystal structure of the anthrax lethal factor. Nature 414229-233. [DOI] [PubMed] [Google Scholar]
- 34.Pelat, T., M. Hust, E. Laffly, F. Condemine, C. Bottex, D. Vidal, M. P. Lefranc, S. Dubel, and P. Thullier. 2007. High-affinity, human antibody-like antibody fragment (single-chain variable fragment) neutralizing the lethal factor (LF) of Bacillus anthracis by inhibiting protective antigen-LF complex formation. Antimicrob. Agents Chemother. 512758-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pezard, C., M. Weber, J. C. Sirard, P. Berche, and M. Mock. 1995. Protective immunity induced by Bacillus anthracis toxin-deficient strains. Infect. Immun. 631369-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puziss, M., L. C. Manning, J. W. Lynch, Barclaye, I. Abelow, and G. G. Wright. 1963. Large-scale production of protective antigen of Bacillus anthracis in anaerobic cultures. Appl. Microbiol. 11330-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salles, I. I., D. E. Voth, S. C. Ward, K. M. Averette, R. K. Tweten, K. A. Bradley, and J. D. Ballard. 2006. Cytotoxic activity of Bacillus anthracis protective antigen observed in a macrophage cell line overexpressing ANTXR1. Cell. Microbiol. 81272-1281. [DOI] [PubMed] [Google Scholar]
- 38.Scobie, H. M., G. J. Rainey, K. A. Bradley, and J. A. Young. 2003. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 1005170-5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scorpio, A., T. E. Blank, W. A. Day, and D. J. Chabot. 2006. Anthrax vaccines: Pasteur to the present. Cell Mol. Life Sci. 632237-2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh, Y., K. R. Klimpel, S. Goel, P. K. Swain, and S. H. Leppla. 1999. Oligomerization of anthrax toxin protective antigen and binding of lethal factor during endocytic uptake into mammalian cells. Infect. Immun. 671853-1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Staats, H. F., S. M. Alam, R. M. Scearce, S. M. Kirwan, J. X. Zhang, W. M. Gwinn, and B. F. Haynes. 2007. In vitro and in vivo characterization of anthrax anti-protective antigen and anti-lethal factor monoclonal antibodies after passive transfer in a mouse lethal toxin challenge model to define correlates of immunity. Infect. Immun. 755443-5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi, A., K. Ogasawara, N. Matsuki, K. Fujinaga, T. Nakaya, K. Ikuta, W. Auwanit, M. Honda, Y. Fukui, T. Sasazuki, K. Iwabuchi, and K. Onoe. 1998. Development of peptide vaccines inducing production of neutralizing antibodies against HIV-1 viruses in HLA-DQ6 mice. Vaccine 161537-1543. [DOI] [PubMed] [Google Scholar]
- 43.Turnbull, P. C., S. H. Leppla, M. G. Broster, C. P. Quinn, and J. Melling. 1988. Antibodies to anthrax toxin in humans and guinea pigs and their relevance to protective immunity. Med. Microbiol. Immunol. 177293-303. [DOI] [PubMed] [Google Scholar]
- 44.Vitale, G., L. Bernardi, G. Napolitani, M. Mock, and C. Montecucco. 2000. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 352739-745. [PMC free article] [PubMed] [Google Scholar]
- 45.Vitale, G., R. Pellizzari, C. Recchi, G. Napolitani, M. Mock, and C. Montecucco. 1998. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 248706-711. [DOI] [PubMed] [Google Scholar]
- 46.Voth, D. E., E. E. Hamm, L. G. Nguyen, A. E. Tucker, Salles, I. I., W. Ortiz-Leduc, and J. D. Ballard. 2005. Bacillus anthracis oedema toxin as a cause of tissue necrosis and cell type-specific cytotoxicity. Cell. Microbiol. 71139-1149. [DOI] [PubMed] [Google Scholar]
- 47.Wang, J. Y., and M. H. Roehrl. 2005. Anthrax vaccine design: strategies to achieve comprehensive protection against spore, bacillus, and toxin. Med. Immunol. 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodrow, P. 2003. Anthrax: forms, symptoms and treatment. Nurs. Stand. 1733-37. [DOI] [PubMed] [Google Scholar]
- 49.Xu, Q., and M. Zeng. 2008. Detoxified lethal toxin as a potential mucosal vaccine against anthrax. Clin. Vaccine Immunol. 15612-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeng, M., Q. Xu, E. D. Hesek, and M. E. Pichichero. 2006. N-fragment of edema factor as a candidate antigen for immunization against anthrax. Vaccine 24662-670. [DOI] [PubMed] [Google Scholar]
- 51.Zhao, P., X. Liang, J. Kalbfleisch, H. M. Koo, and B. Cao. 2003. Neutralizing monoclonal antibody against anthrax lethal factor inhibits intoxication in a mouse model. Hum. Antibodies 12129-135. [PubMed] [Google Scholar]