

Abstract
Aggregation substance proteins encoded by sex pheromone plasmids increase the virulence of Enterococcus faecalis in experimental pathogenesis models, including infectious endocarditis models. These large surface proteins may contain multiple functional domains involved in various interactions with other bacterial cells and with the mammalian host. Aggregation substance Asc10, encoded by plasmid pCF10, is induced during growth in the mammalian bloodstream, and pCF10 carriage gives E. faecalis a significant selective advantage in this environment. We employed a rabbit model to investigate the role of various functional domains of Asc10 in endocarditis. The data suggested that the bacterial load of the infected tissue was the best indicator of virulence. Isogenic strains carrying either no plasmid, wild-type pCF10, a pCF10 derivative with an in-frame deletion of the prgB gene encoding Asc10, or pCF10 derivatives expressing other alleles of prgB were examined in this model. Previously identified aggregation domains contributed to the virulence associated with the wild-type protein, and a strain expressing an Asc10 derivative in which glycine residues in two RGD motifs were changed to alanine residues showed the greatest reduction in virulence. Remarkably, this strain and the strain carrying the pCF10 derivative with the in-frame deletion of prgB were both significantly less virulent than an isogenic plasmid-free strain. The data demonstrate that multiple functional domains are important in Asc10-mediated interactions with the host during the course of experimental endocarditis and that in the absence of a functional prgB gene, pCF10 carriage is actually disadvantageous in vivo.
Enterococcus faecalis has gained notoriety due to the increasing number of cases of enterococcal nosocomial infection, which are complicated by the inherent and acquired antibiotic resistance of this bacterium. Infectious endocarditis, urinary tract and wound infections, and bacteremia are all associated with E. faecalis (16, 25). The high propensity of this organism to transfer antibiotic resistance elements also leads to transfer of these determinants to more virulent bacteria, such as Staphylococcus aureus (35).
Enterococci account for 20% of all bacterial endocarditis cases and are the third leading cause of this disease (14, 15). Ultimately, this infection leads to congestive heart failure and then death. This is due to the focal lesion of this disease, the vegetation, which is comprised of platelets, fibrin, other procoagulant factors, recruited phagocytes, and bacterial cells, typically attached to a valve leaflet. As the vegetation grows, it has the potential to block blood flow around the valve area.
Formation of a nonbacterial thrombotic (NBT) vegetation involves damage on the valve (typically the aortic valve), which can occur through processes such as irregular blood flow caused by valve regurgitation and also through intravenous drug use (17). This damage can also be inflicted by bacteria that have been engulfed by endothelial cells, triggering production of clotting factors (31). Platelets and fibrin are recruited to the damaged site, which composes the NBT vegetation (Fig. 1). Surgical procedures and increased bacterial translocation due to antibiotic-induced bacterial overgrowth (14) can enable enterococci or other bacteria to gain entrance to the bloodstream. These bacteria can adhere to the NBT vegetation, forming a septic vegetation (8). Phagocytes are then recruited to fight off the infection, and often these immune cells engulf bacteria while they are walled inside layers of fibrin (7). Although the majority of enterococcal endocarditis infections probably result from opportunistic infection of previously damaged tissue, as described above, it is possible that endocardiac infection of healthy tissues occurs in some cases (22).
FIG. 1.

NBT vegetation formation, demonstrating interactions between Asc10+ E. faecalis and host cells during formation of the septic vegetation in endocarditis. Platelets, fibrin, and other clotting proteins may comprise part of the vegetation, enabling blood-borne E. faecalis to bind to the growing clot.
The vegetation itself is advantageous for the bacteria, as they are protected from host defenses once they are encased inside fibrin and platelet layers. The lesion is essentially a biofilm, where the physiological state of the bacteria, as well as the extracellular matrix (ECM), may impede the ability of antibiotics to kill the bacteria. In addition, fragments of the vegetation can break off, allowing dissemination of the invading microorganisms to other organs.
An E. faecalis surface protein, aggregation substance, contributes to formation of larger vegetations (4) and mediates adherence to neutrophils (polymorphonuclear leukocytes [PMNs]) (21, 30), renal tubular cells (10), and intestinal epithelial cells. Asc10 is a 137-kDa aggregation substance protein encoded by the prgB gene of the conjugative plasmid pCF10; its expression is triggered by the presence of a peptide pheromone, cCF10, that is produced by plasmid-free cells and induces transfer of pCF10 by conjugation (5, 6). Importantly, Asc10 expression is also induced by a host factor during in vivo growth; pCF10 carriage has a strong selective advantage for enterococci in their hosts under these conditions (3, 9). Plasmid pCF10 is a member of the family of pheromone-responsive plasmids that includes pAD1 and pPD1, each of which encodes its own aggregation substance protein (Asa1 and Asp1, respectively) (34). These proteins share more than 90% identity throughout most of the protein; the exception is a centrally located variable region where the level of identity is 30 to 40% (Asc10 variable region shown in Fig. 2).
FIG. 2.

Schematic maps of the Asc10 mutant proteins expressed by the strains used in the rabbit endocarditis model, highlighting the functional domains of the Asc10 protein. (A) Wild-type full-length Asc10 (pCF10). (B) C-terminal domain deletion variant (pCF10-6) with amino acids 688 to 1138 (nucleotides 2064 to 3414) missing. (C) N-terminal aggregation domain deletion variant (pCF10-4) with amino acids 156 to 358 (nucleotides 468 to 1074) deleted. (D) Central aggregation domain insertion variant (pCF10-1) with 31 amino acids inserted at amino acid 546 (nucleotide 1638). Previous studies have determined that amino acid residues 473 to 683 are an essential component of this central aggregation domain. (E) N-terminal single RGDS → RADS substitution variant (pCF10-3). (F) C-terminal single RGDV → RADV substitution variant (pCF10-7). (G) Double RGD → RAD substitution variant (pCF10-2). RGD motifs occur at amino acid residues 606 and 939, as shown in panel A. SS, signal sequence; aa, amino acids. The diagrams are adapted from diagrams of Waters and Dunny (33). Whether expression of each Asc10 variant resulted in visible clumping of the host bacteria is indicated on the right.
Several functional domains of Asc10 have been identified previously (Fig. 2). A signal sequence is located in the N terminus, and the C terminus contains an LPXTG motif. This motif is present in many gram-positive bacterial surface protein sequences and designates the site that is cleaved by the sortase enzyme, where the protein is attached to the peptidoglycan in the cell wall (13). Two aggregation domains are required for binding to occur between a donor and a recipient during conjugation. The more N-terminal aggregation domain is also known to bind lipoteichoic acid (LTA) (34), while a distinct central domain is also important for aggregation. LTA, a major component of the gram-positive cell wall, has been found to be part of the cognate receptor for Asc10. A set of genes (ebs [enterococcal binding substance]) that are required for this receptor encode products that are involved in LTA biosynthesis (1, 29). Two RGD motifs have been identified in Asc10; in eukaryotic systems these motifs are known to mediate binding to integrins and are involved in other adhesion events, such as those pertaining to the ECM. Evidence suggests that in E. faecalis the RGD motifs mediate interactions with PMNs (30). Exogenously added RGD peptides were able to reduce Asc10+ E. faecalis binding to PMNs; it has been suggested that the presence of Asc10 on the cell surface may alter the interactions between the bacterium and the phagocyte subsequent to engulfment such that the bactericidal functions are reduced.
Previous studies of the functional domains of Asc10 involved expression of cloned mutant alleles of prgB in a nisin-inducible expression vector (33). While it is well established that Asc10 is expressed in vivo in the mammalian bloodstream and during endocarditis infections when the infecting strain carries wild-type pCF10 (3), we were not confident that we could sustain nisin-induced expression from cloned alleles of the gene during experimental endocarditis. Therefore, we used a recently developed allelic exchange system (12) to move mutant prgB alleles into the native context of pCF10 and confirmed that their pheromone-induced expression at the protein level mimicked that of the wild-type gene. In addition, we used allelic exchange to construct an in-frame deletion of prgB in the context of the pCF10 background, making it possible for the first time to test a true null allele of this gene. We then examined the behavior of isogenic strains carrying the pCF10 derivatives in experimental endocarditis. We found that a plasmid-containing strain carrying the prgB null allele was significantly less virulent than an isogenic plasmid-free strain, confirming that a functional prgB gene is required for the previously observed selective advantage of pCF10 carriage during endocarditis caused by E. faecalis (9). The differences in the virulence of strains expressing different variants of Asc10 are consistent with the existence of multiple functional domains in the protein.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Strains used in this study are listed in Table 1. Strains used for endocarditis experiments were constructed in the E. faecalis OG1SSp background (6), as this was the background used in previous studies. Strains were grown in beef heart dialyzable medium containing 5% glucose phosphate for all endocarditis experiments and in Todd-Hewitt (TH) (Becton Dickinson, Sparks, MD) broth for other experiments for approximately 16 h at 37°C. Frozen stocks were maintained at −80°C and were used to start overnight cultures for endocarditis experiments.
TABLE 1.
Strain and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| E. faecalis strains | ||
| OG1SSp | Streptomycin- and spectinomycin-resistant wild-type strain | 6 |
| TX5128 | OG1RF derivative, gelEsprE | 27 |
| Plasmids | ||
| pCF10 | Pheromone-inducible conjugative plasmid | 5 |
| pMSP3535 | Cloning shuttle vector with nisin-inducible promoter | 2 |
| pMSP3609 | Double RGD → RAD mutations in prgB inserted into the pMSP3535 vector | 32 |
| pCF10-1 | Asc10 mutant derivative; 31-amino-acid insertion in central aggregation domain at nucleotide 1638 | 12 |
| pCF10-2 | Asc10 mutant derivative; double RGD mutant with RGD → RAD single-amino-acid substitutions | This study |
| pCF10-3 | Asc10 mutant derivative; N-terminal single RGD mutant (RGDS → RADS) | This study |
| pCF10-4 | Asc10 mutant derivative; N-terminal aggregation domain deletion (nucleotides 468 to 1074) | This study |
| pCF10-5 | Asc10 mutant derivative; double aggregation domain mutant; 31-amino-acid insertion in central aggregation domain and deletion in N-terminal aggregation domain | This study |
| pCF10-6 | Asc10 mutant derivative; C-terminal domain deletion (nucleotides 2064 to 3414) | This study |
| pCF10-7 | Asc10 mutant derivative; C-terminal single RGD mutant (RGDV → RADV) | This study |
| pCF10-8 | prgB deletion mutant retaining first three and last three codons, Asc10− | This study |
| pCWΔ468-1074 | pMSP3535 with in-frame deletion of nucleotides 468 to 1074 in prgB | 34 |
| pCWΔ468-1074/Ω1638 | pMSP3535 carrying double aggregation domain mutant | 34 |
| pCWΔ2064-3414 | pMSP3535 with in-frame deletion of nucleotides 2064 to 3414 in prgB | 34 |
| CK104 | OG1RFΔupp | 12 |
| pVE6007 | Supplies RepA (temperature sensitive), Cmr | 12 |
| pCJK2 | P-upp expression cassette cloned into pORI280 | 12 |
| pCJK47 | Conjugative donor plasmid, carries oriTpCF10 and P-pheS*; pORI280 derivative; Emr | 11 |
| pCJK47ΔprgB | prgB deletion construct with first 9 and last 9 nucleotides, cloned into pCJK47 | This study |
| pCJK2-2 | prgB double RGD mutant cloned into pCJK2 | This study |
| pCJK2-4 | prgB in-frame deletion of nucleotides 468 to 1074 cloned into pCJK2 | This study |
| pCJK2-5 | prgB double aggregation domain Δ468-1074/Ω1638 cloned into pCJK2 | This study |
| pCJK2-6 | prgB in-frame deletion of nucleotides 2064 to 3414 cloned into pCJK2 | This study |
| pCJK2-7 | prgB C-terminal single RGD mutant cloned into pCJK2 | This study |
Construction of Asc10 mutant derivative-expressing E. faecalis strains.
In previous work (33, 34), a series of in-frame insertion and deletion mutations in the prgB gene encoding Asc10 were generated using a TnlacZ/in TnphoA/in transposon system, and oligonucleotide-directed mutagenesis was used to change the two RGD sequences to RAD (singly and in combination). Physical maps of these constructs are shown in Fig. 2. The prgB gene was expressed from the nisin-inducible vector pMSP3535 (33). In the present study, we used a recently developed allelic exchange system (12) to insert the mutant prgB derivatives in the native context into pCF10. Plasmids used for this protocol are listed in Table 1. The pCJK2 plasmid (12) was used to transfer prgB mutations to pCF10 by markerless allelic exchange. pCJK1, pCJK2-2, pCJK2-4, and pCJK2-5 were constructed by inserting the BsrGI/BplI prgB fragment (blunt ended with T4 DNA polymerase) from pMSP3609, pCWΔ468-1074, pCWΔ468-1074/Ω1638, and pCWΔ2064-3414, respectively, into pCJK2 digested with SmaI. pCJK2-7 was created by digesting pMSP3609 with PstI and treating a 1.3-kb fragment with T4 DNA polymerase prior to cloning into SmaI-digested pCJK2. Allelic exchange was carried out as described by Kristich et al. (12) to create pCF10-2, pCF10-3, pCF10-4, pCF10-5, pCF10-6, and pCF10-7. Plasmids pCJK2-2, pCJK2-4, pCJK2-5, pCJK2-6, and pCJK2-7 were electroporated into E. faecalis strain CK104 carrying pVE6007/pCF10. The temperature was increased to 37°C to induce the loss of plasmid pVE6007, making survival of the pCJK2 derivatives dependent on their insertion into pCF10 by homologous recombination. The strains were grown for approximately 20 generations at 37°C and then plated on 5-fluorouracil-containing plates to select for excision of the integrated plasmid. The strains were screened for the desired prgB mutation. An aggregation analysis (12) was used to screen mutations that abolished the aggregation function (pCF10-4, pCF10-5 and pCF10-6). Other pCF10 mutants (pCF10-2, pCF10-3, and pCF10-7) were screened by sequencing prgB fragments amplified by PCR. Either each variant prgB allele was sequenced completely following its insertion into pCF10 or the junctions where homologous recombination occurred were sequenced to verify proper insertion of the prgB variant into pCF10. All endocarditis experiments described below involved the use of derivatives of pCF10 carrying the various prgB alleles in strain OG1SSp.
Construction of E. faecalis prgB deletion mutant.
Two-step PCR was utilized to create an in-frame deletion construct that contained only the first nine and last nine nucleotides of the prgB gene (first three codons and last three codons). This fragment was then subcloned into the pGEM-T Easy plasmid (Promega, Madison, WI) to verify deletion of prgB; the fragment containing the deletion was digested with EcoRI and XbaI and cloned into pCJK47 (11). The resulting pCJK47ΔprgB construct was electroporated into CK104 carrying pVE6007/pCF10, as described above. The temperature was increased to 37°C to induce loss of the temperature-sensitive pVE6007 plasmid, and then pCJK47ΔprgB was forced to integrate into the pCF10 plasmid and the pCJK47 plasmid was excised after growth on p-Cl-phenylalanine for approximately 30 to 40 generations. Colony PCR was used to confirm deletion of the prgB gene in the pCF10 background (pCF10-8), and the correct plasmid construct was transferred from CK104(pCF10-8) into OG1SSp by conjugation. The prgB locus and junction regions were sequenced in both the CK104 and OG1SSp backgrounds, and no extraneous mutations were found. Western blotting and clumping assay analyses were performed to ensure that no surface Asc10 was expressed and that this strain was unable to aggregate.
Rabbit model of endocarditis.
The left carotid artery of New Zealand White rabbits was isolated, and a catheter was inserted until the aortic valve was reached, as previously described (26). Young adult rabbits that weighed 2 to 3 kg were obtained from Bakkom Rabbitry (Red Wing, MN). The catheter remained in place for 2 h to induce damage to the valve and then was removed, after which the neck region was closed and approximately 2 × 109 CFU of E. faecalis was injected intravenously through the marginal ear vein. Four days after this procedure the animals were sacrificed, the hearts were removed, and vegetations were weighed and homogenized for bacterial counting. Homogenates were plated onto TH agar with and without the appropriate antibiotics in order to ensure that resistance markers were not lost during the infection period. The bacterial counts for antibiotic-containing and non-antibiotic-containing plates were similar, indicating that the pCF10 derivatives were not cured during the course of infection. If no visible vegetations were found, all of the aortic valve leaflets were removed and cultured to enumerate the bacteria colonizing the valves in the absence of a visible lesion. The following antibiotics and concentrations were used: streptomycin, 1,000 μg/ml (Cellgro, Herndon, VA); spectinomycin, 250 μg/ml (Sigma-Aldrich, St. Louis, MO); and erythromycin, 10 μg/ml (Sigma). All experiments were conducted under the established guidelines of the University of Minnesota Institutional Animal Care and Use Committee.
Statistical analysis for endocarditis.
Microbial loads (total CFU and CFU/g) were converted to log10 values prior to statistical analysis using one-way analysis of variance followed by Fisher's post hoc test. A P value of <0.05 was considered statistically significant.
ECM protein binding by purified Asc10 protein in a microplate enzyme-linked immunosorbent assay (ELISA).
Purification of wild-type and mutant Asc10 proteins by ethanol precipitation and isoelectric focusing has been described previously (34). Derivatives of E. faecalis strain TX5128 containing plasmids that expressed Asc10 variants (Fig. 2) under a nisin-inducible promoter were utilized. ECM binding by purified Asc10 protein was assayed by using a protocol described elsewhere (20), with some modifications. ECM proteins were resuspended in phosphate-buffered saline (PBS) (1.68 mM KH2PO4, 8.00 mM K2HPO4 [pH 7.4], 0.15 M NaCl) or deionized water, depending on the manufacturer's instructions. Fibrinogen (Calbiochem, San Diego, CA), fibronectin (Roche, Switzerland), vitronectin (Promega, Madison, WI), and von Willebrand factor (Enzyme Research Laboratories, South Bend, IN) were coated onto 96-well 3590 CoStar plates (Corning, Corning, NY). For fibrinogen, fibronectin, and von Willebrand factor, concentrations of 0, 200, 500, 700, 800, and 1,000 ng/well were used; for vitronectin we used 0, 50, 200, 300, 500, and 800 ng/well. The plates were then incubated at 4°C overnight and washed with PBS-0.05% Tween 20 (Sigma). Purified Asc10 protein was layered on the ECM protein (100 μl of a 5-μg/ml solution) for 1 h at 37°C; adherent Asc10 protein was detected with a polyclonal rabbit antibody made against full-length purified Asc10 protein (34). A goat anti-rabbit immunoglobulin G secondary antibody conjugated to horseradish peroxidase (Zymed Laboratories, Inc., San Francisco, CA) was used to bind the adherent primary anti-Asc10 antibody. O-Phenylenediamine substrate (Zymed) containing H2O2 was added (100 μl), and the reaction was stopped with 2.5 M H2SO4 (10 μl). The absorbance at 490 nm was determined spectrophotometrically in order to determine the quantity of Asc10 protein bound to the ECM protein.
Binding of ECM proteins to intact bacterial cells.
E. faecalis cells expressing the mutant Asc10 derivatives were tested for ECM binding using a crystal violet stain-based assay as previously described (19, 28). Bacteria were grown overnight and then washed in PBS twice, and the optical density at 600 nm was adjusted to 1.5. As in the ELISAs described above, derivatives of TX5128 were used. ECM proteins were coated on 3590 CoStar plates at the concentrations specified above. Two percent bovine serum albumin (Sigma) in PBS was used to block unbound sites on the plates for 1 h at 37°C. The plates were washed four times with PBS, and bacteria were added to each well (50 μl; approximately 5.0 × 107 CFU/well). A crystal violet solution (0.5% [wt/vol] crystal violet in H2O; 100 μl; Sigma) was added for 1 min after four washes with PBS. Six washes with PBS were used to remove excess stain, and then 200 μl of citrate buffer (pH 4.3) was added to suspend the bacteria and solubilize the stain. The absorbance at 562 nm was used to determine the relative amounts of stained bacteria adhering to the ECM proteins.
Purification of cell wall fractions for Asc10 mutant strain analysis.
Cell wall fractions were purified from plasmid-free Asc10− strain OG1SSp, the wild-type strain containing pCF10, and strains containing all of the Asc10 mutants (pCF10-1 through pCF10-8). A protocol adapted from the protocol of Kristich et al. (12) was used for this purification. Briefly, overnight cultures of the E. faecalis strains listed above were diluted 1:6 in TH broth containing the appropriate antibiotics. Pheromone (50 ng/ml of cCF10) was added to the strains with the OG1SSp background to stimulate Asc10 expression. The diluted cultures were incubated at 37°C for 2.7 h with aeration and photographed to document the extent of clumping of the Asc10 mutant-expressing strains. Optical densities at 600 nm were determined to ensure that equivalent amounts of cells were used for the cultures.
The pellet from a 1:6-diluted overnight culture was washed with 0.5 ml wash buffer with 1 M KCl (Fisher). Another wash was performed with 0.5 ml of 50 mM wash buffer, which was followed by a final wash in protoplast buffer (wash buffer containing 23% sucrose [Fisher]). The pellet was then resuspended in protoplast buffer containing 10 mg/ml lysozyme (Sigma) and 250 U/ml mutanolysin (Sigma); this mixture was incubated at 37°C for 30 min. The protoplasts were collected by centrifugation (16,060 × g, 1 min) and discarded, while the supernatant was retained and used for Western blot analysis.
Western blot analysis of Asc10 mutant strain cell wall fractions.
Cell wall fractions were mixed with sodium dodecyl sulfate-polyacrylamide gel electrophoresis protein buffer, boiled for 5 min, electrophoresed in a 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, and transferred to a 0.45-μm nitrocellulose membrane (Schleicher and Schuell, Dassel, Germany). A polyclonal anti-Asc10 antibody (1:5,000) was used to detect the presence of Asc10; 2 μl of this antibody was absorbed with 200 μl of a surface extract from TX5128(pMSP3535) to eliminate nonspecific antibodies. The blot was developed using the Pierce (Rockford, IL) SuperSignal West Pico chemiluminescent protocol.
RESULTS
Generation of pCF10 derivatives carrying mutant alleles of prgB.
Our previous genetic analyses of the functional domains of Asc10 were conducted using prgB alleles cloned into the nisin-inducible expression vector pMSP3535 (2) and in vitro assays. As noted in the Introduction, we avoided potential problems of in vivo expression or vector stability during experimental endocarditis by using allelic exchange (12) to move prgB mutations into the native locus of pCF10. We successfully generated pCF10 derivatives encoding production of all of the Asc10 variants shown in Fig. 2 in addition to an in-frame prgB deletion mutant (containing only the first three codons and last three codons), and we used Western blot analysis to confirm that the pheromone-inducible expression of the variant proteins was similar to that observed with wild-type Asc10 (Fig. 3). This gave us confidence that any differences observed between the virulence of mutant strains and the virulence of the wild-type strain in experimental endocarditis would likely be due to functional differences among the proteins rather than to differences in expression.
FIG. 3.
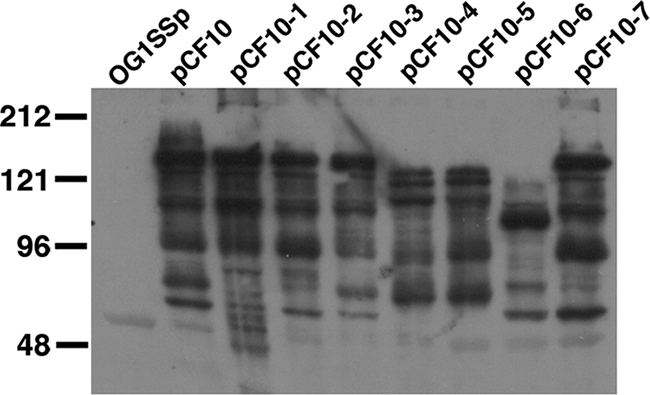
Asc10 surface expression by the Asc10 mutant strains, as determined by Western blot analysis of purified cell wall fractions. All strains used in Western blot analyses had an OG1SSp background. OG1SSp(pCF10) produced mainly a 150-kDa band, which is larger than the predicted 137-kDa band for Asc10. This may have been due to glycosylation or association of LTA with Asc10. The electrophoretic patterns of the Asc10 derivatives, including the multiple banding patterns, have been observed previously (10, 33). The prgB deletion mutant OG1SSp(pCF10-8) was analyzed on a separate Western blot, and no Asc10 surface expression was detected. The smaller bands observed for pCF10-4, pCF10-5, and pCF10-6 were due to large deletions in the prgB gene.
Mutations in prgB reduce the virulence of E. faecalis.
In previously described endocarditis studies, we examined the effects of Asc10 expression on vegetation weights and found that an Asc10+ strain [OG1SSp(pCF10)] produced larger vegetations than Asc10− strains [plasmid-free OG1SSp derivatives]. In the present study, the surgical procedures, the genetic background of the bacterial host strain (OG1SSp), and the infecting doses of bacteria were essentially the same as those used previously, but smaller and younger rabbits were used because larger animals were not available. We determined both the vegetation weights (where distinct vegetations could be found) and the bacterial loads in the infected cardiac tissues as potential indicators of virulence. All data reported here were obtained from rabbits that survived the 4-day course of the experiment. We found that the bacterial load was the most consistent and reproducible indicator of virulence and that strains carrying pCF10 derivatives having either an in-frame deletion of prgB or an allele encoding an Asc10 variant with glycine-to-alanine substitutions in both RGD motifs were much less virulent than all other strains, including an isogenic plasmid-free strain. Other prgB mutations affecting different regions of the protein each had distinct, more modest effects on virulence.
Figure 4 shows representative photographs of dissected hearts from rabbits in these experiments. The specific images were selected to illustrate the appearance of vegetations of various sizes; also shown are infected tissues where no distinct vegetations were observed. In Fig. 5A the bacterial load of the infected hearts is compared to the mass of the recovered vegetations for all of the infected animals used in these experiments. Although we generally found that large vegetations tended to have large numbers of bacteria, there was not a strong quantitative correlation in the pooled data for each bacterial strain between the bacterial load in the infected tissue and the mass of the vegetation. Based on this result, we decided to use the total bacterial load as the primary indicator of the relative ability of each strain to colonize and proliferate within the infected heart tissue.
FIG. 4.
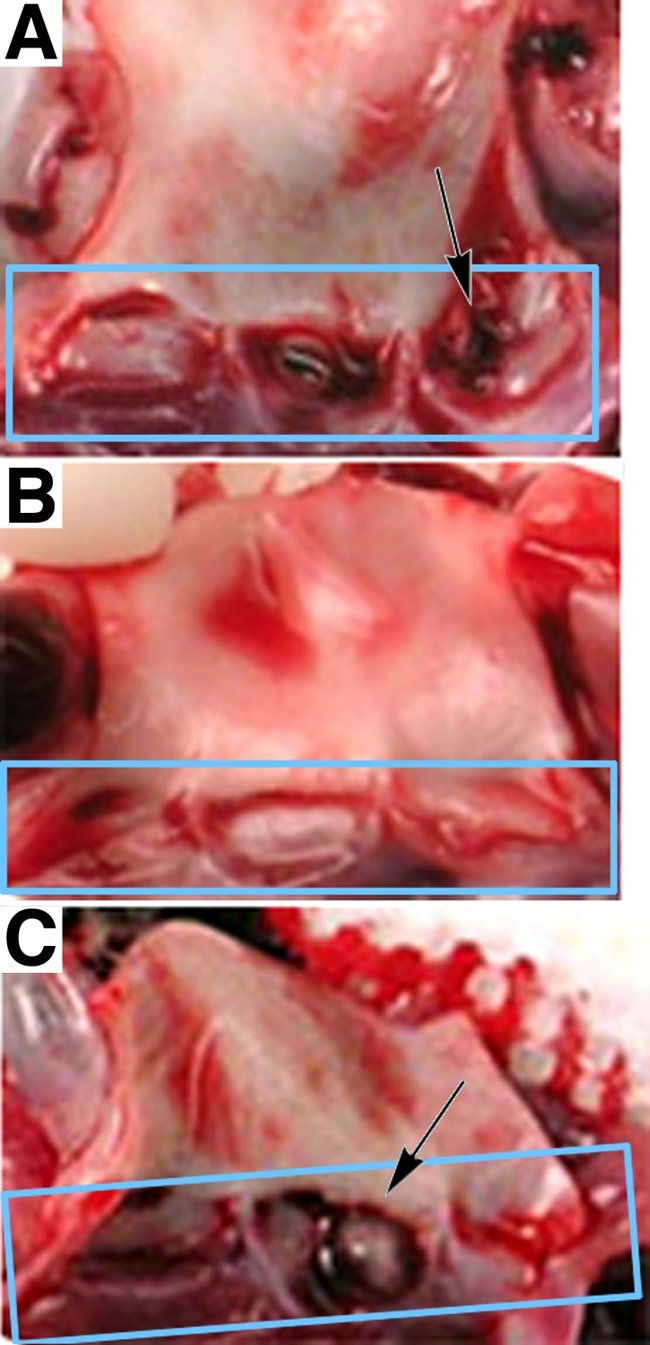
Vegetations on rabbit aortic valves. (A) Aortic valve of a rabbit infected with the central aggregation domain insertion mutant [OG1SSp(pCF10-1)]. The aortic valve is composed of three cup-shaped leaflets, as indicated by the box. The bacterial load of the vegetation removed was 1.50 × 101 CFU. The majority of the vegetation is on the right leaflet (indicated by the arrow). (B) Aortic valve of a rabbit infected with an N-terminal single RGDS mutant [OG1SSp(pCF10-3)]. The valves exhibit no apparent vegetations, and the vegetation bacterial load was 5.30 × 103 CFU. (C) Aortic valve of a rabbit infected with an Asc10− plasmid-free strain (OG1SSp), with one vegetation in the center leaflet (arrow). The vegetation bacterial load was 2.40 × 107 CFU.
FIG. 5.

(A) Plot of vegetation bacterial load (total CFU) versus vegetation weight in a rabbit endocarditis model. E. faecalis cells expressing Asc10 mutant derivatives (2 × 109 CFU) were injected intravenously after catheter placement in the carotid artery for 2 h; the catheter was removed prior to microbe injection. All vegetations on the aortic valve of each rabbit were pooled to obtain the vegetation weight and bacterial load. When no vegetations were found, the valves were removed and plated to determine the valve bacterial load. The low R2 value (0.27) indicates that there is no correlation between the sets of values. (B) Arithmetic and geometric mean vegetation bacterial loads in the rabbit endocarditis model, expressed as log10 total CFU. The dashed line indicates the minimum level of detection (1.93 log10 total CFU). *, P ≤ 0.01 for a comparison with the wild-type Asc10+ strain.
In Fig. 5B, the cumulative results of all experimental infections for the total bacterial loads are shown (the CFU/g of infected tissue from the same experiments are shown in Fig. S1 in the supplementary material). Of note were the findings for the Asc10− prgB deletion mutant OG1SSp(pCF10-8), whose bacterial load was significantly lower than those of the wild-type [OG1SSp(pCF10)] and plasmid-free (OG1SSp) strains. This deletion mutant colonized the vegetations at levels that were approximately 1/100,000 those of the strain carrying wild-type pCF10 and 1/10,000 those of the plasmid-free strain. In spite of the lack of a strong statistical correlation between bacterial loads and vegetation weights, it is noteworthy that the mean vegetation weight for the prgB deletion strain was 5.35 mg, which was the lowest weight recorded for the Asc10 mutant strains (compared to 49.2 mg for the wild-type pCF10 strain).
A strain carrying an in-frame deletion (plasmid pCF10-6) resulting in the loss of a 450-amino-acid region of Asc10 proximal to the wall-anchoring C-terminal domain exhibited virulence that was indistinguishable from that of the strain carrying wild-type pCF10, while other less drastic changes in Asc10 had much greater effects on virulence. This indicates that amino acid residues 688 to 1138 of Asc10 play a minimal role in the pathogenic effects of the protein.
The mutant exhibiting the lowest virulence in the endocarditis model was the double RGD variant [OG1SSp(pCF10-2)]. This result was striking because the prgB point mutations resulting in the single-amino-acid substitutions changing RGD to RAD represent the most subtle changes that we made in the protein (the other variants all had significant insertions or deletions in the protein). The aggregation domain variants [OG1SSp(pCF10-1) and OG1SSp(pCF10-4)] were less virulent than strains expressing the wild-type protein, but they exhibited a higher level of virulence than the double RGD mutant. Moreover, cells with the double RGD mutation were still able to clump, which also indicates that bacterial aggregation itself is not the sole determinant of Asc10-mediated virulence. The N-terminal single RGDS mutation [OG1SSp(pCF10-3)] resulted in bacterial loads that were approximately 1/10,000 those observed in infections with a strain expressing wild-type Asc10 [OG1SSp(pCF10)], whereas the bacterial loads of the double RGD mutant [OG1SSp(pCF10-2)] were less than 1/100,000 those of the wild-type strain and the virulence was essentially equivalent to the virulence of the strain carrying the prgB null allele. Mutation of the C-terminal RGDV domain had a more modest, but still significant effect on virulence, producing bacterial loads that were about 1/100 those associated with wild-type Asc10. This suggests that both the RGD motifs contribute to virulence but that the N-terminal motif may be more important. Since in the predicted topology of the protein the N terminus projects away from the cell and the C terminus is more buried in the thick cell envelope, this observation is not surprising. Possible explanations for the fact that several strains with mutant alleles of prgB were less virulent than the plasmid-free strain are considered in the Discussion.
ECM proteins are not involved in initial adherence of Asc10-mediated E. faecalis to vegetations.
Since it was previously shown that Asa1 mediated E. faecalis binding to the ECM proteins fibronectin, vitronectin, thrombospondin, and collagen type I (24), Asc10 interactions with these ECM proteins were investigated in this study. These ECM proteins could serve as the initial substrates for bacterial attachment in the valve-vegetation environment, enhancing the ability of Asc10+ E. faecalis to form the septic vegetation. Fibrinogen, fibronectin, vitronectin, and von Willebrand factor binding was tested using an ELISA in order to investigate potential interactions between purified Asc10 protein and these ECM proteins (Fig. 6). Asc10 binding at levels above background levels was not observed with the wild-type protein. However, some Asc10 variants, such as the N-terminal aggregation domain deletion variant (Fig. 2), were found to bind at much higher levels than the wild-type protein to immobilized ECM proteins (Fig. 6). It is possible that the changes in the protein structure of the Asc10 variants allowed for binding via a domain masked in the wild-type protein, but it is not known whether such variants could be produced from the full-length protein during in vivo growth. Adherence was further tested with E. faecalis cells expressing the mutant derivatives of Asc10, and no binding attributable to Asc10 was demonstrated with any of the strains. These cumulative data do not suggest strongly that Asc10 mediates bacterial binding to these ECM proteins in vivo.
FIG. 6.

Adherence of mutant Asc10 purified proteins to ECM proteins. The amounts of bound Asc10 proteins were measured by ELISA, and increasing absorbance represented larger amounts of Asc10 protein adhering to the ECM substrate. (A) Asc10 binding to fibrinogen. The N-terminal aggregation domain deletion variant protein (encoded by pCF10-4), but not the wild-type protein, was found to bind fibrinogen. (B) The N-terminal aggregation domain deletion (encoded by pCF10-4) and double aggregation domain (encoded by pCF10-5) variant proteins, but not the wild-type protein, were able to bind vitronectin. (C) The central aggregation domain insertion variant protein (encoded by pCF10-1), but not the wild-type protein, bound von Willebrand factor. The data in each graph are representative of the results of at least three experiments.
Glycosaminoglycans (GAGs) are also significant components of the valve tissue; they are found in the inner layers between single sheets of endothelial cells. Thus, it is possible that when there is valvular damage, GAGs could be exposed to serve as a binding substrate for infecting E. faecalis cells. The GAGs heparan sulfate and chondroitin sulfate were investigated for binding to E. faecalis cells. In immobilized GAG ELISAs, binding was demonstrated with whole E. faecalis cells (see Fig. S2 in the supplementary material). However, no significant differences between Asc10+ strain GAG binding and Asc10− strain GAG binding were found, suggesting that GAGs are also not involved in the initial Asc10-mediated E. faecalis adherence in the vegetation environment. Asc10-independent binding of E. faecalis to GAGs could play a role in vegetation formation by Asc10− strains.
DISCUSSION
Asc10 domains involved in the endocarditis disease process have not been characterized previously, which was the main motivation for this study. Previous studies have indicated that Asc10+ E. faecalis is able to induce large vegetations in New Zealand White rabbits (9), although smaller vegetations were still found in Asc10− plasmid-free (OG1SSp) E. faecalis-infected animals; Hirt et al. (9) observed a difference between the vegetation weights produced by infection with a strain carrying pCF10 and the vegetation weights produced by infection with a plasmid-free strain larger than the difference observed in this study. These workers also (9) examined isogenic strains carrying pCF10, a pCF10 derivative with a transposon insertion in prgB, or no plasmid and used vegetation weight as the indicator of virulence, but they did not measure vegetation bacterial loads. In the current study we examined a larger number of strains, including the strains tested previously (9) and a prgB deletion mutant. Although in this study the infected hearts with larger vegetations generally had higher bacterial loads (Fig. 5A), there was not a quantitative correlation between vegetation weights and bacterial loads. Nevertheless, our data qualitatively agree with the results described in the previous report (9), as in both studies the pCF10-containing strain induced larger vegetations and resulted in larger bacterial loads than the Asc10− strains. In the case of the pCF10-containing strain, all the infected hearts had bacterial loads larger than 106 CFU/g, while numerous rabbits infected with the prgB deletion mutant and several other prgB mutants had bacterial loads of <104 CFU/g (see Fig. S1b in the supplemental material). The smaller vegetations observed by Hirt et al. (9) in rabbits infected with a plasmid-free strain could have resulted from the fact that the animals in the present study were younger and smaller or from the fact that we used outbred rabbits, which might show variability in susceptibility to such infections. Furthermore, these experiments examined only a single endpoint at which virulence was assessed, and it is possible that some of the animals with high bacterial counts but small or no vegetations might have developed vegetations if they had been examined after longer periods of time.
This is the first time that a complete in-frame prgB deletion mutant has been employed in the rabbit endocarditis model, and the vegetation weight and bacterial load obtained with this mutant were dramatically decreased compared to the vegetation weight and bacterial load obtained with the strain carrying pCF10. The fact that a pCF10 derivative carrying a prgB null allele was much less virulent than a plasmid-free strain suggests that carriage of the pCF10 plasmid may suppress expression of other chromosomally encoded adhesins or that abundant expression of other surface proteins encoded by pCF10, such as Sec10 (encoded by prgA on the pCF10 plasmid), could mask the exposure of chromosomal surface adhesins or other proteins that could contribute to vegetation formation and share functional redundancy with Asc10. In view of the fact that abundant expression of Sec10 can essentially coat the entire cell surface (18), it is quite conceivable that cells carrying pCF10 variants expressing either no Asc10 or defective Asc10 could be less virulent than plasmid-free cells. Further experimentation is required to evaluate this possibility.
In a previous functional analysis of Asc10, the protein was expressed from cloned prgB alleles using a nisin-inducible expression system (33). Preliminary unpublished data suggested that the plasmid vector used previously might be unstable during experimental infections, so in this study we used a recently developed allelic exchange system (12) to move mutant alleles back into the native context within pCF10, where strong in vivo induction of prgB expression during in vivo growth has been demonstrated (3, 9). Western blot analysis of the strains used for these studies indicated that all of the Asc10 variants used in the experiments were expressed at similar levels and were stable enough to be detected readily (Fig. 3). Also, all of the mutants with mutations outside the previously identified aggregation domains exhibited bacterial aggregation functions, suggesting that the overall structure of the protein was intact in most cases. We cannot completely rule out the possibility that some of the phenotypic effects of the mutations tested could have resulted from reduced protein stability in vivo. However, we noted that the large deletion associated with pCF10-6 had no effect on virulence, whereas point mutations resulting in RGD-to-RAD changes, which could be considered the most subtle changes made in the protein, had the greatest effects on virulence. Therefore, we believe that most of the changes observed resulted from disruption of specific functional domains rather than from gross, nonspecific alterations in protein stability.
The simplest explanation for the cumulative results of this study is that two different functional domains of Asc10 affect the virulence of the E. faecalis bacterium in the host during experimental endocarditis (Fig. 7). Disruption of the previously identified N-terminal and central domains involved in bacterial aggregation and cell wall LTA binding had a significant effect on virulence in these studies. Previous results suggested that Asc10 mediates binding to fibrin, and our attempts to identify Asc10-mediated binding activities with other host receptors (ECM proteins and GAGs) (see Fig. S2 in the supplemental material) did not reveal any additional ligands for Asc10 binding. Recent experiments employing porcine heart valves as substrates for in vitro adherence (O. Chuang, C. Wells, and G. Dunny, unpublished data) suggested that the aggregation domains are involved in attachment to endocardiac tissue, presumably using either fibrin or a so-far-unidentified receptor. Thus, it appears that the contribution of the aggregation domains to virulence in experimental endocarditis involves initial attachment to host tissues, but further analysis is required to confirm this.
FIG. 7.

Proposed functions of Asc10 domains. SS, signal sequence; TNF-α, tumor necrosis factor alpha; IL-1, interleukin-1; IL-6, interleukin-6.
Our results appear to conflict with data from another study examining the interaction of ECM proteins and aggregation substance protein. Rozdzinski et al. found that aggregation substance was able to mediate interactions with ECM proteins (24). However, a different background strain was used (OG1X instead of OG1RF), and the aggregation substance protein examined was Asa1. Asa1 shares 90% identity with Asc10, excluding a variable region where the identity is only 30 to 40%; it is possible that the variable domain of Asa1 mediates the interactions with the ECM proteins. In our studies, we utilized a background strain (TX5128) which does not express the gelatinase or serine proteases and which facilitates purification of full-length protein; these proteases in a wild-type strain may process the Asc10 protein, resulting in exposure of ECM-binding domains. These issues remain unresolved but merit further study.
We think that the RGD motifs are involved in a second, independent function that enhances virulence. The mechanistic nature of this function is currently unknown, although our group has previously shown that these motifs are not involved in adherence and internalization of Asc10+ E. faecalis by intestinal epithelial cells (32, 34). However, there is evidence that the RGD motifs have a role in Asc10-mediated resistance to killing following ingestion of bacteria by PMNs, even though Asc10-expressing bacteria may attach more efficiently to the PMN surface (21). Recently, we observed Asc10-mediated suppression of expression of host immune function genes in experimental endocarditis, and it may be speculated that the effects of the RGD mutations observed in the present study are related to a role of these motifs in suppression of the initial host response to infection of the cardiac tissue. These motifs may also somehow promote the anti-inflammatory environment that is seen in Asc10+ E. faecalis-infected rabbits (P. M. Schlievert, O. N. Chuang, M. L. Peterson, S. M. Grindle, L. C. Case, and G. M. Dunny, submitted for publication). As these motifs mediate binding to PMNs or other professional phagocytes, it is possible that by increasing opportunities for Asc10+ E. faecalis to become enclosed in phagosomes, the bacteria are better able to interfere with the phagocyte's normal ability to kill the invading organisms. Once intracellular, Asc10+ E. faecalis may block expression of inflammatory cytokines, such as tumor necrosis factor alpha, interleukin-1β, and interleukin-6.
The possibility that Asc10 has immunomodulatory properties was also suggested in a previous study by Schlievert et al. (26). The dual-function model for the role of Asc10 in virulence could account for the fact that some of the mutants were less virulent than a plasmid-free strain. If a mutant retained the ability to attach to phagocytes but was not protected from killing, it would be less virulent than a strain not expressing Asc10. Alternatively, expression of an Asc10 variant could reduce or mask the surface expression of another chromosomally encoded surface virulence factor, such as the Ace protein (23), without providing a positive contribution to virulence.
Supplementary Material
Acknowledgments
This research was supported by NIH grant HL51987 and by MinnCResT training fellowship T32DE07288.
We acknowledge Laura Case and John Mleziva for assistance during the endocarditis procedures and Tim Leonard for work on images.
Editor: V. J. DiRita
Footnotes
Published ahead of print on 27 October 2008.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Bensing, B. A., and G. M. Dunny. 1993. Cloning and molecular analysis of genes affecting expression of binding substance, the recipient-encoded receptor(s) mediating mating aggregate formation in Enterococcus faecalis. J. Bacteriol. 1757421-7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryan, E. M., T. Bae, M. Kleerebezem, and G. M. Dunny. 2000. Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44183-190. [DOI] [PubMed] [Google Scholar]
- 3.Chandler, J. R., H. Hirt, and G. M. Dunny. 2005. A paracrine peptide sex pheromone also acts as an autocrine signal to induce plasmid transfer and virulence factor expression in vivo. Proc. Natl. Acad. Sci. USA 10215617-15622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow, J. W., L. A. Thal, M. B. Perri, J. A. Vazquez, S. M. Donabedian, D. B. Clewell, and M. J. Zervos. 1993. Plasmid-associated hemolysin and aggregation substance production contribute to virulence in experimental enterococcal endocarditis. Antimicrob. Agents Chemother. 372474-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunny, G., M. Yuhasz, and E. Ehrenfeld. 1982. Genetic and physiological analysis of conjugation in Streptococcus faecalis. J. Bacteriol. 151855-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunny, G. M., B. L. Brown, and D. B. Clewell. 1978. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc. Natl. Acad. Sci. USA 753479-3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durack, D. T. 1975. Experimental bacterial endocarditis. IV. Structure and evolution of very early lesions. J. Pathol. 11581-89. [DOI] [PubMed] [Google Scholar]
- 8.Durack, D. T., and P. B. Beeson. 1972. Experimental bacterial endocarditis. I. Colonization of a sterile vegetation. Br. J. Exp. Pathol. 5344-49. [PMC free article] [PubMed] [Google Scholar]
- 9.Hirt, H., P. M. Schlievert, and G. M. Dunny. 2002. In vivo induction of virulence and antibiotic resistance transfer in Enterococcus faecalis mediated by the sex pheromone-sensing system of pCF10. Infect. Immun. 70716-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreft, B., R. Marre, U. Schramm, and R. Wirth. 1992. Aggregation substance of Enterococcus faecalis mediates adhesion to cultured renal tubular cells. Infect. Immun. 6025-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kristich, C. J., J. R. Chandler, and G. M. Dunny. 2007. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57131-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kristich, C. J., D. A. Manias, and G. M. Dunny. 2005. Development of a method for markerless genetic exchange in Enterococcus faecalis and its use in construction of a srtA mutant. Appl. Environ. Microbiol. 715837-5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazmanian, S. K., H. Ton-That, and O. Schneewind. 2001. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 401049-1057. [DOI] [PubMed] [Google Scholar]
- 14.McCormick, J. K., H. Hirt, G. M. Dunny, and P. M. Schlievert. 2000. Pathogenic mechanisms of enterococcal endocarditis. Curr. Infect. Dis. Rep. 2315-321. [DOI] [PubMed] [Google Scholar]
- 15.Megran, D. W. 1992. Enterococcal endocarditis. Clin. Infect. Dis. 1563-71. [DOI] [PubMed] [Google Scholar]
- 16.Moellering, R. C., Jr. 1992. Emergence of Enterococcus as a significant pathogen. Clin. Infect. Dis. 141173-1176. [DOI] [PubMed] [Google Scholar]
- 17.Moreillon, P., and Y. A. Que. 2004. Infective endocarditis. Lancet 363139-149. [DOI] [PubMed] [Google Scholar]
- 18.Olmsted, S. B., S. L. Erlandsen, G. M. Dunny, and C. L. Wells. 1993. High-resolution visualization by field emission scanning electron microscopy of Enterococcus faecalis surface proteins encoded by the pheromone-inducible conjugative plasmid pCF10. J. Bacteriol. 1756229-6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peacock, S. J., N. P. Day, M. G. Thomas, A. R. Berendt, and T. J. Foster. 2000. Clinical isolates of Staphylococcus aureus exhibit diversity in fnb genes and adhesion to human fibronectin. J. Infect. 4123-31. [DOI] [PubMed] [Google Scholar]
- 20.Perkins, S., E. J. Walsh, C. C. Deivanayagam, S. V. Narayana, T. J. Foster, and M. Hook. 2001. Structural organization of the fibrinogen-binding region of the clumping factor B MSCRAMM of Staphylococcus aureus. J. Biol. Chem. 27644721-44728. [DOI] [PubMed] [Google Scholar]
- 21.Rakita, R. M., N. N. Vanek, K. Jacques-Palaz, M. Mee, M. M. Mariscalco, G. M. Dunny, M. Snuggs, W. B. Van Winkle, and S. I. Simon. 1999. Enterococcus faecalis bearing aggregation substance is resistant to killing by human neutrophils despite phagocytosis and neutrophil activation. Infect. Immun. 676067-6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rantz, L. A., and W. M. M. Kirby. 1943. Enterococcic infections. An evaluation of the importance of fecal streptococci and related organisms in the causation of human disease. Arch. Intern. Med. 71516-528. [Google Scholar]
- 23.Rich, R. L., B. Kreikemeyer, R. T. Owens, S. LaBrenz, S. V. Narayana, G. M. Weinstock, B. E. Murray, and M. Hook. 1999. Ace is a collagen-binding MSCRAMM from Enterococcus faecalis. J. Biol. Chem. 27426939-26945. [DOI] [PubMed] [Google Scholar]
- 24.Rozdzinski, E., R. Marre, M. Susa, R. Wirth, and A. Muscholl-Silberhorn. 2001. Aggregation substance-mediated adherence of Enterococcus faecalis to immobilized extracellular matrix proteins. Microb. Pathog. 30211-220. [DOI] [PubMed] [Google Scholar]
- 25.Schaberg, D. R., D. H. Culver, and R. P. Gaynes. 1991. Major trends in the microbial etiology of nosocomial infection. Am. J. Med. 9172S-75S. [DOI] [PubMed] [Google Scholar]
- 26.Schlievert, P. M., P. J. Gahr, A. P. Assimacopoulos, M. M. Dinges, J. A. Stoehr, J. W. Harmala, H. Hirt, and G. M. Dunny. 1998. Aggregation and binding substances enhance pathogenicity in rabbit models of Enterococcus faecalis endocarditis. Infect. Immun. 66218-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh, K. V., X. Qin, G. M. Weinstock, and B. E. Murray. 1998. Generation and testing of mutants of Enterococcus faecalis in a mouse peritonitis model. J. Infect. Dis. 1781416-1420. [DOI] [PubMed] [Google Scholar]
- 28.Styriak, I., A. Laukova, C. Fallgren, and T. Wadstrom. 1999. Binding of selected extracellular matrix proteins to enterococci and Streptococcus bovis of animal origin. Curr. Microbiol. 39327-0335. [DOI] [PubMed] [Google Scholar]
- 29.Trotter, K. M., and G. M. Dunny. 1990. Mutants of Enterococcus faecalis deficient as recipients in mating with donors carrying pheromone-inducible plasmids. Plasmid 2457-67. [DOI] [PubMed] [Google Scholar]
- 30.Vanek, N. N., S. I. Simon, K. Jacques-Palaz, M. M. Mariscalco, G. M. Dunny, and R. M. Rakita. 1999. Enterococcus faecalis aggregation substance promotes opsonin-independent binding to human neutrophils via a complement receptor type 3-mediated mechanism. FEMS Immunol. Med. Microbiol. 2649-60. [DOI] [PubMed] [Google Scholar]
- 31.Veltrop, M. H., H. Beekhuizen, and J. Thompson. 1999. Bacterial species- and strain-dependent induction of tissue factor in human vascular endothelial cells. Infect. Immun. 676130-6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waters, C. M., M. H. Antiporta, B. E. Murray, and G. M. Dunny. 2003. Role of the Enterococcus faecalis GelE protease in determination of cellular chain length, supernatant pheromone levels, and degradation of fibrin and misfolded surface proteins. J. Bacteriol. 1853613-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waters, C. M., and G. M. Dunny. 2001. Analysis of functional domains of the Enterococcus faecalis pheromone-induced surface protein aggregation substance. J. Bacteriol. 1835659-5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waters, C. M., H. Hirt, J. K. McCormick, P. M. Schlievert, C. L. Wells, and G. M. Dunny. 2004. An amino-terminal domain of Enterococcus faecalis aggregation substance is required for aggregation, bacterial internalization by epithelial cells and binding to lipoteichoic acid. Mol. Microbiol. 521159-1171. [DOI] [PubMed] [Google Scholar]
- 35.Weigel, L. M., D. B. Clewell, S. R. Gill, N. C. Clark, L. K. McDougal, S. E. Flannagan, J. F. Kolonay, J. Shetty, G. E. Killgore, and F. C. Tenover. 2003. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 3021569-1571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.