

Abstract
The intracellular gram-negative bacterial pathogen Salmonella enterica serovar Typhimurium gains entry into nonphagocytic cells by manipulating the assembly of the host actin cytoskeleton. S. enterica serovar Typhimurium entry requires a functional type III secretion system, a conduit through which bacterial effector proteins are directly translocated into the host cytosol. We and others have previously reported the enhancement of tyrosine kinase activities during Salmonella serovar Typhimurium infection; however, neither specific kinases nor their targets have been well characterized. In this study, we investigated the roles of the cellular Abelson tyrosine kinase (c-Abl) and the related protein Arg in the context of serovar Typhimurium infection. We found that bacterial internalization was inhibited by more than 70% in cells lacking both c-Abl and Arg and that treatment of wild-type cells with a pharmaceutical inhibitor of the c-Abl kinase, STI571 (imatinib), reduced serovar Typhimurium invasion efficiency to a similar extent. Bacterial infection led to enhanced phosphorylation of two previously identified c-Abl substrates, the adaptor protein CT10 regulator of kinase (CrkII) and the Abelson-interacting protein Abi1, a component of the WAVE2 complex. Furthermore, overexpression of the nonphosphorylatable form of CrkII resulted in decreased invasion. Taken together, these findings indicate that c-Abl is activated during S. enterica serovar Typhimurium infection and that its phosphorylation of multiple downstream targets is functionally important in bacterial internalization.
The intracellular bacterial pathogen Salmonella enterica serovar Typhimurium can effectively invade intestinal epithelial cells by inducing a dynamic reorganization of the host actin cytoskeleton. This reorganization is achieved by injection of an array of bacterial effector proteins through a molecular syringe apparatus, referred to as the type III secretion system (T3SS) (18). These secreted effectors include molecules that can directly modulate actin filament assembly and other that coordinate cytoskeleton reorganization through manipulation of host signaling pathways (for a review, see reference 28). For example, the effector protein SopE promotes activation of the Rho family GTPase Rac1, which is necessary for the extension of actin-rich membrane protrusions that engulf the attached bacteria.
Previous work has shown that host tyrosine kinases are also activated as a consequence of Salmonella infection (30), but the roles of specific kinases or their substrates in Salmonella internalization remain poorly understood. c-Src has been shown to function in the internalization of Shigella flexneri (15) and other bacteria (Listeria monocytogenes [38] and Yersinia pseudotuberculosis [29]). However, we recently found that neither c-Src nor the related kinases Fyn and Yes played a significant role in Salmonella internalization (35). Although the nonreceptor tyrosine kinase focal adhesion kinase (FAK) is necessary for Salmonella invasion, its function in this context is structural and does not require its kinase domain (35).
The Abelson family of nonreceptor tyrosine kinases contains two members, c-Abl and the closely related kinase Arg. These ubiquitously expressed kinases are unique in their possession of actin binding domains, which allow their direct interaction with both globular and filamentous actin (43, 45). Both c-Abl and Arg have been shown to act as signaling intermediates between surface receptors, such as integrins and growth factor receptors, and the intracellular cytoskeletal network. Perhaps not surprisingly, many bacterial and viral pathogens have been shown to exploit Abl signaling pathways for their pathogenesis (11, 40, 41). For instance, c-Abl is required for the intracellular motility of Shigella flexneri, where it is recruited to comet tails and phosphorylates the actin nucleation-promoting factor N-WASP (8). Similarly, c-Abl facilitates the formation of actin pedestals during enteropathogenic Escherichia coli infection by phosphorylating and binding to the translocated intimin receptor Tir (2).
As noted above, the secreted Salmonella effector protein SopE promotes activation of the Rho family GTPase Rac1 protein, which is necessary for the cytoskeletal rearrangements that drive bacterial internalization (13, 31). Rac1-induced actin polymerization requires intermediate players such as the heptameric Arp2/3 complex (12) and the Arp2/3-activating WAVE2 complex (23, 36). We have previously shown that depletion of either WAVE2 or another component of the complex, Abi1 (Abelson-interacting protein 1), using RNA interference (RNAi) significantly reduced Salmonella entry (36). Since Abi1 is often associated with Abl family members, we hypothesized that Abl kinases also contribute to the signal-mediated cytoskeleton reorganization induced during S. enterica serovar Typhimurium invasion.
Here we report that c-Abl becomes selectively enriched at sites of active serovar Typhimurium entry and that cells lacking both c-Abl and Arg are more resistant than wild-type (WT) cells to serovar Typhimurium infection. Furthermore, blocking Abl kinase activities in epithelial cells with the pharmacological inhibitor STI571 (imatinib) also inhibits Salmonella infection. Mechanistically, c-Abl-mediated tyrosine phosphorylation of both CrkII and Abi1 is enhanced during host cell invasion, and inhibition of CrkII phosphorylation impairs bacterial entry. Collectively, our data support a role for Abl kinases in Salmonella serovar Typhimurium pathogenesis.
MATERIALS AND METHODS
Bacterial strains.
S. enterica serovar Typhimurium WT strain SL1344 and its isogenic entry-deficient derivative VV341 (22), and the Salmonella pathogenicity island 2 (SPI-2) strain deficient for T3SS (ssaV) (19) have been described previously. Bacteria were made invasive as previously described by Lee and Falkow (26). Briefly, a single colony was inoculated into LB broth and grown for 8 h under aerobic conditions and then diluted back to 1:1,000 under oxygen-limiting conditions (i.e., not shaken) for overnight growth. Bacteria were harvested the following morning when their density reached ∼5 × 108 to 7 × 108 CFU/ml.
Cell culture.
Epithelial MDCK and HeLa cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 2 mM l-glutamine, 1 mM sodium pyruvate, 4.5 g/liter glucose, and antibiotics. Mouse embryonic fibroblasts (MEFs) lacking c-Abl and Arg and corresponding isogenic WT MEFs were gifts from A. J. Koleske (Yale University, New Haven, CT). MEFs were maintained in the same medium described for epithelial cells. All cells were kept at 37°C with 5% CO2.
Plasmids, antibodies, and chemical inhibitors.
The hemagglutinin (HA)-tagged c-Abl construct and the Abl inhibitor STI571 were generously provided by M. A. Schwartz (University of Virginia, Charlottesville, VA). myc-CrkII and the myc-CrkII Y221F mutant were gifts from K. Vuori (Burnham Institute, La Jolla, CA). FLAG-tagged WT Rac1 and N17 Rac1 were provided by Ian Macara (University of Virginia). The DsRed plasmid was a gift from J. Brumell (University of Toronto, Ontario, Canada). Mouse monoclonal anti-Abl (8E9; 1:250) and anti-Crk (1:1,000) antibodies were obtained from BD Biosciences (San Jose, CA). Monoclonal anti-HA (16B12; 1:1,000) antibody was purchased from Covance (Berkeley, CA). Monoclonal anti-myc (9E10; 1:1,000) antibody was purified from hybridoma supernatants by the University of Virginia hybridoma facility. Polyclonal antiphosphotyrosine 221 CrkII (1:1,000) was purchased from Cell Signaling (Danvers, MA). Rac1 monoclonal antibody (1:1,000) was purchased from Upstate Biotechnology (Lake Placid, NY). Monoclonal Abi1 antibody was a gift from G. Scita (IFOM, Milan, Italy). A combination of 4G10 (1:1,000) and PY99 (1:1,000) antiphosphotyrosine antibodies was used for detecting phospho-Abi1 and were purchased from Upstate (Charlottesville, VA) and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. Fluorescein isothiocyanate-phalloidin was purchased from Molecular Probes (Eugene, OR). Alexa 647-conjugated phalloidin (1:50) was purchased from Invitrogen (Carlsbad, CA). Polyclonal rabbit antilipopolysaccharide antibody (anti-LPS; 1:500) was obtained from Difco. Polyclonal goat anti-rabbit and anti-mouse antibodies conjugated with Alexa 488 fluorophore (1:500) were both purchased from Molecular Probes (Eugene, OR). Texas Red-conjugated goat anti-rabbit antibody (1:1,500) was purchased from Jackson ImmunoResearch (West Grove, PA). Goat anti-mouse secondary antibodies conjugated to horseradish peroxidase were obtained from Amersham (Piscataway, NJ). Blots were visualized using Pierce (Rockford, IL) SuperSignal ECL reagents.
Imatinib treatment.
Where indicated, cells were preincubated with 10 μM STI571 in the tissue culture medium described above, prior to infection, at 37°C with 5% CO2. Cells were then washed once in Hanks' buffered saline solution (HBSS) and equilibrated in 10 μM STI571-HBSS. Infections were also carried out in the presence of 10 μM STI571.
Transient transfection.
Where noted, ectopic expression was achieved by transient transfection using Lipofectamine 2000 purchased from Invitrogen (Carlsbad, CA). Transfection was done by following the manufacturer's recommended protocol.
RNAi.
HeLa cells were transfected with either predesigned SMARTpool short interfering RNA (siRNA) oligonucleotides targeting human Abl1 (GenBank accession number NM_007313) or nontargeting siRNA no. 2 oligonucleotides purchased from Dharmacon (Lafayette, CO). Cells were transfected at 24 h after plating and again at 48 h, using Lipofectamine RNAiMax reagent from Invitrogen (Carlsbad, CA) according to the manufacturer's protocol. Cells were then infected at 72 h postplating and assayed using the gentamicin resistance assay described below.
Gentamicin resistance assays.
The gentamicin resistance assay has been described previously (13). Briefly, cells were seeded at 2 × 105 cells/well onto 24-well culture dishes 18 h prior to infection. They were then infected at a multiplicity of infection (MOI) of 50 (or as indicated) for 60 min with the WT SL144 strain diluted in HBSS. Infected cells were then treated with 500 μg/ml gentamicin for 90 min prior to lysing them in 1% Triton X-100. CFU were enumerated by plating aliquots of lysates onto MacConkey agar. Values were normalized to the total number of internalized S. enterica serovar Typhimurium bacteria in the control sample.
Immunoprecipitation and Western blotting.
HeLa cells were serum starved for 4 to 18 h prior to infection. Cells were then infected with WT SL1344 or its isogenic strains (VV341 or the ssaV mutant) for the indicated time, followed by lysis in RIPA lysis buffer (50 mM Tris [pH 7.4], 1% NP-40, 150 mM NaCl, 0.5% deoxycholate, 10% glycerol, 0.1% sodium dodecyl sulfate [SDS]) supplemented with 1 mM sodium vanadate, 50 mM sodium fluoride, and a cocktail of protease inhibitors (0.1 mM phenylmethylsulfonyl fluoride and 1 μg/ml each of pepstatin, leupeptin, and antipain). Abi1 phosphorylation was assayed as described previously (9). Briefly, cells were serum starved overnight, infected for 30 min, and then lysed in buffer containing 10 mM sodium phosphate, 1 mM EDTA, 100 mM NaCl, 2 mM EGTA, 10 mM sodium fluoride, 1 mM sodium vanadate, 10 mM Na2P2O7, 0.5% deoxycholate, 0.05% SDS, 5% glycerol, 1% NP-40, and protease inhibitors. After they were precleared with Sepharose CL4B beads (Sigma, St. Louis, MO), the lysates were immunoprecipitated with the indicated antibodies for 4 to 24 h at 4°C. Whole-cell lysate controls represented 1/10 of the total lysate. Samples were loaded onto 10 to 13% SDS-polyacrylamide gels and probed using the indicated antibodies. Each experiment was performed independently at least three times.
Immunofluorescence microscopy.
One day prior to infection, cells were seeded onto glass coverslips. After 30 min of infection, cells were fixed with 4% paraformaldehyde, followed by blocking and permeabilization in 10% normal goat serum (NGS) with 0.2% saponin in phosphate-buffered saline (PBS). Primary and secondary antibodies were each diluted in blocking buffer and incubated with samples for 30 min. After being washed, coverslips were mounted using Fluoromount G. Images were acquired with a ×60 objective on a Nikon Eclipse E800 microscope (Melville, NY) and a QImaging Retiga cooled charge-coupled device camera (Burnaby, BC, Canada).
Dual-color immunofluorescence microscopy.
Details of the dual-color immunofluorescence microscopy method have been described previously (35). Briefly, cells were infected for 30 min and fixed in 2% paraformaldehyde, followed by blocking in 10% NGS-PBS (in the absence of saponin). Extracellular bacteria were stained with rabbit anti-LPS, followed by Alexa 488-conjugated anti-rabbit immunoglobulin G. Cell membranes were then permeabilized using 10% NGS-PBS with 0.2% saponin. Total (extracellular and intracellular) bacteria were then labeled by incubation with rabbit anti-LPS antibody, followed by Texas Red-conjugated goat anti-rabbit immunoglobulin G. Under these conditions, extracellular bacteria appeared yellow, while the intracellular bacteria pool remained red.
Rac1 activation assay.
The GTP-Rac1 pull-down assay was done as described previously (13). Infected cells were lysed in 50 mM Tris-HCl (pH 7.5), 2 mM MgCl2, 0.1 M NaCl, 1% NP-40, and 10% glycerol with protease inhibitors. Active Rac1 was pulled down with a fusion of glutathione S-transferase (GST) to the p21-binding domain of Pak (PBD) for 30 min, washed four times in lysis buffer, and resuspended in 2× SDS sample buffer. Samples were resolved by 13% SDS-polyacrylamide gel electrophoresis.
Statistical analysis.
The two-tailed Student t test was used to analyze all data, where applicable. A P value of <0.05 was considered statistically significant.
RESULTS
c-Abl and/or Arg is necessary for efficient S. enterica serovar Typhimurium infection.
c-Abl and Arg are nonreceptor tyrosine kinases that play an integral role in regulating actin cytoskeleton dynamics. To determine whether c-Abl is recruited to the sites of Salmonella invasion, endogenous c-Abl was detected in HeLa cells infected with WT S. enterica serovar Typhimurium (strain SL1344) expressing DsRed. At 30 min postinfection, cells were fixed and processed for immunofluorescence microscopy. c-Abl was detected by using monoclonal anti-Abl antibody (8E9) and Alexa 647-conjugated phalloidin to detect filamentous actin. As shown in Fig. 1A, c-Abl became concentrated at the sites of active bacterial internalization, where it colocalized with filamentous actin. This localization was not due to nonspecific binding of the anti-Abl antibody, as staining of HeLa cells expressing HA-tagged c-Abl also revealed the same association with the invasive bacteria (Fig. 1B).
FIG. 1.

Abl family kinases are required for efficient Salmonella serovar Typhimurium internalization. (A and B) HeLa cells were infected for 30 min with WT S. enterica serovar Typhimurium strain SL1344 expressing DsRed and then fixed and stained for either endogenous c-Abl (A) or exogenous HA-tagged c-Abl (B) (green). Sites of active invasion were detected with Alexa 647-conjugated phalloidin (blue). (C) WT MEFs or isogenic cells lacking c-Abl and Arg (Abl−/− Arg−/−) were infected with SL1344 for 30 min. Internalization efficiency was quantified by using the two-color immunofluorescence assay described in Materials and Methods. The number of intracellular bacteria per cell per field in WT cells was set to a value of 1, and internalization efficiency in Abl−/− Arg−/− cells is expressed as a fraction of that normalized value. Values and standard deviations were derived from the average of three independent experiments (P < 0.021). (D) HeLa cells were either depleted of endogenous c-Abl by using siRNA oligonucleotides (siAbl), mock depleted with nontargeting siRNA (scramble), or not transfected (mock). Infection and quantification of invasion efficiency were done as described in Materials and Methods. Values are normalized to the total number of internalized bacteria in the mock-treated control cells. P < 0.05. IB, immunoblotting.
To determine if Abl family kinases are necessary for Salmonella infection, we measured the efficiency of bacterial internalization in MEFs derived from mice lacking both c-Abl and Arg (25), using a two-color immunofluorescence-based assay as previously described (35). Briefly, infected cells were fixed and extracellular bacteria were stained prior to permeabilization by using an anti-LPS antibody, followed by Alexa 488-conjugated secondary antibody (Fig. 1A and B). Cells were then permeabilized, and total bacteria were labeled with anti-LPS and Texas Red-conjugated secondary antibodies (Fig. 1A and B). As shown in Fig. 1C, the infection of c-Abl−/− Arg−/− MEFs was reduced by 76% (±15% standard deviation [SD]) relative to that of MEFs derived from WT mice. Qualitatively similar results were obtained using a standard gentamicin resistance assay (data not shown).
Because it is formally possible that c-Abl−/− Arg−/− MEFs may have acquired adaptive mutations in the course of selection that could affect bacterial entry independently of c-Abl/Arg, we also measured internalization efficiency in HeLa cells depleted of endogenous c-Abl, using siRNA. As shown in Fig. 1D, the extent of inhibition was quantitatively similar to that observed for c-Abl−/− Arg−/− MEFs. Together, these data suggest that Abl kinases have an important role in Salmonella invasion.
We have previously shown that another nonreceptor tyrosine kinase, FAK, is necessary for Salmonella internalization into host cells but that its kinase activity is not required, suggesting that FAK acts as a scaffold in this context (35). To determine if c-Abl kinase activity is necessary for Salmonella entry, we made use of a pharmacological inhibitor specific for Abl family kinases, STI571, also known as imatinib (Gleevec) (5). As shown in Fig. 2A, pretreatment of HeLa cells with 10 μM STI571 reduced Salmonella internalization by approximately 65% (±14% SD), similar to the inhibition observed for cells lacking both c-Abl and Arg (Fig. 1C and D). Furthermore, we also demonstrated that this impaired invasion was not specific to HeLa cells, as it also occurred in MDCK cells (Fig. 2B). Importantly, STI571 had no effect on bacterial replication in broth culture (data not shown), nor did it substantially interfere with serovar Typhimurium adherence to the cell surface (data not shown). It should be noted that STI571 has been shown to inhibit other tyrosine kinases (6). To address this, we depleted c-Abl by using siRNA oligonucleotides and pretreated these cells with 10 μM STI571. Following 60 min of infection, internalized bacteria were then enumerated by a standard gentamicin resistance assay. We found that knockdown of c-Abl alone or treatment of cells with STI571 alone reduced bacterial internalization to similar extents (data not shown). However, the combination of c-Abl knockdown and STI571 treatment led to an additional, statistically significant inhibition of bacterial uptake. We conclude that while c-Abl is an important regulator of S. enterica serovar Typhimurium entry, other kinases targeted by STI571 (including possibly Arg) also contribute to Salmonella internalization.
FIG. 2.

Abl kinase activities are required for S. enterica serovar Typhimurium internalization. (A) HeLa cells were pretreated with 10 μM STI571 for 60 min prior to infection for 1 h with SL1344 at an MOI of ∼50. Infected cells were incubated for 90 min with gentamicin and then lysed in 1% Triton X-100. CFU were enumerated by plating aliquots of lysates on MacConkey agar. Values were normalized to the total number of S. enterica serovar Typhimurium bacteria in the control sample. Data represent the means ± standard deviations for triplicate determinations (P < 0.035). (B) MDCK cells were pretreated with STI571 as described for panel A and infected for 30 min. Internalized bacteria were enumerated using the two-color immunofluorescence assay as described in Materials and Methods. Data represent the means ± standard errors of the means of three independent experiments (P < 0.02).
Rac1 activation occurs independently of c-Abl kinase activity.
To determine if the inhibition of bacterial internalization was due to impaired actin reorganization, HeLa cells were infected in the presence or absence of STI571 and stained for filamentous actin by using fluorescent phalloidin. Because the treatment of cells with STI571 induced such a pronounced decrease in bacterial uptake, we were surprised to find that it did not noticeably impair the formation of actin-rich foci around attached bacteria and that no obvious differences were detected in either the sizes or the morphologies of these structures (Fig. 3A). Normal actin ruffles were also observed in HeLa cells depleted of c-Abl, suggesting that c-Abl is not directly involved in the initial steps of Salmonella invasion (Fig. 3B).
FIG. 3.

STI571 does not inhibit Salmonella serovar Typhimurium-induced ruffle formation or Rac1 GTPase activation. (A) HeLa cells were pretreated with 10 μM STI571 for 60 min. Cells were then infected with SL1344 expressing DsRed for 30 min. Actin was visualized using fluorescein isothiocyanate-conjugated phalloidin. (B) Endogenous c-Abl was depleted from HeLa cells, using siRNA oligonucleotides. Cells were then infected for 30 min with SL1344 expressing DsRed and processed for immunofluorescence microscopy as described for panel A. (C) HeLa cells were pretreated with 10 μM STI571 as described above. At the indicated time point postinfection (pi), cells were lysed, and active Rac1 (Rac1-GTP) was precipitated with GST-PBD beads as described in Materials and Methods. Bands representing GST-PBD and Rac1-GTP are indicated. WCL, whole-cell lysate; IB, immunoblotting.
Both Salmonella and Shigella induce activation of Rho family GTPase Rac1 to trigger cytoskeleton reorganization. The activation of Rac1 by S. flexneri, which is due to activation of the host Rac1 guanine nucleotide exchange factor Dock180 protein, is inhibited by STI571 (7). However, S. enterica serovar Typhimurium activates Rac1 directly using the secreted effector proteins SopE/E2 (13, 21). To determine if Rac1 activation is inhibited by STI571 in the context of Salmonella infection, we monitored the levels of Rac1-GTP by using a well-characterized pull-down assay (13). Briefly, cells were infected with serovar Typhimurium in the presence or absence of 10 μM STI571 and then lysed, and active Rac1 was recovered from the lysates by using a GST fusion construct containing the Rac1 binding domain of the serine-threonine kinase PAK1 (GST-PBD). We found that the levels of Rac1 activation were similar in both untreated and STI571-pretreated cells (Fig. 3C), suggesting that the inhibition observed for Salmonella serovar Typhimurium internalization is not due to impaired translocation of SopE or the downstream activation of Rac1.
To confirm that the formation of actin foci observed in STI571-treated cells was dependent on Rac1 activation, we ectopically expressed epitope-tagged WT or dominant negative (N17) Rac1 in HeLa cells prior to STI571 treatment. Cells were then infected for 30 min and processed for immunofluorescence microscopy. As depicted in Fig. 4A, actin ruffles were readily visualized in cells expressing WT Rac1, both in the presence and in the absence of the Abl kinase inhibitor. In contrast, cells expressing N17 Rac1 had little to no F-actin recruitment in either control or STI571-treated cells (Fig. 4B). Collectively, these data indicate that Rac1 does not require c-Abl kinase activities for recruitment to the plasma membrane, activation, or the induction of cytoskeleton rearrangements at the early steps of Salmonella entry.
FIG. 4.

Actin focus formation is dependent on Rac1 but not c-Abl. HeLa cells expressing FLAG-tagged WT Rac1 (A) or Rac1 N17 (B) were pretreated with 10 μM STI571 for 60 min. Cells were then infected with SL1344 expressing DsRed for 30 min, fixed, and stained for Rac1 (green) and F-actin (blue).
The CrkII adaptor molecule is phosphorylated during early stages of serovar Typhimurium infection.
The best-characterized substrate of c-Abl is the adaptor protein CrkII, which consists of an N-terminal SH2 domain and two C-terminal SH3 domains (17, 44). c-Abl has been shown to phosphorylate CrkII at a conserved tyrosine residue, Y221, which resides in the linker region between the two SH3 domains, and is the only kinase known to phosphorylate this site. To determine if CrkII associates with c-Abl during Salmonella infection, we first examined CrkII localization in infected cells. HeLa cells expressing myc-CrkII were infected for 30 min with the WT Salmonella strain and then fixed and processed for immunofluorescence microscopy. As shown in Fig. 5A, CrkII was clearly present at active areas of Salmonella internalization, where it colocalized with filamentous actin. This observation is in agreement with previous findings by Boyle et al. (4), who demonstrated the localization of endogenous CrkII to Salmonella serovar Typhimurium invasion sites.
FIG. 5.

Phosphorylation of CrkII at Y221 is induced by S. enterica serovar Typhimurium infection and requires an intact T3SS. (A) HeLa cells were infected with SL1344 expressing DsRed for 30 min. myc-CrkII (green) and actin-rich ruffles (blue) were visualized with monoclonal anti-myc antibody and Alexa 647-conjugated phalloidin, respectively. (B) At the indicated time points postinfection, HeLa cell lysates were collected and immunoprecipitated (IP) with a mouse anti-Crk antibody, followed by immunoblotting (IB) with rabbit anti-pY221-CrkII. WT SL1344 bacteria were used at an MOI of ∼50, while the VV341 strain was used at an MOI of ∼100. WCL, whole-cell lysates. (C) HeLa cells were infected with SL1344 or isogenic mutant strains (VV341 or the ssaV mutant) for 20 min, all at an MOI of ∼50. Lysates were collected and processed as described in the legend to panel B. (D) HeLa cells, either untransfected (control) or expressing WT CrkII or CrkII Y221F, were infected for 60 min with SL1344 at an MOI of ∼50, and internalization was quantified by using the gentamicin resistance assay described in Materials and Methods. Data shown represent an average of three independent experiments. *, P < 0.009.
We next determined whether Salmonella infection resulted in enhanced phosphorylation of CrkII at Y221. For this purpose, HeLa cell lysates were harvested at 10, 20, 40, and 60 min postinfection, and endogenous CrkII was immunoprecipitated using an anti-CrkII antibody. Phosphorylation at Y221 was then detected by immunoblotting using an antibody specific for CrkII phosphorylated at Y221. As shown in Fig. 5B, infection with Salmonella serovar Typhimurium induced a significant increase in CrkII Y221 phosphorylation at the 10-min time point, which remained elevated for 60 min postinfection. To confirm that CrkII phosphorylation is dependent on c-Abl in this context, we examined CrkII phosphorylation in cells depleted of c-Abl by RNAi. HeLa cells depleted of c-Abl exhibited dramatically reduced basal levels of CrkII phosphorylation, which did not increase in response to serovar Typhimurium infection (data not shown). This indicates that c-Abl is the primary kinase responsible for CrkII phosphorylation in both uninfected and infected cells.
To determine if CrkII phosphorylation is dependent on the SPI-1-encoded invasion machinery, we used an isogenic SPI-1 T3SS-deficient strain (VV341). Since this strain is intrinsically less adherent to the cell surface than the WT SL1344 strain, we determined the titer for the VV341 strain inoculum to identify an MOI that provided an equivalent number of adherent bacteria (data not shown), which proved to be approximately double that of SL1344. Accordingly, HeLa cells were infected with VV341 bacteria at an MOI of 100, while the WT SL1344 strain was used at an MOI of 50. Under these conditions, we found that CrkII phosphorylation was significantly reduced in cells infected with the noninvasive serovar Typhimurium strain relative to that in cells infected with the WT strain, suggesting the importance of SPI-1 effectors in c-Abl kinase activation (Fig. 5B). In contrast to the VV341 bacteria, the strain that was defective in the assembly of the SPI-2 T3SS (ssaV) induced CrkII phosphorylation as effectively as did the WT strain (Fig. 5C). Furthermore, if WT SL1344 bacteria were grown to induce maximal SPI-1 expression by using aerobic conditions, increased CrkII phosphorylation was detected within 5 min of infection (data not shown).
We next sought to determine if CrkII Y221 phosphorylation is a prerequisite for bacterial internalization. For this assay, HeLa cells were transfected with either myc-tagged WT CrkII or the nonphosphorylatable mutant (CrkII Y221F), and internalization was quantified by using a standard gentamicin resistance assay. As shown in Fig. 5D, the expression of WT CrkII had no effect on internalization efficiency, relative to that of mock-transfected controls. In contrast, infection of cells expressing the CrkII Y221F mutant was reduced by 30% (±11%). Given that the transfection efficiency of HeLa cells in this assay is approximately 50% (data not shown), this value probably underestimates the impact of the Y221F mutant's expression on bacterial entry. Taken together, these data suggest that phosphorylation of the CrkII adaptor protein by Abl tyrosine kinases plays an important role in S. enterica serovar Typhimurium internalization.
Salmonella infection stimulates the c-Abl-dependent phosphorylation of Abi1.
We have previously shown that Salmonella infection induces translocation of the WASP-like protein WAVE2 to the plasma membrane, where it activates the Arp2/3 complex to promote the formation of branched actin filaments (36). WAVE2 is part of a multiprotein complex containing the Rac-binding protein PIR121, the adaptor proteins Abi1 and Nap1, and an associated protein, HSPC300 (23). In that study, we demonstrated that RNAi-mediated depletion of Abi1, which was originally identified as a binding partner for c-Abl, dramatically reduced the efficiency of Salmonella infection. Importantly, recent evidence indicates that both WAVE2 and Abi1 are phosphorylated by Abl and that this is necessary for the translocation of the WAVE2 complex to the plasma membrane (27). These observations led us to hypothesize that c-Abl might also function in modulating the WAVE2/Abi1 complex during bacterial infection, as it does in other cellular contexts (37, 14).
To test this hypothesis, HeLa cells were infected with WT SL1344 or strain VV341 for 30 min in the presence or absence of 10 μM STI571. Cells were then lysed, and endogenous Abi1 was immunoprecipitated, followed by immunoblotting with antiphosphotyrosine antibodies (Fig. 6). Compared to the uninfected control, Salmonella infection induced a readily detectable increase in Abi1 phosphorylation. Both basal and Salmonella-induced Abi1 phosphorylation were completely blocked by incubation of the cells with STI571, indicating that this modification is due to Abl-mediated phosphorylation. Furthermore, phosphorylation was not induced by the noninvasive strain VV341, indicating that it is dependent on active invasion (Fig. 6, lane 5). These data indicate that phosphorylation of Abi1 is enhanced in response to serovar Typhimurium infection and, together with our previous data, suggest that this is an important signaling event in bacterial internalization.
FIG. 6.
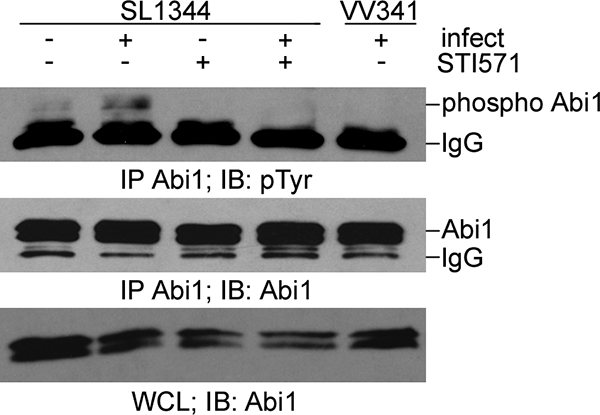
Abi1 is tyrosine phosphorylated during infection in an Abl-dependent manner. HeLa cells were pretreated with 10 μM STI571 (STI571) prior to infection with strain SL1344 or VV341 for 30 min. Abi1 immunoprecipitates were immunoblotted (IB) for tyrosine phosphorylation using a combination of 4G10 and PY99 antibodies as described in Materials and Methods. IP, immunoprecipitation; IgG, immunoglobulin G.
DISCUSSION
Abl tyrosine kinases play an important role in actin cytoskeleton remodeling in response to various extracellular stimuli. Therefore, it is not surprising that bacterial pathogens, such as Helicobacter pylori (41, 32), Shigella flexneri (8, 7), and enteropathogenic E. coli (40), have evolved mechanisms to exploit these conserved molecules for their pathogenic purposes. In the present study, we investigated whether Abl kinases are also involved in Salmonella pathogenesis. We found that Salmonella infection leads to the recruitment of c-Abl to sites of bacterial entry and triggers Abl kinase activation through a mechanism that requires an intact bacterial invasion machinery. Moreover, both cells derived from c-Abl−/− Arg−/− mice and cells depleted of c-Abl by siRNA are poorly infected by S. enterica serovar Typhimurium. Similarly, treatment of cells with the pharmacological inhibitor STI571 inhibited bacterial internalization to the same extent as deletion/depletion of Abl and Arg. Taken together, these findings strongly suggest that Abl kinases are important regulators of Salmonella entry into host cells (Fig. 7).
FIG. 7.

A role for Salmonella-induced activation of c-Abl kinase in bacterial entry. Upon attachment, S. enterica serovar Typhimurium induces c-Abl kinase activation through an as-yet-unknown mechanism. Abl activation is SPI-1 dependent and may involve one or more secreted effector proteins (?, green oval). Effectors could activate Abl directly or indirectly through an upstream host protein. Alternatively, bacterial attachment may directly activate host proteins (?, blue oval) that then activate Abl. Once activated, Abl then triggers phosphorylation of Abi1 (leading to enhanced actin polymerization), as well as the adaptor molecule CrkII at tyrosine residue 221. The role of CrkII in Salmonella internalization remains unknown.
Comparisons between Salmonella and Shigella.
Both Salmonella and Shigella induce a form of “triggered” phagocytosis that requires the function of the Rho family GTPase Rac1 protein. While both pathogens use translocated effector proteins to stimulate Rac1 activation in host cells, the mechanisms used to achieve this activation differ. In the case of S. flexneri, an endogenous Rac1 gauanine nucleotide exchange factor, Dock180, is stimulated by the effector protein IpgB1 (20). Importantly, Rac1 activation by Shigella infection is inhibited by STI571, indicating that Dock180 lies downstream of c-Abl/Arg in a signaling pathway leading to Rac1 activation (7). However, Salmonella infection activates Rac1 directly by using the secreted effector proteins SopE/E2 (21). In agreement with this, and in contrast to Shigella infection, we found that Rac1 activation induced by Salmonella infection was not inhibited by STI571, suggesting that other components that function either downstream or in parallel with Rac1 are regulated by Abl.
How does c-Abl promote Salmonella internalization?
Perhaps the best-characterized substrate of the Abl kinase is the adaptor protein CrkII, which is part of an evolutionarily conserved signaling module that functions in cell migration, invasion, phagocytosis, and survival (10). CrkII has a simple modular domain organization consisting of a single N-terminal SH2 domain that binds phosphotyrosines and two C-terminal SH3 domains that bind proline-rich domains on other proteins. The related protein CrkL lacks the N-terminal SH3 domain. CrkII is phosphorylated by c-Abl on a conserved tyrosine residue (Y221) located in the linker region between the two SH3 domains, resulting in an intramolecular interaction between the N-terminal SH2 domain and phospho-Y221 (17, 33). This phosphorylation is thought to induce a “closed” or inhibitory conformation that impairs the association of CrkII with several of its binding partners, including the scaffolding protein p130Cas (10, 24, 33, 34). Inhibition of CrkII Y221 phosphorylation by genetic deletion of Abl/Arg or treatment of cells with STI571 enhances the coupling of Crk to p130Cas and promotes cell migration (24). Conversely, expression of an activated c-Abl mutant uncouples CrkII from p130Cas and inhibits migration (24). Paradoxically, Abl and Arg are activated by integrin ligation, which is necessary for motility, and phosphorylated CrkII is concentrated at the leading edge of migrating cells (1, 42). It is thought that this phosphorylated pool may become rapidly dephosphorylated by local tyrosine phosphatases, allowing dynamic association/dissociation of the Crk/Cas complex (10).
Burton and colleagues have shown that infection of cells with S. flexneri induced phosphorylation of CrkII in an Abl-dependent manner and that bacterial invasion efficiency was significantly reduced by the expression of the nonphosphorylatable CrkII Y221F mutant (7). Similarly, expression of a CrkII SH2 domain mutant that blocks phosphotyrosine binding (R38V) was found to dramatically reduce internalization of both S. flexneri (3) and Y. pseudotuberculosis (46). Here we show that CrkII phosphorylation is similarly induced by Salmonella serovar Typhimurium infection and that expression of the Y221F mutant inhibits bacterial internalization. CrkII phosphorylation occurs early in the infection process (Fig. 5B), suggesting that it has an upstream role in the assembly of the phagocytic apparatus (Fig. 7).
We have previously shown that p130Cas plays an important role in the internalization of S. enterica serovar Typhimurium, where it is necessary for proper assembly of the phagocytic machinery (35). p130Cas is itself phosphorylated by c-Abl. Intriguingly, reconstitution of p130Cas−/− cells with mutant constructs lacking the Crk-binding domain failed to restore invasion, unlike WT p130Cas cells (35), suggesting that the p130Cas-CrkII interaction has an important function in Salmonella-induced cytoskeletal reorganization. We hypothesize that the dynamic association/dissociation of this complex, mediated by c-Abl and/or Arg, is necessary for completion of the phagocytic process. In agreement with this hypothesis, it has been shown that treatment of human monocytes with STI571 impairs their ability to phagocytose both opsonized and nonopsonized particles (16).
It should be noted that, using an RNAi approach, Boyle et al. (4) found that HeLa cells lacking CrkII showed no defects in Salmonella serovar Typhimurium invasion, which seems to contradict the data presented here (Fig. 5D). Those authors hypothesized that CrkII may function redundantly with other related adaptors. It is likely that CrkII Y221F functions as a dominant negative inhibitor, interacting with its binding partners in a manner that is not regulated by c-Abl and impairing its dynamic interaction with other signaling components. In cells depleted of endogenous CrkII, such inhibition would not be apparent.
Another target of the Abl kinase is the WAVE2 complex, which promotes actin filament branching by activating the Arp2/3 complex. WAVE2 exists in cells as part of a multiprotein complex with the adaptor proteins Abi1 and Nap1, the Rac-binding subunit PIR121, and another protein, HSPC300 (23). Abi1 was originally identified as a binding partner for c-Abl, and both Abi1 and WAVE2 are phosphorylated by Abl, enhancing the ability of the WAVE2 complex to promote actin polymerization (27, 39). We have previously shown that Abi1 and WAVE2 are necessary for efficient Salmonella serovar Typhimurium invasion (36). Here we show that Abi1 is tyrosine phosphorylated in response to infection with serovar Typhimurium and that this phosphorylation is abrogated by treatment of cells with STI571, indicating that it is dependent on c-Abl and/or Arg (Fig. 6). However, it appears that inhibition of this phosphorylation does not impair formation of actin-rich foci at sites of bacterial attachment (see below), suggesting that it is not necessary for membrane ruffle formation. Whether it is required for a later stage of Salmonella internalization remains to be determined.
Because the internalization of Salmonella serovar Typhimurium was substantially inhibited (∼70%) in cells lacking both c-Abl and Arg (Fig. 1C) and also following pharmacological blockade of both kinases by STI571 (Fig. 2), we initially assumed that this inhibition would result from impaired cytoskeleton reorganization. However, we were surprised to discover that the actin-rich foci formed under these conditions were similar in both size and morphology to those observed in control cells (Fig. 3A and B). This finding is in contrast to our previous observations for cells lacking FAK, where foci were not detectable at all, or in cells lacking p130Cas, where foci were abnormally large but the actin within them was aberrantly organized (35). It is possible that the loss of c-Abl activity results in aberrant actin assembly at a level that is below the resolving power of fluorescence microscopy. Alternatively, c-Abl may selectively regulate a late step in bacterial entry such as phagosome closure. Clearly, further research will be needed to elucidate the role of Abl in Salmonella pathogenesis.
Acknowledgments
We thank Martin Schwartz (University of Virginia, Charlottesville, VA) for reagents and helpful discussions; Kristina Vuori (Burnham Institute for Medical Research, La Jolla, CA) for the CrkII Y221F construct; John Brumell (University of Toronto, Canada) for the DsRed plasmid; Giorgio Scita (IFOM, Milan, Italy) for Abi1 antibody; and Anthony Koleske (Yale University, CT) for c-Abl−/− Arg−/− MEFs. We also thank the members of the Casanova laboratory for support.
This research was funded by a National Institutes of Health grant (DK58536) to J.E.C. and NRSA Cancer and Infectious Disease training grants to K.T.L.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 20 October 2008.
REFERENCES
- 1.Abassi, Y. A., and K. Vuori. 2002. Tyrosine 221 in Crk regulates adhesion-dependent membrane localization of Crk and Rac and activation of Rac signaling. EMBO J. 214571-4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bommarius, B., D. Maxwell, A. Swimm, S. Leung, A. Corbett, W. Bornmann, and D. Kalman. 2007. Enteropathogenic Escherichia coli Tir is an SH2/3 ligand that recruits and activates tyrosine kinases required for pedestal formation. Mol. Microbiol. 631748-1768. [DOI] [PubMed] [Google Scholar]
- 3.Bougnères, L., S. E. Girardin, S. A. Weed, A. V. Karginov, J. C. Olivo-Marin, J. T. Parsons, P. J. Sansonetti, and G. T. Van Nhieu. 2004. Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J. Cell Biol. 166225-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle, E. C., N. F. Brown, J. H. Brumell, and B. B. Finlay. 2007. Src homology domain 2 adaptors affect adherence of Salmonella enterica serovar Typhimurium to non-phagocytic cells. Microbiology 1533517-3526. [DOI] [PubMed] [Google Scholar]
- 5.Buchdunger, E., J. Zimmermann, H. Mett, T. Meyer, M. Müller, B. J. Druker, and N. B. Lydon. 1996. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 56100-104. [PubMed] [Google Scholar]
- 6.Buchdunger, E., C. L. Cioffi, N. Law, D. Stover, S. Ohno-Jones, B. J. Druker, and N. B. Lydon. 2000. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 295139-145. [PubMed] [Google Scholar]
- 7.Burton, E. A., R. Plattner, and A. M. Pendergast. 2003. Abl tyrosine kinases are required for infection by Shigella flexneri. EMBO J. 225471-5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burton, E. A., T. N. Oliver, and A. M. Pendergast. 2005. Abl kinases regulate actin comet tail elongation via an N-WASP-dependent pathway. Mol. Cell Biol. 258834-8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campa, F., N. Machuy, A. Klein, and T. Rudel. 2006. A new interaction between Abi-1 and betaPIX involved in PDGF-activated actin cytoskeleton reorganisation. Cell Res. 16759-770. [DOI] [PubMed] [Google Scholar]
- 10.Chodniewicz, D., and R. L. Klemke. 2004. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim. Biophys. Acta 169263-76. [DOI] [PubMed] [Google Scholar]
- 11.Coyne, C. B., and J. M. Bergelson. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124119-131. [DOI] [PubMed] [Google Scholar]
- 12.Criss, A. K., and J. E. Casanova. 2003. Coordinate regulation of Salmonella enterica serovar Typhimurium invasion of epithelial cells by the Arp2/3 complex and Rho GTPases. Infect. Immun. 712885-2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Criss, A. K., D. M. Ahlgren, T. S. Jou, B. A. McCormick, and J. E. Casanova. 2001. The GTPase Rac1 selectively regulates Salmonella invasion at the apical plasma membrane of polarized epithelial cells. J. Cell Sci. 1141331-1341. [DOI] [PubMed] [Google Scholar]
- 14.Dai, Z., and A. M. Pendergast. 1995. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes Dev. 92569-2582. [DOI] [PubMed] [Google Scholar]
- 15.Dehio, C., M. C. Prévost, and P. J. Sansonetti. 1995. Invasion of epithelial cells by Shigella flexneri induces tyrosine phosphorylation of cortactin by a pp60c-src-mediated signalling pathway. EMBO J. 142471-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dewar, A. L., K. V. Doherty, T. P. Hughes, and A. B. Lyons. 2005. Imatinib inhibits the functional capacity of cultured human monocytes. Immunol. Cell Biol. 8348-56. [DOI] [PubMed] [Google Scholar]
- 17.Feller, S. M., B. Knudsen, and H. Hanafusa. 1994. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J. 132341-2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galán, J. E., and H. Wolf-Watz. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444567-573. [DOI] [PubMed] [Google Scholar]
- 19.Guy, R. L., L. A. Gonias, and M. A. Stein. 2000. Aggregation of host endosomes by Salmonella requires SPI2 translocation of SseFG and involves SpvR and the fms-aroE intragenic region. Mol. Microbiol. 371417-1435. [DOI] [PubMed] [Google Scholar]
- 20.Handa, Y., M. Suzuki, K. Ohya, H. Iwai, N. Ishijima, A. J. Koleske, Y. Fukui, and C. Sasakawa. 2007. Shigella IpgB1 promotes bacterial entry through the ELMO-Dock180 machinery. Nat. Cell Biol. 9121-128. [DOI] [PubMed] [Google Scholar]
- 21.Hardt, W. D., L. M. Chen, K. E. Schuebel, X. R. Bustelo, and J. E. S. Galán. 1998. Typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93815-826. [DOI] [PubMed] [Google Scholar]
- 22.Hueck, C. J., M. J. Hantman, V. Bajaj, C. Johnston, C. A. Lee, and S. I. Miller. 1995. Salmonella typhimurium secreted invasion determinants are homologous to Shigella Ipa proteins. Mol. Microbiol. 18479-490. [DOI] [PubMed] [Google Scholar]
- 23.Innocenti, M., A. Zucconi, A. Disanza, E. Frittoli, L. B. Areces, A. Steffen, T. E. Stradal, P. P. Di Fioere, M. F. Carlier, and G. Scita. 2004. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat. Cell Biol. 6319-327. [DOI] [PubMed] [Google Scholar]
- 24.Kain, K. H., and R. L. Klemke. 2001. Inhibition of cell migration by Abl family tyrosine kinases through uncoupling of Crk-CAS complexes. J. Biol. Chem. 27616185-16192. [DOI] [PubMed] [Google Scholar]
- 25.Koleske, A. J., A. M. Gifford, M. L. Scott, M. Nee, R. T. Bronson, K. A. Miczek, and D. Baltimore. 1998. Essential roles for Abl and Arg tyrosine kinases in neurulation. Neuron 211259-1272. [DOI] [PubMed] [Google Scholar]
- 26.Lee, C. A., and S. Falkow. 1990. The ability of Salmonella to enter mammalian cells is affected by bacterial growth state. Proc. Natl. Acad. Sci. USA 874304-4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leng, Y., J. Zhang, K. Badour, E. Arpaia, S. Freeman, P. Cheung, M. Siu, and K. Siminovitch. 2005. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc. Natl. Acad. Sci. USA 1021098-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ly, K. T., and J. E. Casanova. 2007. Mechanisms of Salmonella entry into host cells. Cell Microbiol. 92103-2111. [DOI] [PubMed] [Google Scholar]
- 29.McGee, K., M. Zettl, M. Way, and M. Fällman. 2001. A role for N-WASP in invasin-promoted internalisation. FEBS Lett. 50959-65. [DOI] [PubMed] [Google Scholar]
- 30.Murli, S., R. O. Watson, and J. E. Galán. 2001. Role of tyrosine kinases and the tyrosine phosphatase SptP in the interaction of Salmonella with host cells. Cell Microbiol. 3795-810. [DOI] [PubMed] [Google Scholar]
- 31.Patel, J. C., and J. E. Galán. 2005. Manipulation of the host actin cytoskeleton by Salmonella: all in the name of entry. Curr. Opin. Microbiol. 810-15. [DOI] [PubMed] [Google Scholar]
- 32.Poppe, M., S. M. Feller, G. Romer, and S. Wessler. 2007. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 263462-3472. [DOI] [PubMed] [Google Scholar]
- 33.Ren, R., Z. S. Ye, and D. Baltimore. 1994. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev. 8783-795. [DOI] [PubMed] [Google Scholar]
- 34.Rosen, M. K., T. Yamazaki, G. D. Gish, C. M. Kay, T. Pawson, and L. E. Kay. 1995. Direct demonstration of an intramolecular SH2-phosphotyrosine interaction in the Crk protein. Nature 374477-479. [DOI] [PubMed] [Google Scholar]
- 35.Shi, J., and J. E. Casanova. 2006. Invasion of host cells by Salmonella typhimurium requires focal adhesion kinase and p130Cas. Mol. Biol. Cell 174698-4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi, J., G. Scita, and J. E. Casanova. 2005. WAVE2 signaling mediates invasion of polarized epithelial cells by Salmonella typhimurium. J. Biol. Chem. 28029849-29855. [DOI] [PubMed] [Google Scholar]
- 37.Shi, Y., K. Alin, and S. P. Goff. 1995. Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes Dev. 92583-2597. [DOI] [PubMed] [Google Scholar]
- 38.Sousa, S., D. Cabanes, L. Bougnères, M. Lecuit, P. Sansonetti, G. Tran-Van-Nhieu, and P. Cossart. 2007. Src, cortactin and Arp2/3 complex are required for E-cadherin-mediated internalization of Listeria into cells. Cell Microbiol. 92629-2643. [DOI] [PubMed] [Google Scholar]
- 39.Stuart, J. R., F. H. Gonzalez, H. Kawai, and Z. M. Yuan. 2006. c-Abl interacts with the WAVE2 signaling complex to induce membrane ruffling and cell spreading. J. Biol. Chem. 28131290-31297. [DOI] [PubMed] [Google Scholar]
- 40.Swimm, A., B. Bommarius, Y. Li, D. Cheng, P. Reeves, M. Sherman, D. Veach, W. Bornmann, and D. Kalman. 2004. Enteropathogenic Escherichia coli use redundant tyrosine kinases to form actin pedestals. Mol. Biol. Cell 153520-3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tammer, I., S. Brandt, R. Hartig, W. König, and S. Backert. 2007. Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 1321309-1319. [DOI] [PubMed] [Google Scholar]
- 42.Ting, A. Y., K. H. Kain, R. L. Klemke, and R. Y. Tsien. 2001. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc. Natl. Acad. Sci. USA 9815003-15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Etten, R. A., P. K. Jackson, D. Baltimore, M. C. Sanders, P. T. Matsudaira, and P. A. Janmey. 1994. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J. Cell Biol. 124325-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang, B., T. Mysliwiec, S. M. Feller, B. Knudsen, H. Hanafusa, and G. D. Kruh. 1996. Proline-rich sequences mediate the interaction of the Arg protein tyrosine kinase with Crk. Oncogene 131379-1385. [PubMed] [Google Scholar]
- 45.Wang, Y., A. L. Miller, M. S. Mooseker, and A. J. Koleske. 2001. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc. Natl. Acad. Sci. USA 9814865-14870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weidow, C. L., D. S. Black, J. B. Bliska, and A. H. Bouton. 2000. CAS/Crk signalling mediates uptake of Yersinia into human epithelial cells. Cell. Microbiol. 2549-560. [DOI] [PubMed] [Google Scholar]