

Abstract
The protective antigen (PA) component of anthrax toxin binds the I domain of the receptor ANTXR1. Integrin I domains convert between open and closed conformations that bind ligand with high and low affinities, respectively; this process is regulated by signaling from the cytoplasmic domains. To assess whether intracellular signals might influence the interaction between ANTXR1 and PA, we compared two splice variants of ANTXR1 that differ only in their cytoplasmic domains. We found that cells expressing ANTXR1 splice variant 1 (ANTXR1-sv1) bound markedly less PA than did cells expressing a similar level of the shorter splice variant ANTXR1-sv2. ANTXR1-sv1 but not ANTXR1-sv2 associated with the actin cytoskeleton, although disruption of the cytoskeleton did not affect binding of ANTXR-sv1 to PA. Introduction of a cytoplasmic domain missense mutation found in the related receptor ANTXR2 in a patient with juvenile hyaline fibromatosis impaired actin association and increased binding of PA to ANTXR1-sv1. These results suggest that ANTXR1 has two affinity states that may be modulated by cytoplasmic signals.
Anthrax toxin is comprised of three proteins that assemble into toxic complexes on the surfaces of host cells (2, 32). Protective antigen (PA) binds to either of two structurally related cellular receptors and is then cleaved by a furin-like protease to release a 20-kDa amino-terminal fragment (8, 17, 31, 43). The remaining PA63 fragment oligomerizes to form a ring-shaped heptamer that binds the catalytic moieties of the toxin, edema factor and lethal factor (LF) (19, 28, 30). The assembled toxin complex is internalized by receptor-mediated endocytosis and is trafficked into a low-pH endosome where the PA63 heptamer converts from a prepore to a membrane-inserted pore, allowing translocation of edema factor and LF into the cytosol (1, 18, 29, 37).
The two anthrax toxin receptors, ANTXR1 (ATR/TEM8) and ANTXR2 (CMG2), are widely expressed in human tissues, and both are predicted to have multiple isoforms from alternative splicing (8, 9, 43). Both receptors are thought to be involved in cell matrix interactions since the extracellular domain of ANTXR1 was shown to bind collagen type I and to immunoprecipitate with the C5 domain of collagen α3, while that of ANTXR2 was shown to bind collagen type IV and laminin (5, 15, 33, 49). ANTXR1 functions as an adhesion molecule, as it was demonstrated to mediate cell spreading via an actin-dependent mechanism (49).
The extracellular von Willebrand factor type A or integrin-inserted domain of ANTXR1/2 binds PA (8, 21, 41, 43). This domain is also found in a variety of other proteins, including integrins, and often mediates protein-protein interactions (50). Ligand binding by integrins is modulated by structural changes in the I domain that convert it between a low-affinity “closed” conformation and a high-affinity “open” conformation (45). The conversion from a closed to an open conformation alters the coordination of a divalent cation by residues in the I domain that comprise the metal ion-dependent adhesion site (MIDAS) (12, 22, 46). The divalent cation has a higher electrophilicity in the open conformation, which facilitates binding of an acidic residue in the ligand (12).
Structural studies revealed that the MIDAS metal of the ANTXR2 I domain binds the acidic residue D683 in PA and that the complexed I domain resembles the open conformation of the αM integrin I domain (20, 21, 41). Although a structure of the ANTXR1 I domain has not been solved, MIDAS residues DXSXS…T…D are conserved between ANTXR1 and ANTXR2, and biochemical studies suggest that PA binds ANTXR1 in a similar manner to binding of ANTXR2 (7, 42). Mutation of the amino-terminal residue of the ANTXR1 MIDAS motif, D50, disrupts metal coordination and was shown to reduce binding to PA (7). Furthermore, mutation of T118, which is predicted to prevent adoption of the open confirmation, or mutation of D683 in PA impairs the interaction (7, 39). Although it has been hypothesized that all I domains that contain a perfect MIDAS motif can undergo an integrin-like conformational switch (6), there are no data that demonstrate whether the wild-type I domain of either ANTXR1 or ANTXR2 can exist in a closed conformation.
There are three isoforms of ANTXR1, namely, ANTXR1-sv1, ANTXR1-sv2, and ANTXR1-sv3 (44). ANTXR1-sv3 does not contain a transmembrane domain, so this variant does not function as an anthrax toxin receptor. ANTXR1-sv1 and ANTXR1-sv2 have identical extracellular domains (consisting of an I domain and a membrane-proximal region) and transmembrane domains but have different cytoplasmic tails (44). The cytoplasmic tail of ANTXR1-sv1 contains 221 amino acids, and that of ANTXR1-sv2 contains 25 amino acids; the first 21 amino acids of the tails are identical, but the next 4 differ between variants (44).
Here we investigated functional differences between ANTXR1-sv1 and ANTXR1-sv2. Cells that expressed ANTXR1-sv1 bound less PA than did cells that expressed ANTXR1-sv2. Moreover, we found that ANTXR1-sv1 but not ANTXR1-sv2 interacted with the actin cytoskeleton, although disruption of the actin cytoskeleton by use of latrunculin A did not increase the binding of PA to ANTXR1-sv1. Introducing a point mutation into the ANTXR1-sv1 cytoplasmic domain did, however, disrupt the receptor's interaction with the cytoskeleton and increased binding to PA. These results suggest that an adaptor binds the ANTXR1-sv1 cytoplasmic domain and may be responsible for modulating PA binding affinity and linkage of the receptor to the actin cytoskeleton. Thus, this work provides evidence that the ANTXR1 I domain is regulated cytoplasmically.
MATERIALS AND METHODS
Proteins.
PA and LF were purified as described previously (3, 16). PASSSR is a furin-resistant mutant of PA that has been described previously (4). For flow cytometric assays, PASSSRK563C was labeled with Alexa fluor 488 maleimide according to the manufacturer's instructions (Invitrogen).
Plasmids and transfection.
The ANTXR1-sv1-HA plasmid has been described previously (13). Briefly, a hemagglutinin (HA) tag was inserted into pcDNA3 (Invitrogen) between ApaI and NheI restriction sites. The ANTXR1-sv1 and ANTXR1-sv2 genes were amplified using the forward primer 5′-CGCGGATCCGCCATGGCCACGGCGG-3′ and the reverse primers 5′-CGCTCTAGAGACAGAAGGCCTTGGAGG-3′ and 5′-CGCTCTAGATTTTATTTTATTTTCCTCACTCTC-3′, respectively. PCR products were digested using BamHI and XbaI, and the resulting product was ligated into pcDNA3-HA. QuikChange site-directed mutagenesis (Stratagene) was used according to the manufacturer's instructions to introduce mutations into ANTXR1. The T118A mutation in ANTXR1-sv1 and ANTXR1-sv2 was introduced using the oligonucleotide 5′-CTGCCAGGAGGAGACGCCTACATGCATGAAGG-3′ and its complement. The ANTXR1-sv1 tail truncations were amplified from the pcDNA3-ANTXR1-sv1-HA plasmid by use of the forward primer 5′-GACTCACTATAGGGAGACC-3′ and the following reverse primers: 5′-GCGTCTAGACAACTTAGCACCTTCTTCTGTGG-3′ for ANTXR1-sv11-410 and 5′-GCGTCTAGACATCTTGACTCTTGCATTCTTTG-3′ for ANTXR1-sv11-420. The PCR products were digested with BamHI and XbaI and ligated into the pcDNA3-HA plasmid. The ANTXR2489 gene was amplified using the forward primer 5′-GAGACCCAAGCTTGCCGCCATGGTGGCGGAGC-3′ and the reverse primer 5′-CGCTCTAGAAGCAGTTAGCTCTTTCTCAATAC-3′. The PCR product was digested using HindIII and XbaI, and the resulting product was ligated into pcDNA3-HA. The Y381C mutation in ANTXR2 was introduced using the oligonucleotide 5′-GATGCTTCCTATTGTGGTGGTCGAGGG-3′ and its complement. The Y383C mutation in ANTXR1-sv1 was introduced using the oligonucleotide 5′-GACGCCTCTTATTGTGGTGGGAGAGGC-3′ and its complement. The ANTXR1-sv1-EGFP and ANTXR1-sv2-EGFP plasmids were described previously (43). CHOR1.1 and U20S cells were transfected using Superfect (Qiagen) or polyethyleneimine according to the manufacturer's instructions.
Cell culture.
CHOR1.1 receptor-negative cells were maintained in F12 medium with 10% fetal bovine serum (Invitrogen) and 1× penicillin-streptomycin (Sigma). For CHOR1.1 cells stably expressing ANTXR1, the same medium was used, with the addition of 0.25 mg/ml G418. U20S cells were maintained in HG-DME medium supplemented with 10% fetal bovine serum and 1× penicillin-streptomycin and 0.25 mg/ml G418.
Biotinylation assays.
Cells were treated with 1 mg/ml EZ-Link sulfo-NHS-LC-biotin (Pierce) for 2 h on ice and then washed twice with TBS (20 mM Tris, pH 7.6, 140 mM NaCl) to quench unreacted biotin prior to lysis with EBC lysis buffer (50 mM Tris-HCl, pH 8, 0.1 M NaCl, 0.5% [vol/vol] NP-40, 50 μg/ml phenylmethylsulfonyl fluoride). ImmunoPure immobilized streptavidin beads (Pierce) were added to normalized protein lysates and rotated at 4°C for 2 h. The beads were washed three times with lysis buffer, and the biotinylated proteins were eluted from the beads with sodium dodecyl sulfate (SDS) sample buffer and then subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to nitrocellulose, and biotinylated ANTXR1-HA proteins were detected using polyclonal anti-HA antibody (Sigma).
PA binding assays.
Cells were seeded into 24-well plates and grown overnight at 37°C. Confluent monolayers were treated with 10−8 M PASSSR at 4°C for 2 h in F12 medium supplemented with 1% bovine serum albumin (BSA) and buffered with 20 mM HEPES, pH 8. The cells were washed three times with phosphate-buffered saline (PBS) prior to lysis with lysis buffer. Equal amounts of total protein lysates (40 μg) were subjected to SDS-PAGE (7.5% polyacrylamide gel). Proteins were transferred to nitrocellulose, and PA was detected using anti-PA antibody raised against full-length PA in rabbits (a gift from R. J. Collier); β-actin was detected using monoclonal anti-β-actin antibody clone AC-15 (Sigma).
For binding assays performed in suspension, cells were treated with PBS supplemented with 10 mM EDTA until cells detached from the wells. Cells were then washed with PBS to remove the EDTA, and PASSSR was added in TBS, pH 7.4, supplemented with either 2 mM EDTA or 2 mM MnSO4. Cells were rotated for 2 h at 4°C and processed as described above.
For kinetic association assays (Table 1), confluent monolayers of CHOR1.1 cells stably expressing either ANTXR1-sv1-HA or ANTXR1-sv2-HA were exposed to PBS supplemented with 10 mM EDTA until cells detached. Cells were distributed equally into tubes and washed twice with PBS to remove any EDTA. Cells were suspended in F12 medium supplemented with 20 mM HEPES, pH 8.0, and 1% BSA and treated with either 50 nM of PASSSR for 0, 15, 30, 45, 60, 90, 120, 240, or 360 min or 150 nM of PASSSR for 0, 5, 10, 15, 20, 25, 30, 60, or 360 min. Cells were then washed with PBS prior to lysis with EBC lysis buffer. Forty micrograms of total protein lysate was separated by SDS-PAGE and transferred to a nitrocellulose membrane. PA was detected using anti-PA antibody, and levels were quantified using a Kodak 4000MM Pro gel image station and Kodak 1D image analysis software. Levels of bound PA were averaged from four independent experiments and plotted as percentages, with the value for 360 min with 150 μM of PASSSR set to 100%. Curves were globally fitted to a kinetic association model by use of PRISM software. Data for association rate constants (kon) and dissociation rate constants (koff) were derived from the association curves, and the equilibrium binding constant (Kd) was determined from the equation Kd = koff/kon.
TABLE 1.
Constants derived from kinetic association assaysa
| Variant | Association rate constant (kon) (M−1 min−1) | Dissociation rate constant (koff) (min−1) | Equilibrium binding constant (Kd) (M) |
|---|---|---|---|
| ANTXR1-sv1 | 4.5 × 105 ± 5.8 × 104 | 9.9 × 10−3 ± 2.5 × 10−3 | 2.1 × 10−8 ± 6 × 10−9 |
| ANTXR1-sv2 | 5.5 × 105 ± 6.9 × 104 | 9.1 × 10−3 ± 2.8 × 10−3 | 1.6 × 10−8 ± 5 × 10−9 |
Data are means ± standard errors of the means. The P values for the differences in kon, koff, and Kd are 0.30, 0.84, and 0.54, respectively.
For the experiment shown in Fig. 4, cells were exposed to PBS supplemented with 10 mM EDTA until cells detached from the wells. The suspension cells were then incubated with 1 μM latrunculin A or an equivalent volume of dimethyl sulfoxide (DMSO) for 45 min with shaking at 37°C. Cells were treated with 10−8 M PASSSR in TBS, pH 7.4, with 2 mM MnSO4 in the presence of latrunculin A or DMSO for 2 h at 4°C prior to lysis.
FIG. 4.
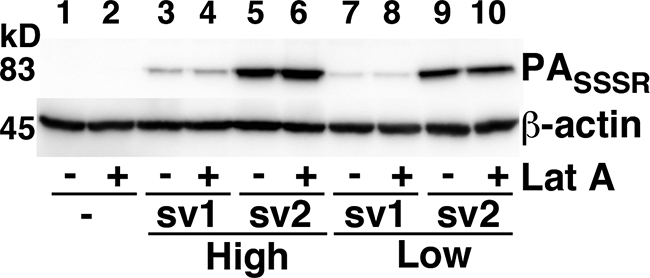
Disruption of the actin cytoskeleton does not affect binding of PA to ANTXR1-sv1 or ANTXR1-sv2. Receptor-negative cells or cells expressing high or low levels of ANTXR1-sv1 or ANTXR1-sv2 were treated with latrunculin A or DMSO vehicle at 37°C for 45 min and then incubated with 10−8 M PASSSR for 2 h at 4°C. Cells were washed with PBS and lysed, and the lysates were subjected to Western blotting with anti-PA antibody.
Flow cytometric analysis.
Cells were detached from wells by use of PBS supplemented with 10 mM EDTA and then washed with ice-cold PBS. Cells were treated with or without 10−8 M Alexa fluor-labeled PASSSR in F12 medium supplemented with 1% BSA and buffered with 20 mM HEPES, pH 8, in suspension for 2 h, with rotation, at 4°C. Cells were washed three times with PBS and then resuspended in PBS supplemented with 1% BSA. Flow cytometric analysis was performed using a FACSCalibur flow cytometer (BD Biosciences), and all data were analyzed using FlowJo software (Tree Star Inc.).
MEK1 cleavage assays.
Confluent monolayers of cells in 24-well plates were treated with 2 × 10−8 M trypsin-nicked PA and 5 × 10−9 M LF for 0, 15, 30, 45, or 60 min at 37°C prior to lysis with lysis buffer. Forty micrograms of total protein lysate was separated by SDS-PAGE and transferred to nitrocellulose. Western blots were performed using rabbit polyclonal anti-MEK1 (Upstate).
Coimmunoprecipitation assays.
U20S cells were stably transfected with pcDNA-HA, pcDNA-ANTXR1-sv1-HA, or pcDNA-sv2-HA. Cells from two 10-cm plates were scraped into 1 ml of EBC lysis buffer (50 mM Tris, pH 8, 120 mM NaCl, 0.5% [vol/vol] NP-40, 50 μg/ml phenylmethylsulfonyl fluoride) and rotated at 4°C for 1 h. Lysates were clarified by centrifugation, and protein concentrations were determined using the Bradford assay. Two microliters of rabbit anti-HA antibody (Sigma) was added to lysates, and the mixtures were rotated at 4°C for 2 h. Approximately 30 μl of BSA-blocked protein A Sepharose beads (Amersham) was added to the lysates, and the mixtures were rotated at 4°C for 1 h. Beads were washed twice with 1 ml of EBC lysis buffer and then washed three times with NETN buffer (20 mM Tris, pH 8, 1 mM EDTA, 900 mM NaCl, 0.5% NP-40). Proteins were eluted with SDS loading dye, subjected to SDS-PAGE, and stained with silver.
Actin association assays.
Cells were scraped into PHEM (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2, pH 6.9) buffer and then lysed in PHEM buffer supplemented with 0.15% Triton X-100 for 5 min on ice prior to centrifugation at 16,000 × g for 30 min at 4°C. The supernatant (soluble fraction) was collected, while the pellet (insoluble fraction) was resuspended in SDS sample buffer. Equal volumes of supernatant and the solubilized pellets were subjected to SDS-PAGE and Western blotting.
Fluorescence recovery after photobleaching (FRAP).
Cells were seeded onto 25-mm glass coverslips and grown overnight to ∼80% confluence. Samples were placed in Attofluor chambers (Molecular Probes) and viewed with a confocal laser microscope (Zeiss LSM 510 Meta), using a 60× oil immersion objective lens. Two equal areas (for bleached and control regions) at the edges of the same cell were defined. After three basal readings, the selected region was irreversibly bleached using the 488-nm laser line, resulting in a 50% to 70% reduction in fluorescence intensity. Recovery of fluorescence was then monitored over a 375-s period at 10-s intervals by scanning the selected regions at low laser power to minimize photobleaching during sampling. The fluorescence of bleached areas was normalized to that of the corresponding control (unbleached) region at each time point to correct for possible drift or photobleaching incurred during low-light sampling. Recovery curves were fit to single-phase exponential association.
RESULTS
Binding of PA to ANTXR1-sv1 and ANTXR1-sv2.
To determine if there are functional differences between ANTXR1-sv1 and ANTXR1-sv2, we made stable cell lines expressing either ANTXR1-sv1-HA or ANTXR1-sv2-HA epitope-tagged fusion proteins in receptor-negative CHO-K1 cells (CHOR1.1). The surface expression levels of the receptors were assessed by treating cells with a non-membrane-permeative biotinylation reagent, precipitating the biotinylated proteins, and then probing the precipitates for the receptors by Western blotting. Clones that expressed a high level or a low level of each variant (Fig. 1A, compare lanes 2 and 3 for high expression and lanes 4 and 5 for low expression) were selected for further analysis. To assess binding of PA to the receptor splice variants, cells were incubated with furin-resistant PASSSR on ice for 2 h to allow binding to the receptors while preventing processing and internalization; the amount of bound PASSSR was determined by Western blot assays. Cells that expressed ANTXR1-sv2 bound >4 times more PA than did cells that expressed similar levels of ANTXR1-sv1 (Fig. 1B, compare lane 2 with lane 3 and lane 4 with lane 5). Indeed, cells that expressed the lower level of ANTXR1-sv2 bound more PA than did cells that expressed the higher level of ANTXR1-sv1 (Fig. 1B, compare lanes 2 and 5).
FIG. 1.

Cells that express ANTXR1-sv1 bind less PA than do cells that express ANTXR1-sv2. (A) Receptor-negative CHOR1.1 cells and CHOR1.1 cells stably transfected with either ANTXR1-sv1-HA or ANTXR1-sv2-HA were treated with a non-membrane-permeative biotinylation reagent. Biotinylated proteins were precipitated with streptavidin-agarose and analyzed by Western blotting using anti-HA antibody. (B) Cells were incubated with a furin-insensitive mutant of PA, PASSSR, for 2 h on ice. The cells were washed with PBS and lysed, and the lysates were subjected to Western blotting with anti-PA antibody. Blots were also probed for β-actin to ensure equal loading. (C) Cells were treated with EDTA until they detached from the wells. The cells were then washed with TBS and resuspended in buffer containing either EDTA or MnSO4, as indicated. The cells were incubated with PASSSR for 2 h at 4°C and then washed, and the amount of bound PASSSR was assessed by Western blotting. Blots were also probed for β-actin to ensure equal loading. (D) CHOR1.1 cells (green) and cells stably transfected with ANTXR1-sv1-HA (top panel; blue), ANTXR1-sv2-HA (top panel; red), ANTXR1-sv1-T118A-HA (bottom panel; blue), and ANTXR1-sv2-T118A-HA (bottom panel; red) were incubated with Alexa fluor-labeled PASSSR in suspension for 2 h at 4°C and then washed, and the amount of bound PASSSR was assessed by flow cytometry. As a negative control, cells expressing ANTXR1-sv2-HA were not incubated with Alexa fluor 488-labeled PASSSR (orange).
To confirm that the interactions between PA and the ANTXR1 splice variants were dependent on divalent cations, PA binding assays were conducted using EDTA-treated cells. Cells were treated with EDTA, which caused them to detach from the well, and the suspended cells were incubated with PASSSR in buffer containing either EDTA or MnSO4. Cells expressing ANTXR1-sv2-HA (Fig. 1C, lanes 9 and 15) bound more PASSSR than did cells expressing ANTXR1-sv1-HA (Fig. 1C, lanes 6 and 12) in the presence of MnSO4. Neither receptor variant was able to bind PASSSR in the presence of EDTA (Fig. 1C, lanes 5, 8, 11, and 14), and no detectable PASSSR bound untransfected CHOR1.1 cells (Fig. 1C, lanes 1 to 3). Surface biotinylation assays conducted in suspension confirmed that similar levels of ANTXR1-sv1-HA and ANTXR1-sv2-HA were expressed on the surfaces of the “high-expression” cells and that similar levels were detected on the “low-expression” cells (data not shown).
There could be various reasons that a smaller amount of PASSSR was detected on cells expressing ANTXR1-sv1-HA and a larger amount was detected on cells expressing the shorter splice variant. One possibility is that the affinity of PA for ANTXR1-sv1-HA is lower than that for ANTXR1-sv2-HA. A second possibility is that a subpopulation of ANTXR1-sv1-HA is in a high-affinity state that binds PA but that most ANTXR1-sv1-HA is in a low-affinity state that does not stably bind PA in this assay. In the latter case, the affinities measured would be similar if the high-affinity conformations of ANTXR1-sv1 and ANTXR1-sv2 were the same and if the sensitivity of the assay was too low to detect binding of PA to the low-affinity state of ANTXR1-sv1. To distinguish between these possibilities, we performed kinetic binding experiments to measure the affinity of PA for cells expressing either ANTXR1-sv1-HA or ANTXR1-sv2-HA. There was no significant difference between the equilibrium binding constants determined using the two cell lines (Table 1). This suggests, therefore, that less PA binds to cells expressing ANTXR1-sv1 than to those expressing ANTXR1-sv2 because a smaller fraction of the ANTXR1-sv1 receptor is in a high-affinity state.
We next sought to determine if the difference in binding of PASSSR to ANTXR1-sv1 and to ANTXR1-sv2 resulted from a difference in their I-domain conformations. We mutated residue T118 to alanine in both splice variants so that both receptors would be in the closed (low-affinity) conformation. Stable cell lines expressing similar levels of either ANTXR1-sv1-T118A-HA or ANTXR1-sv2-T118A-HA fusion protein, as determined by surface biotinylation assays, were then compared using flow cytometry for the ability to bind Alexa fluor 488-labeled PASSSR. This flow cytometry assay was used because its sensitivity was sufficient to detect binding of PA to the mutant receptors. As before, cells that expressed ANTXR1-sv2-HA bound more PASSSR than did cells that expressed ANTXR1-sv1-HA (Fig. 1D, top panel). In contrast, there was no difference in the levels of PASSSR binding between cells that expressed ANTXR1-sv1-T118A-HA and those that expressed ANTXR1-sv2-T118A-HA (Fig. 1D, bottom panel). Taken together, these results indicate that more PA binds to cells that express ANTXR1-sv2 than to cells that express ANTXR1-sv1 because more of the ANTXR1-sv2 receptors have open I domains.
Differential rates of cleavage of MEK1 by lethal toxin in cells expressing ANTXR1 variants.
We next assessed whether the expression of different ANTXR1 splice variants would influence the rate of cleavage of an intracellular target of lethal toxin, MEK1. Since LF cleaves an amino-terminal fragment from MEK1, toxin activity was monitored as a loss of the MEK1 band in a Western blot probed with an antibody that recognizes the amino-terminal region of MEK1 (Fig. 2A). The MEK1 signal diminished over time in toxin-treated cells expressing either ANTXR1-sv1 or ANTXR1-sv2 but not in receptor-negative CHOR1.1 cells. Approximately 50% of MEK1 was cleaved in cells expressing ANTXR1-sv2 within 15 min, and ∼90% was cleaved by 30 min (Fig. 2B, open circles). In comparison, MEK1 cleavage was delayed in cells expressing ANTXR1-sv1, with only ∼25% of MEK1 cleaved at 15 min and ∼90% cleaved at 60 min (Fig. 2B, open triangles). These results suggest that the higher level of PA bound to cells that express ANTXR1-sv2 than to cells that express ANTXR1-sv1 leads to an increased rate of intoxication.
FIG. 2.
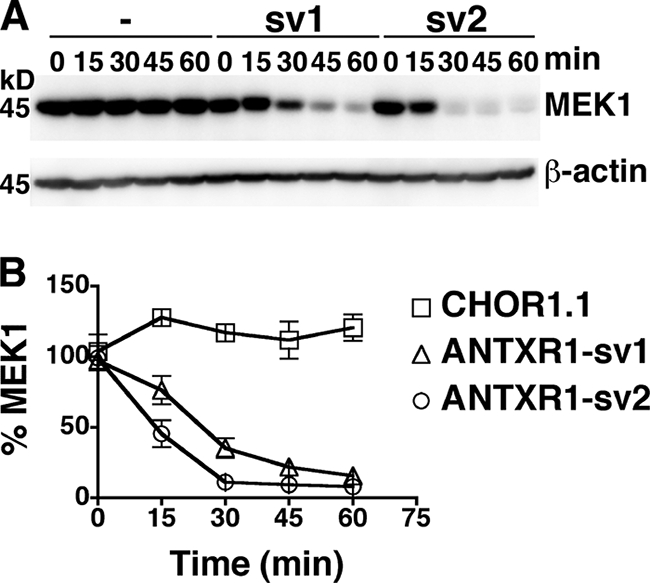
Increased rate of LF-dependent cleavage of MEK1 in cells expressing ANTXR1-sv2 compared to that in cells expressing ANTXR1-sv1. (A) CHOR1.1 cells (lanes 1 to 5) or cells stably expressing either ANTXR1-sv1-HA (lanes 6 to 10) or ANTXR1-sv2-HA (lanes 11 to 15) were treated with lethal toxin at 37°C for the indicated times. Cells were lysed, and the lysates were subjected to Western blotting with anti-MEK1 antibody. Blots were also probed for β-actin to ensure equal loading. (B) The levels of MEK1 from panel A were averaged for three independent experiments. Error bars represent standard errors of the means.
Actin associates with ANTXR1-sv1 but not with ANTXR1-sv2.
Since ANTXR1-sv1 and ANTXR1-sv2 differ only in the sequence of their cytoplasmic domains, we reasoned that the observed difference in PA binding between the splice variants might be due to proteins that differentially interact with the cytoplasmic domains. To determine if the receptor variants interact with other proteins, we performed coimmunoprecipitation assays using human osteosarcoma cells stably transfected with either pcDNA3-ANTXR1-sv1-HA, pcDNA3-ANTXR1-sv2-HA, or pcDNA3-HA. A human cell line was used in these experiments to facilitate the identification of proteins by mass spectrometry. Anti-HA antibodies were used to immunoprecipitate the HA-tagged receptors, and coimmunoprecipitated proteins were separated by SDS-PAGE and visualized by silver staining. An ∼45-kDa protein was immunoprecipitated from cells that expressed ANTXR1-sv1-HA but not from those that expressed ANTXR1-sv2-HA or HA (Fig. 3A). This protein was identified as actin by mass spectrometry analysis and confirmed by Western blotting (data not shown).
FIG. 3.

ANTXR1-sv1 associates with the actin cytoskeleton. (A) U20S cells were stably transfected with ANTXR1-sv1-HA, ANTXR1-sv2-HA, or pcDNA3-HA. Cells were lysed, and the lysates were mixed with anti-HA antibody. Antibody and associated proteins were precipitated with protein A-Sepharose, subjected to SDS-PAGE, and stained with silver. The asterisk indicates an ∼45-kDa protein that coimmunoprecipitates with ANTXR1-sv1-HA but not with ANTXR1-sv2-HA or HA alone. (B) FRAP curves for CHOR1.1 cells expressing either ANTXR1-sv1-EGFP (red squares) or ANTXR1-sv2-EGFP (blue triangles) and treated with either latrunculin A (right panel) or DMSO vehicle (left panel). The data were averaged over seven independent experiments. Error bars represent standard errors of the means. (C) CHOR1.1 cells or cells expressing ANTXR1-sv1-HA or ANTXR1-sv2-HA were subjected to 0.15% Triton X-100 extraction for 5 min on ice and centrifuged for 30 min at 4°C. Proteins that fractionated in the pellet (P) or supernatant (S) were subjected to Western blotting with anti-HA and anti-β-actin antibodies. (D) Cells expressing ANTXR1-sv1 were pretreated with latrunculin A or DMSO vehicle for 45 min at 37°C prior to Triton X-100 extraction as described for panel C.
Since proteins associated with the actin cytoskeleton exhibit low lateral mobility on the cell surface (23, 24, 38), we performed FRAP assays to compare the mobilities of ANTXR1-sv1 and ANTXR1-sv2. Confocal microscopy was used to follow the dynamics in real time of ANTXR1-sv1-EGFP or ANTXR1-sv2-EGFP in stably transfected CHOR1.1 cells. A region of interest at the edge of a cell was subjected to fluorescence photobleaching, and recovery of fluorescence was monitored every 10 s for 375 s by measuring the fluorescence in the region, using a low laser power. FRAP curves revealed that surface ANTXR1-sv2-EGFP receptors repopulated the photobleached region, with a recovery half-life (t1/2) of ∼7 s (Fig. 3B, left panel, blue triangles). In contrast, only ∼75% recovery was observed in cells expressing ANTXR1-sv1-EGFP (red squares), with a t1/2 of ∼17 s. Full recovery with a t1/2 of ∼6 s was observed for both ANTXR1-sv1-EGFP and ANTXR1-sv2-EGFP when cells were first treated with an actin-depolymerizing drug, latrunculin A (Fig. 3B, right panel). These results are consistent with the notion that ANTXR1-sv1 is associated with the actin cytoskeleton.
To further establish that ANTXR1-sv1 binds the actin cytoskeleton, we conducted actin association assays, using CHOR1.1 cells that express either ANTXR1-sv1-HA or ANTXR1-sv2-HA. Cells were solubilized at 4°C in buffer containing 0.15% Triton X-100, and the resulting lysates were centrifuged. Proteins that associate with the insoluble actin cytoskeleton are pelleted under these conditions, while those not associated with the cytoskeleton largely remain in the supernatant. This assay revealed that the majority of ANTXR1-sv1-HA was found in the pellet fraction (Fig. 3C), indicating that it was associated with the cytoskeleton. In contrast, ANTXR1-sv2-HA was distributed between the insoluble and soluble fractions (Fig. 3C). As expected, treatment of cells with latrunculin A prior to Triton X-100 extraction caused most of the actin to fractionate in the supernatant (Fig. 3D). Likewise, treatment of cells with latrunculin A shifted the majority of ANTXR1-sv1-HA into the soluble fraction (Fig. 3D).
Disruption of the actin cytoskeleton does not affect binding of PA to ANTXR1-sv1.
To determine whether the difference between the amounts of PA bound to ANTXR1-sv1 and ANTXR1-sv2 could be attributed to differential association of the variants with the cytoskeleton, we performed a binding assay using cells that were treated with latrunculin A. ANTXR1-sv1-HA- and ANTXR1-sv2-HA-expressing cells were left untreated or were treated with latrunculin A prior to incubation with PASSSR. The amount of bound PASSSR was assessed by Western blot assays. We found that ANTXR1-sv1-HA-expressing cells bound similar amounts of PA in the presence and absence of latrunculin A (Fig. 4, compare lane 3 with lane 4 and lane 7 with lane 8); similar results were observed with ANTXR1-sv2-HA-expressing cells (Fig. 4, lanes 5, 6, 9, and 10), indicating that disruption of the actin cytoskeleton did not affect the amount of PA bound to either receptor variant.
A conserved region of the ANTXR1-sv1 cytoplasmic domain mediates cytoskeleton association and influences binding of PA.
We next sought to determine whether distinct regions of the cytoplasmic domain were responsible for linking ANTXR1-sv1 to the cytoskeleton and for controlling the binding of PA to the extracellular I domain. We made C-terminal truncation constructs to shorten the ANTXR1-sv1 tail and transiently transfected them into CHOR1.1 cells. The majority of ANTXR1-sv11-420-HA was observed in the pellet fraction, indicating an association with the cytoskeleton, whereas ANTXR1-sv11-410-HA was found in the soluble fraction (Fig. 5A). As observed previously, ANTXR1-sv2-HA (amino acids 1 to 368) was distributed between the pellet and soluble fractions, while wild-type ANTXR1-sv1-HA (amino acids 1 to 564) was found predominantly in the pellet fraction.
FIG. 5.
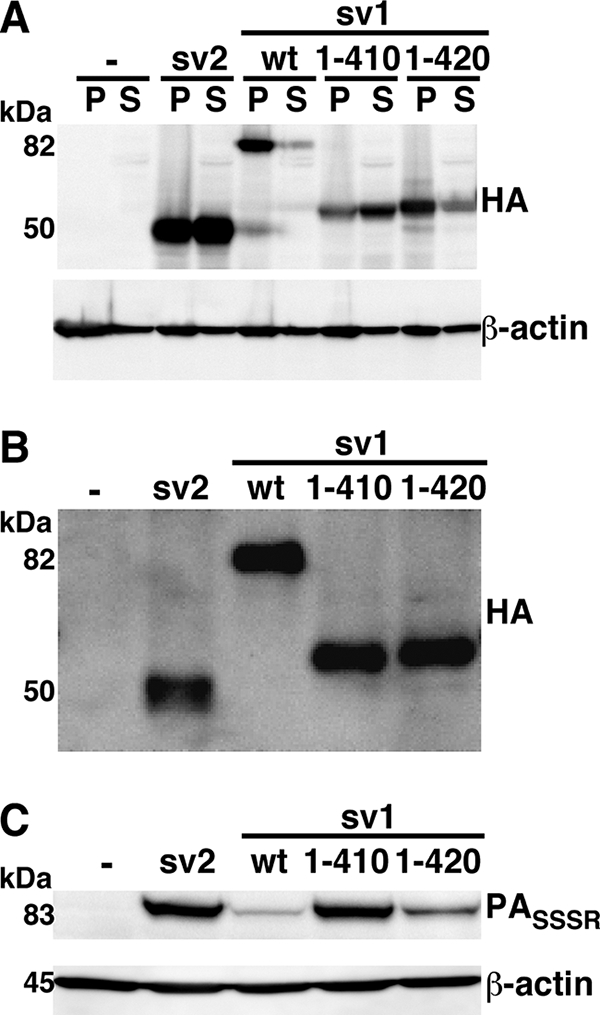
ANTXR1-sv1 requires amino acids 1 to 420 for actin association and PA binding regulation. (A) CHOR1.1 cells transiently transfected with either wild-type ANTXR1-sv1 or ANTXR1-sv1 truncation mutants were subjected to 0.15% Triton X-100 extraction for 5 min on ice and centrifuged for 30 min at 4°C. Proteins that fractionated in the pellet (P) or supernatant (S) were subjected to Western blotting with anti-HA and anti-β-actin antibodies. (B) CHOR1.1 cells expressing either wild-type ANTXR1-sv1 or ANTXR1-sv1 truncation mutants were treated with a non-membrane-permeative biotinylation reagent. Biotinylated proteins were precipitated with streptavidin-agarose and analyzed by Western blotting using anti-HA antibody. (C) Cells were incubated with PASSSR for 2 h on ice. The cells were then washed with PBS and lysed, and the lysates were subjected to Western blotting with anti-PA antibody. Blots were probed for β-actin to ensure equal loading. Blots are representative of three independent experiments.
Binding assays were then conducted to localize the region of the cytoplasmic domain that causes the reduction of PA binding to ANTXR1-sv1. CHOR1.1 cells transiently expressing the wild-type and truncation constructs were subjected to surface biotinylation assays to ensure similar surface expression of each receptor (Fig. 5B). As observed previously, cells expressing ANTXR1-sv2-HA bound more PASSSR than did cells expressing wild-type ANTXR1-sv1-HA (Fig. 5C). Cells expressing ANTXR1-sv11-410-HA bound levels of PASSSR similar to those in cells expressing ANTXR1-sv2-HA, while ANTXR1-sv11-420-HA cells bound levels of PASSSR similar to those in cells expressing wild-type ANTXR1-sv1-HA (Fig. 5C). These results indicate that the regions of the cytoplasmic domain that affect PA binding and actin association overlap. Interestingly, an ∼50-amino-acid region that is highly conserved in both ANTXR1 (amino acids 370 to 417) and ANTXR2 proteins across various species is intact in ANTXR1-sv11-420 but is truncated in ANTXR1-sv11-410 (14).
A point mutation in ANTXR1-sv1 increases PA binding and disrupts actin association.
Mutations in ANTXR2 (CMG2) are found in patients with juvenile hyaline fibromatosis (JHF) and infantile systemic hyalinosis (11, 14). One JHF-associated mutation, Y381C, maps to the cytosolic domain of ANTXR2 within the ∼50-amino-acid conserved region (14). We speculated that this mutation and the analogous mutation in ANTXR1, Y383C, might impair the ability of the receptors to associate with the actin cytoskeleton. An actin association assay was performed using CHOR1.1 cells transiently transfected with ANTXR2-HA or ANTXR2-Y381C-HA. We found that ANTXR2-HA was present mostly in the insoluble pellet, while ANTXR2-Y381C-HA was found mostly in the supernatant (Fig. 6A). Next, we transiently transfected CHOR1.1 cells with either ANTXR1-sv1-HA or ANTXR1-sv1-Y383C-HA. An actin association assay revealed that wild-type ANTXR1-sv1-HA was present mostly in the insoluble pellet fraction, whereas ANTXR1-sv1-Y383C-HA was found mostly in the soluble fraction (Fig. 6B). Thus, the Y383C mutation decreases the association of ANTXR1-sv1 with the actin cytoskeleton.
FIG. 6.
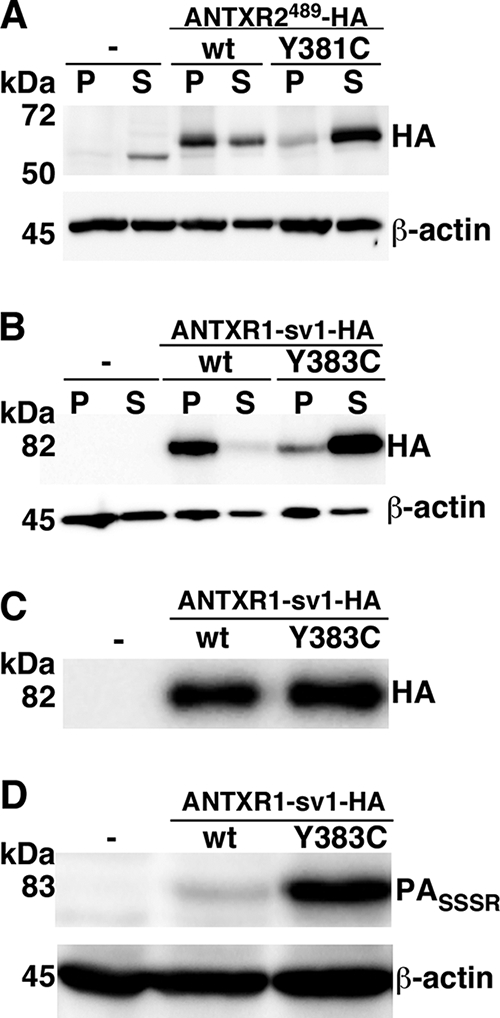
ANTXR1-sv1-Y383C binds more PA than ANTXR1-sv1 does and does not associate with the actin cytoskeleton. (A) CHOR1.1 cells or cells transiently expressing ANTXR2489-HA or the JHF-linked mutant, ANTXR2489-Y381C-HA, were subjected to 0.15% Triton X-100 extraction for 5 min on ice and centrifuged for 30 min at 4°C. Proteins that fractionated in the pellet (P) or supernatant (S) were subjected to Western blotting with anti-HA and anti-β-actin antibodies. (B) CHOR1.1 cells or cells expressing ANTXR1-sv1-HA or ANTXR1-sv1-Y383C-HA were subjected to 0.15% Triton X-100 extraction for 5 min on ice and centrifuged for 30 min at 4°C. Proteins that fractionated in the pellet (P) or supernatant (S) were subjected to Western blotting with anti-HA and anti-β-actin antibodies. (C) Receptor-negative CHOR1.1 cells or CHOR1.1 cells transiently transfected with either ANTXR1-sv1-HA or ANTXR1-sv1-Y383C-HA were treated with a non-membrane-permeative biotinylation reagent. Biotinylated proteins were precipitated with streptavidin-agarose and analyzed by Western blotting using anti-HA antibody. (D) Cells were incubated with PASSSR for 2 h on ice. The cells were then washed with PBS and lysed, and the lysates were subjected to Western blotting with anti-PA antibody. Blots were also probed for β-actin to ensure equal loading. Blots are representative of three independent experiments.
We next performed a binding assay to assess the effect of this mutation on the interaction between ANTXR1-sv1 and PA. Wild-type ANTXR1-sv1-HA or ANTXR1-sv1-Y383C-HA was transiently transfected into CHOR1.1 cells, and the amounts of PASSSR bound to cells were determined by a Western blot assay. Cells that expressed the Y383C mutant were able to bind more PA than were cells that expressed a similar level of wild-type ANTXR1-sv1 (Fig. 6C and D).
DISCUSSION
This study demonstrates functional differences between two splice variants of ANTXR1 that differ only in the sequences and lengths of their cytoplasmic domains. We have demonstrated that cells that express ANTXR1-sv1 bind less PA than do cells that express a similar level of ANTXR1-sv2 and that only ANTXR1-sv1 associates with the actin cytoskeleton. Cleavage of MEK1, a cytosolic target of lethal toxin, occurs at a higher rate in cells that express ANTXR1-sv2, presumably because a larger amount of PA binds these cells.
Structural studies have demonstrated that PA binds a conformation of the ANTXR2 I domain that is structurally most similar to the open conformation of the αM I domain (20, 21, 41). The αM I domain can convert between open and closed conformations, modulating the affinity for ligand, but there is no structural evidence that indicates that the ANTXR1 and ANTXR2 I domains can exist in closed conformations. Indeed, an isoleucine residue which is conserved in all α-integrin I domains and stabilizes the closed conformation of αM by inserting into a hydrophobic pocket aligns with a polar residue in ANTXR1 and ANTXR2 (21). This suggested that the closed conformation of the ANTXR1 and ANTXR2 I domains might not be stable (21). Our work, however, provides evidence that ANTXR1 can exist in high-affinity and low-affinity conformations analogous to the states observed in integrin I domains. Although the central residues implicated in the structural conversion are conserved between the ANTXR1 and ANTXR2 I domains, it remains unknown whether the ANTXR2 I domain can exist in two affinity states.
Many integrins are expressed in a default low-affinity state, but the conformation of the I domain can open in response to signals mediated through the cytoplasmic domain of the receptor, in a process referred to as inside-out signaling (27, 45). Activation of integrin heterodimers occurs when talin binds to the cytosolic domain of the β subunit. It is thought that binding of talin alters the interaction between the transmembrane domains of the α and β subunits and that this is how the signal is transmitted across the cell membrane to open the I domain (36, 47). If the different affinity states of ANTXR1 also result from differences in transmembrane domain interactions, this would imply that ANTXR1 exists as heterodimers and/or homodimers. LRP6 has been reported to be a coreceptor of ANTXR1 (48), although subsequent studies have indicated that it is not required for intoxication (40, 52). We have shown previously that the transmembrane domain of ANTXR1 homodimerizes and that HA- and T7-tagged ANTXR1 proteins coimmunoprecipitate (13). The involvement of transmembrane domain interactions in the activation of ANTXR1, however, remains to be established.
Activation or inactivation of integrins can occur through mutations in the cytosolic domains that affect the interaction between the α and β subunits or between the β cytosolic domain and talin (26, 34, 35, 51). We found that a point mutation in the cytosolic domain of ANTXR1-sv1 (Fig. 5C) or deletion of the domain (M. Y. Go, unpublished data) converted the receptor from a low-affinity state to a high-affinity state and also impaired the ability of ANTXR1-sv1 to interact with actin. Since disruption of the cytoskeleton with latrunculin A did not affect the activation state of ANTXR1-sv1, we speculate that an adaptor protein both inactivates ANTXR1-sv1 and links the receptor to the cytoskeleton. The hypothesis that a single adaptor protein (or complex) is responsible for both actin association and I domain regulation is supported by the observation that a point mutation in the cytoplasmic domain affects both activities. We did not, however, detect talin in ANTXR1-sv1 immunoprecipitates (data not shown).
We explored the cytoskeletal association of ANTXR1 by using live-cell imaging. Consistent with the coimmunoprecipitation assays, which did not show an association between ANTXR1-sv2 and actin, FRAP assays indicated that the t1/2 values for ANTXR1-sv2 were similar in cells that had intact and disrupted cytoskeletons (t1/2, ∼6 to 7 s). In contrast, ANTXR1-sv1 required a longer time than ANTXR1-sv2 to reach maximal recovery (t1/2, ∼17 s), and the recovery plateaued at only ∼75%. In cells treated with latrunculin A, ANTXR1-sv1 exhibited similar dynamics to those of ANTXR1-sv2. These results suggest that ∼25% of ANTXR1-sv1 is immobilized by the cytoskeleton and that diffusion is limited by the cytoskeleton for most of the remaining fraction.
Although we have not directly addressed the possible influence of the cytoskeleton on toxin internalization, we note that association of the cytoskeleton with the receptor is not required for uptake because ANTXR1-sv2 can mediate intoxication (Fig. 2). It remains to be determined whether ANTXR1-sv1 must dissociate from the cytoskeleton before internalization occurs, and if so, whether PA influences this process. There is also a possibility that cytoskeleton-associated receptors are internalized through a different pathway from that for receptors that are not associated with the cytoskeleton. Future studies are required to better understand the potential role of the cytoskeleton in toxin uptake.
ANTXR1-sv1 is upregulated in tumor-associated endothelial cells and may function in angiogenesis (10). With potential implications for angiogenesis, a previous study demonstrated that the association of ANTXR1-sv1 with the cytoskeleton mediates cell spreading (49). The cytoskeleton linkage is likely to be functionally important, since spreading was inhibited by pharmacological disruption of the cytoskeleton. Our results suggest, therefore, that neither ANTXR1-sv2 nor ANTXR1-sv1-Y383C mediates cell spreading effectively because of a reduced association with the cytoskeleton. It is not clear whether the ANTXR2 Y381C mutant (Y381 in ANTXR2 aligns with Y383 in ANTXR1) found in a patient with JHF would also be defective at spreading. Another JHF-associated mutation, located in the I domain of ANTXR2, decreased adhesion of fibroblasts to laminin (11). In contrast, an infantile systemic hyalinosis-associated truncation mutant appeared to increase surface expression of ANTXR2 (25). It is difficult to conceive how the effects of these mutations lead to the clinical manifestations of these diseases.
Acknowledgments
This research was supported by NIH grant RO1 AI067683. M.Y.G. received support from the Ontario Graduate Scholarship Program. J.M. holds the Canada Research Chair in Bacterial Pathogenesis.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 20 October 2008.
REFERENCES
- 1.Abrami, L., S. H. Leppla, and F. G. van der Goot. 2006. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J. Cell Biol. 172309-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abrami, L., N. Reig, and F. G. van der Goot. 2005. Anthrax toxin: the long and winding road that leads to the kill. Trends Microbiol. 1372-78. [DOI] [PubMed] [Google Scholar]
- 3.Batty, S., E. M. Chow, A. Kassam, S. D. Der, and J. Mogridge. 2006. Inhibition of mitogen-activated protein kinase signalling by Bacillus anthracis lethal toxin causes destabilization of interleukin-8 mRNA. Cell. Microbiol. 8130-138. [DOI] [PubMed] [Google Scholar]
- 4.Beauregard, K. E., R. J. Collier, and J. A. Swanson. 2000. Proteolytic activation of receptor-bound anthrax protective antigen on macrophages promotes its internalization. Cell. Microbiol. 2251-258. [DOI] [PubMed] [Google Scholar]
- 5.Bell, S. E., A. Mavila, R. Salazar, K. J. Bayless, S. Kanagala, S. A. Maxwell, and G. E. Davis. 2001. Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 1142755-2773. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya, A. A., M. L. Lupher, Jr., D. E. Staunton, and R. C. Liddington. 2004. Crystal structure of the A domain from complement factor B reveals an integrin-like open conformation. Structure 12371-378. [DOI] [PubMed] [Google Scholar]
- 7.Bradley, K. A., J. Mogridge, G. Jonah, A. Rainey, S. Batty, and J. A. Young. 2003. Binding of anthrax toxin to its receptor is similar to alpha integrin-ligand interactions. J. Biol. Chem. 27849342-49347. [DOI] [PubMed] [Google Scholar]
- 8.Bradley, K. A., J. Mogridge, M. Mourez, R. J. Collier, and J. A. Young. 2001. Identification of the cellular receptor for anthrax toxin. Nature 414225-229. [DOI] [PubMed] [Google Scholar]
- 9.Bradley, K. A., and J. A. Young. 2003. Anthrax toxin receptor proteins. Biochem. Pharmacol. 65309-314. [DOI] [PubMed] [Google Scholar]
- 10.Carson-Walter, E. B., D. N. Watkins, A. Nanda, B. Vogelstein, K. W. Kinzler, and B. St. Croix. 2001. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 616649-6655. [PubMed] [Google Scholar]
- 11.Dowling, O., A. Difeo, M. C. Ramirez, T. Tukel, G. Narla, L. Bonafe, H. Kayserili, M. Yuksel-Apak, A. S. Paller, K. Norton, A. S. Teebi, V. Grum-Tokars, G. S. Martin, G. E. Davis, M. J. Glucksman, and J. A. Martignetti. 2003. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am. J. Hum. Genet. 73957-966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emsley, J., C. G. Knight, R. W. Farndale, M. J. Barnes, and R. C. Liddington. 2000. Structural basis of collagen recognition by integrin alpha2beta1. Cell 10147-56. [DOI] [PubMed] [Google Scholar]
- 13.Go, M. Y., S. Kim, A. W. Partridge, R. A. Melnyk, A. Rath, C. M. Deber, and J. Mogridge. 2006. Self-association of the transmembrane domain of an anthrax toxin receptor. J. Mol. Biol. 360145-156. [DOI] [PubMed] [Google Scholar]
- 14.Hanks, S., S. Adams, J. Douglas, L. Arbour, D. J. Atherton, S. Balci, H. Bode, M. E. Campbell, M. Feingold, G. Keser, W. Kleijer, G. Mancini, J. A. McGrath, F. Muntoni, A. Nanda, M. D. Teare, M. Warman, F. M. Pope, A. Superti-Furga, P. A. Futreal, and N. Rahman. 2003. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am. J. Hum. Genet. 73791-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hotchkiss, K. A., C. M. Basile, S. C. Spring, G. Bonuccelli, M. P. Lisanti, and B. I. Terman. 2005. TEM8 expression stimulates endothelial cell adhesion and migration by regulating cell-matrix interactions on collagen. Exp. Cell Res. 305133-144. [DOI] [PubMed] [Google Scholar]
- 16.Kassam, A., S. D. Der, and J. Mogridge. 2005. Differentiation of human monocytic cell lines confers susceptibility to Bacillus anthracis lethal toxin. Cell. Microbiol. 7281-292. [DOI] [PubMed] [Google Scholar]
- 17.Klimpel, K. R., S. S. Molloy, G. Thomas, and S. H. Leppla. 1992. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 8910277-10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krantz, B. A., R. A. Melnyk, S. Zhang, S. J. Juris, D. B. Lacy, Z. Wu, A. Finkelstein, and R. J. Collier. 2005. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 309777-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lacy, D. B., H. C. Lin, R. A. Melnyk, O. Schueler-Furman, L. Reither, K. Cunningham, D. Baker, and R. J. Collier. 2005. A model of anthrax toxin lethal factor bound to protective antigen. Proc. Natl. Acad. Sci. USA 10216409-16414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lacy, D. B., D. J. Wigelsworth, R. A. Melnyk, S. C. Harrison, and R. J. Collier. 2004. Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. USA 10113147-13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacy, D. B., D. J. Wigelsworth, H. M. Scobie, J. A. Young, and R. J. Collier. 2004. Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 1016367-6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, J. O., L. A. Bankston, M. A. Arnaout, and R. C. Liddington. 1995. Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure 31333-1340. [DOI] [PubMed] [Google Scholar]
- 23.Lippincott-Schwartz, J., N. Altan-Bonnet, and G. H. Patterson. 2003. Photobleaching and photoactivation: following protein dynamics in living cells. Nat. Cell Biol. 2003(Suppl.)S7-S14. [PubMed] [Google Scholar]
- 24.Lippincott-Schwartz, J., E. Snapp, and A. Kenworthy. 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2444-456. [DOI] [PubMed] [Google Scholar]
- 25.Liu, S., H. J. Leung, and S. H. Leppla. 2007. Characterization of the interaction between anthrax toxin and its cellular receptors. Cell. Microbiol. 9977-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu, C. F., and T. A. Springer. 1997. The alpha subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1. J. Immunol. 159268-278. [PubMed] [Google Scholar]
- 27.Luo, B. H., and T. A. Springer. 2006. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 18579-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melnyk, R. A., K. M. Hewitt, D. B. Lacy, H. C. Lin, C. R. Gessner, S. Li, V. L. Woods, Jr., and R. J. Collier. 2006. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J. Biol. Chem. 2811630-1635. [DOI] [PubMed] [Google Scholar]
- 29.Miller, C. J., J. L. Elliott, and R. J. Collier. 1999. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry 3810432-10441. [DOI] [PubMed] [Google Scholar]
- 30.Mogridge, J., K. Cunningham, D. B. Lacy, M. Mourez, and R. J. Collier. 2002. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc. Natl. Acad. Sci. USA 997045-7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molloy, S. S., P. A. Bresnahan, S. H. Leppla, K. R. Klimpel, and G. Thomas. 1992. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 26716396-16402. [PubMed] [Google Scholar]
- 32.Mourez, M. 2004. Anthrax toxins. Rev. Physiol. Biochem. Pharmacol. 152135-164. [DOI] [PubMed] [Google Scholar]
- 33.Nanda, A., E. B. Carson-Walter, S. Seaman, T. D. Barber, J. Stampfl, S. Singh, B. Vogelstein, K. W. Kinzler, and B. St. Croix. 2004. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI). Cancer Res. 64817-820. [DOI] [PubMed] [Google Scholar]
- 34.O'Toole, T. E., Y. Katagiri, R. J. Faull, K. Peter, R. Tamura, V. Quaranta, J. C. Loftus, S. J. Shattil, and M. H. Ginsberg. 1994. Integrin cytoplasmic domains mediate inside-out signal transduction. J. Cell Biol. 1241047-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Toole, T. E., D. Mandelman, J. Forsyth, S. J. Shattil, E. F. Plow, and M. H. Ginsberg. 1991. Modulation of the affinity of integrin alpha IIb beta 3 (GPIIb-IIIa) by the cytoplasmic domain of alpha IIb. Science 254845-847. [DOI] [PubMed] [Google Scholar]
- 36.Partridge, A. W., S. Liu, S. Kim, J. U. Bowie, and M. H. Ginsberg. 2005. Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J. Biol. Chem. 2807294-7300. [DOI] [PubMed] [Google Scholar]
- 37.Rainey, G. J., D. J. Wigelsworth, P. L. Ryan, H. M. Scobie, R. J. Collier, and J. A. Young. 2005. Receptor-specific requirements for anthrax toxin delivery into cells. Proc. Natl. Acad. Sci. USA 10213278-13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reits, E. A., and J. J. Neefjes. 2001. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat. Cell Biol. 3E145-E147. [DOI] [PubMed] [Google Scholar]
- 39.Rosovitz, M. J., P. Schuck, M. Varughese, A. P. Chopra, V. Mehra, Y. Singh, L. M. McGinnis, and S. H. Leppla. 2003. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J. Biol. Chem. 27830936-30944. [DOI] [PubMed] [Google Scholar]
- 40.Ryan, P. L., and J. A. Young. 2008. Evidence against a human cell-specific role for LRP6 in anthrax toxin entry. PLoS ONE 3e1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santelli, E., L. A. Bankston, S. H. Leppla, and R. C. Liddington. 2004. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature 430905-908. [DOI] [PubMed] [Google Scholar]
- 42.Scobie, H. M., J. M. Marlett, G. J. Rainey, D. B. Lacy, R. J. Collier, and J. A. Young. 2007. Anthrax toxin receptor 2 determinants that dictate the pH threshold of toxin pore formation. PLoS ONE 2e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scobie, H. M., G. J. Rainey, K. A. Bradley, and J. A. Young. 2003. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 1005170-5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scobie, H. M., and J. A. Young. 2005. Interactions between anthrax toxin receptors and protective antigen. Curr. Opin. Microbiol. 8106-112. [DOI] [PubMed] [Google Scholar]
- 45.Shimaoka, M., J. Takagi, and T. A. Springer. 2002. Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 31485-516. [DOI] [PubMed] [Google Scholar]
- 46.Shimaoka, M., T. Xiao, J. H. Liu, Y. Yang, Y. Dong, C. D. Jun, A. McCormack, R. Zhang, A. Joachimiak, J. Takagi, J. H. Wang, and T. A. Springer. 2003. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 11299-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wegener, K. L., A. W. Partridge, J. Han, A. R. Pickford, R. C. Liddington, M. H. Ginsberg, and I. D. Campbell. 2007. Structural basis of integrin activation by talin. Cell 128171-182. [DOI] [PubMed] [Google Scholar]
- 48.Wei, W., Q. Lu, G. J. Chaudry, S. H. Leppla, and S. N. Cohen. 2006. The LDL receptor-related protein LRP6 mediates internalization and lethality of anthrax toxin. Cell 1241141-1154. [DOI] [PubMed] [Google Scholar]
- 49.Werner, E., A. P. Kowalczyk, and V. Faundez. 2006. Anthrax toxin receptor 1/tumor endothelium marker 8 mediates cell spreading by coupling extracellular ligands to the actin cytoskeleton. J. Biol. Chem. 28123227-23236. [DOI] [PubMed] [Google Scholar]
- 50.Whittaker, C. A., and R. O. Hynes. 2002. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell 133369-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ylanne, J., Y. Chen, T. E. O'Toole, J. C. Loftus, Y. Takada, and M. H. Ginsberg. 1993. Distinct functions of integrin alpha and beta subunit cytoplasmic domains in cell spreading and formation of focal adhesions. J. Cell Biol. 122223-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Young, J. J., J. L. Bromberg-White, C. Zylstra, J. T. Church, E. Boguslawski, J. H. Resau, B. O. Williams, and N. S. Duesbery. 2007. LRP5 and LRP6 are not required for protective antigen-mediated internalization or lethality of anthrax lethal toxin. PLoS Pathog. 3e27. [DOI] [PMC free article] [PubMed] [Google Scholar]