

Abstract
ICP0, an α (immediate-early) protein of herpes simplex virus 1, performs at least two key functions. It blocks inhibition of viral-gene expression by interferon, a function dependent on the degradation of the ND10 components PML and SP100 by the ubiquitin ligase expressed by the RING finger (RF), and it blocks silencing of viral DNA mediated by the HDAC1/2-CoREST-REST complex. In the latter case, a mutant CoREST lacking the HDAC1 binding site compensates totally or in part for the absence of ICP0 in a cell-type-dependent manner. Here, we compare the phenotypes of an ICP0 mutant containing disabling amino acid substitutions in the RF with those of a mutant with substitutions in the CoREST binding site (R8507). We report the following: (i) the onset of replication of both mutants was delayed, but the RF mutant yields did not reach wild-type virus levels even as late as 48 h after infection, and (ii) in infected cells, PML is rapidly degraded by wild-type virus, with some delay by the R8507 mutant, and is spared by the RF mutant. The translocation of ICP0 to the cytoplasm is impaired in cells infected with the RF mutant or delayed in cells infected with the R8507 mutant. Finally, in contrast to wild-type viruses, both mutants are inhibited by alpha or gamma interferon. The results indicate that both sets of events, the degradation of PML and the blocking of silencing, are interdependent and in large measure dependent on events in the ND10 nuclear bodies.
Infected cell protein 0 (ICP0) of herpes simplex virus 1 (HSV-1) is largely dispensable for viral replication in cells infected at high virus/cell ratios but is essential in cells infected at low ratios or in experimental animal systems. ΔICP0 mutants are hypersensitive to interferon (24) and at low multiplicities of infection are arrested after the expression of α (immediate-early) genes (31, 34). In transfected cells, the most prominent phenotype of ICP0 is that of a promiscuous transactivator primarily of genes introduced by infection or transfection, although it does not bind to DNA (reviewed in references 10 and 30). The 775-residue ICP0 is encoded by a spliced RNA of three exons. A prominent feature of the protein is a RING finger (RF) domain located in the sequences encoded by exon 2. Extensive studies carried out in several laboratories have shown that the primary functions of ICP0, those of blocking interferon and those enabling transition from α to β (early) gene expression, are encoded in different domains of ICP0. Briefly, the RF domain acts as a ubiquitin ligase (1, 11) that has been shown to degrade PML and SP100 (3, 8) and that is linked to the degradation of other proteins (e.g., DNA-dependent protein kinase and CNP-C) (5, 18, 26). PML is a major component and regulatory protein of nuclear domain 10 (ND10), a nuclear body containing SP100, Daxx, and numerous other proteins (23, 25). The link to anti-interferon activity stems from the observation that in murine PML+/+ cells, HSV-1 is inhibited by alpha interferon (IFN-α) or IFN-γ. These interferons have a minimal effect on viral replication in sibling PML−/− cells (2).
The transition from α- to β-gene expression involves blocking the silencing of viral DNA by the HDAC1/2-CoREST-REST complex (7, 9), which also includes lysine-specific demethylase 1 (16, 32). Thus, a G+C-rich sequence contained near the C terminus of ICP0 is conserved at the N terminus of CoREST. A sequence downstream of the conserved site binds CoREST (9). In infected cells, ICP0 dislodges the CoREST/REST complex from HDAC1/2, and both sets of proteins are then translocated from the nucleus to the cytoplasm (7). Evidence that this process is required for the transition from α-gene to downstream gene expression is based on the observation that a truncated CoREST lacking the N-terminal domain, including the HDAC1 binding site inserted in place of ICP0, compensates completely or in part for the absence of ICP0 in a cell-type-dependent manner (9).
The objective of the studies described here was to study the phenotype of a mutant lacking the binding site for CoREST (R8507) and to compare the properties of the mutant with those of a mutant in the RF domain. It is relevant to this report that in wild-type virus-infected cells ICP0 initially accumulates in ND10 structures. Within a few hours, PML is degraded and ICP0 begins to fill the nucleus, and between 5 and 7 h after infection, ICP0 is translocated to the cytoplasm (14, 20). We report that the degradation of PML is blocked in RF mutant-infected cells and delayed in cells infected with the R8507 mutant. Both mutants are impaired in the ability to block interferon. The studies point to ND10 as the principal site in which the functions of ICP0 must be expressed for optimal replication in the cells tested in this study.
MATERIALS AND METHODS
Cells and viruses.
Vero and HeLa cells originally obtained from the American Type Culture Collection were grown in Dulbecco's modified Eagle's medium supplemented with 5% fetal bovine serum. Human embryonic lung (HEL) fibroblasts immortalized by transduction with telomerase, a kind gift from Thomas E. Shenk (Princeton University), were grown as described above but in medium supplemented with 10% serum. U2OS cells were grown in McCoy 5A medium (Gibco-BRL) supplemented with 10% serum. The C116G/C156A mutant, a kind gift from Saul Silverstein (Columbia University), carries the designated amino acid substitutions in a cDNA copy of ICP0 (17). It is referred to throughout this report as the RF mutant. The parent virus, carrying a cDNA copy of ICP0 [HSV-1(vCPc0)], was also obtained from Saul Silverstein (28). The titers of all viruses were determined on U2OS cells as previously described (9).
Construction of recombinant viruses.
The objective of the procedures described below was to introduce four sets of amino acid substitutions into the CoREST binding site in ICP0. A HindIII-XbaI fragment containing the ICP0 gene from pRB8521 (9) was cloned into pcDNA3.1(+) to generate pRB8525. The 3.3-kb SacI-MluI fragment from pRB8525 was substituted for the XhoI-MluI fragment of ICP0 in pRB8521 to generate pRB8526. This substitution eliminated the SalI site in the multiple cloning sites and left the SalI site at the 3′ end of ICP0 as a single SalI cut for future cloning. To replace the intron containing the ICP0 gene with ICP0 cDNA within its genomic context, an NcoI-BglII fragment with a cDNA copy of ICP0 from MTS1-α0PL (19) was cloned into NcoI-PstI-digested pRB8521 to generate pRB8527. The NcoI-SalI fragment of ICP0 cDNA was digested from pRB8527 to replace the NcoI-SalI fragment in pRB8526. This generated pRB8528 containing an ICP0 cDNA in the context of ICP0 5′ and 3′ flanking sequences. The 5′ flanking sequence beyond the NcoI site in pRB8528 is approximately 0.7 kb in length. To ensure recombination, an Asc I-NcoI (∼1.2-kb) fragment from pRB115 was recovered and cloned into HindIII-NcoI-digested pRB8528 to extend the 5′ flanking sequence, resulting in pRB8529.
An NruI-SalI fragment from the C-terminal end of the ICP0 coding region was subcloned between EcoRV and SalI sites to serve as a template for site-directed mutagenesis. Primers 5′-GCCTGCCCATCCTGGCAATGGCAACGGGGAACATCG-3′ and 5′-CGATGTTCCCCGTTGCCATTGCCAGGATGGGCAGGC-3′ were designed for D671A/E673A substitutions. Primers 5′-GAAACATGGCGACCGCACTGGCAGCCGCGGTCCCCG-3′ and 5′-CGGGGACCGCGGCTGCCAGTGCGGTCGCCATGTTTC-3′ were designed for the R693A/R695A mutation. Primers 5′-GTCCCCGGCTGGAGCGCAGCAACCCTGCTCCCCG AG-3′ and 5′-CTCGGGGAGCAGGGTTGCTGCGCTCCAGCCGGGGAC-3′ were designed for the R703A/R704A mutation. Primers 5′-GAGACCGCGGGTAACGCAGTGATGCCCCCCG AG-3′ and 5′-CTCGGGGGGCATCACTGCGTTACCCGCGGTCTC-3′ were designed for the H714A mutation. Mutations were generated with a QuickChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's manual. Restriction fragments containing D671A/E673A, R693A/R695A, R703/R704A, and H714A mutations were cut out by MluI plus SalI and ligated into the MluI-SalI-digested pRB8529, generating pRB8531 (D671A/E673A), pRB8532 (R693A/R695A), pRB8533 (R703A/R704A), and pRB8534 (H714A). The PmeI-PstI fragments from pRB8531, pRB8532, pRB8533, and pRB8534 were then cloned into SacI-PstI-digested pKO5 and generated pKO8531, pKO8532, pKO8533, and pKO8534. A ClaI-MscI fragment containing a tetracycline resistance (Tcr) gene was cut from pACYC184 and cloned between the BamHI and HpaI sites of pRB8521. The resultant pRB8535 was digested with XhoI-XbaI, and the Tcr fragment flanked by ICP0 sequence was cloned into pKO5 to generate pKO8535.
All the unmatched ends of the clones listed above were blunt ended.
Competent cells of Escherichia coli strain RR1 harboring bacterial accessory chromosome 8502 (BAC8502) (9) were electroporated with pKO8535 and incubated at 43°C on Lennox LB (Invitrogen, Carlsbad, CA) plates containing zeocin (25 μg/ml) and chloramphenicol (20 μg/ml). The colonies were diluted and plated on Lennox LB plates containing chloramphenicol (20 μg/ml) and 5% sucrose. The colonies grown on the sucrose plates were screened by colony hybridization. The recombinant BAC8503 obtained had one copy of ICP0 replaced by an ampicillin resistance (Ampr) gene and the other copy of ICP0 replaced by a Tcr gene. Competent cells of RR1 harboring BAC8503 (Ampr/Tcr) were electroporated with the pKO8531, pKO8532, pKO8533, or pKO8534 plasmid. After the selection process, BAC8507 (Ampr/ICP0 cDNA-D671A/E673A), BAC8508 (Ampr/ICP0 cDNA-R693A/R695A), BAC8509 (Ampr/ICP0 cDNA-R703A/R704A), and BAC8510 (Ampr/ICP0 cDNA-H714A) were obtained.
Plasmid DNAs isolated from BAC8507, BAC8508, BAC8509, and BAC8510 were transfected into rabbit skin cells. Plaques obtained on RSC cells were purified three times on the Vero cells. Viruses tend to carry copies of the same genes in the repeat sequences. Viral DNAs were isolated from individual plaques, and recombinant viruses R8507 (both copies of ICP0-cDNA-D671A/E673A), R8508 (both copies of ICP0 cDNA-R693A/R695A), R8509 (both copies of ICP0 cDNA-R703A/R704A), and R8510 (both copies of ICP0 cDNA-H714A) were identified by Southern analysis and amplified for further experiments. The final constructs were sequenced to verify that no adventitious mutations were introduced into ICP0 during mutagenesis and assembly of the recombinant viruses.
Immunoprecipitation.
HeLa cells grown in 75-cm2 flasks were harvested 7 h after mock infection or exposure to virus and lysed in 1 ml of lysis buffer (10 mM Tris, pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 1 mM dithiothreitol, 0.5% NP-40, 0.1 mM NaVO4, 10 mM NaF, 10 mM glycerol phosphate, 1× protease inhibitor cocktail). After brief sonication and centrifugation, the cell debris was discarded. The supernatant fluids were reacted with a protein A-Sepharose CL-4B 50% slurry (GE Healthcare, Pittsburgh, PA) at 4°C for 20 min. On removal of the beads, a half volume of each lysate was reacted with 1 μl of polyclonal anti-ICP0 antibody overnight at 4°C. The lysate-antibody mixtures were incubated with 50 μl of protein A-Sepharose CL-4B 50% slurry at 4°C for 1 h. The beads were then rinsed four times with the same buffer and eluted by 1× sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer.
Immunoblots.
Total cell lysates or immunoprecipitates were electrophoretically separated on denaturing polyacrylamide gels and transferred to preequilibrated polyvinylidene difluoride membranes (Millipore, Billerica, MA). The membranes were blocked in TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Tween 20) containing 5% nonfat dry milk and reacted at 4°C overnight with primary antibody in TBST/5% dry milk, rinsed, and reacted with alkaline phosphatase- or peroxidase-conjugated secondary antibodies (Sigma, St. Louis, MO). The membranes were rinsed and developed with either BCIP (5-bromo-4-chloro-3-indolylphosphate) plus nitroblue tetrazolium (Denville Scientific, Inc., Metuchen, NJ) or enhanced chemiluminescence Western blotting detection reagents (GE Healthcare, Pittsburgh, PA) according to the manufacturer's instructions. In experiments involving immune precipitation, the amount of the input represented 5% of the amount of lysates that was reacted with the antibody.
Confocal microscopy.
HEL cells seeded on four-well slides (Thermo Scientific, Waltham, MA) were mock infected or virus infected as described in Results. The cells were then fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked in PBS containing 1% bovine serum albumin and 10% horse serum. The slides were then reacted with primary antibodies and stained with fluorescein isothiocyanate-conjugated goat anti-rabbit (Sigma, St. Louis, MO) and Texas red-conjugated goat anti-mouse (Invitrogen, Carlsbad, CA) secondary antibodies or Alexa 488-conjugated goat anti-mouse (Invitrogen, Carlsbad, CA) and Alexa 594-conjugated goat anti-rabbit (Invitrogen, Carlsbad, CA) secondary antibodies. The images were taken with a Zeiss LSM410 confocal microscope.
Antibodies.
Polyclonal and monoclonal anti-PML (catalog no. sc-5621 and sc-966) and polyclonal anti-Daxx (sc-7152) antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Polyclonal anti-CoREST (catalog no. 07-455) and monoclonal anti-HDAC1 (catalog no. 05-614) were from Upstate Biotechnology (Millipore, Billerica, MA). Monoclonal anti-CoREST antibody (catalog no. 612146) was from BD Biosciences (San Jose, CA). The polyclonal ICP0 antibody has been described elsewhere (15).
RESULTS
Construction and characterization of HSV-1 mutants carrying amino acid substitutions in the binding site of CoREST in ICP0.
We constructed and verified the sequences of four ICP0 genes carrying amino acid substitutions in the binding site of CoREST (Fig. 1). These mutants were R8507 (D671A/E673A), R8508 (R693A/R695A), R8509 (R703A/R704A), and R8510 (H714A). In the first experiment, the titers of the stocks made in Vero cells were determined in Vero and U20S cells. Included in the titrations was a stock of RF mutant virus obtained from Saul Silverstein (C156A/C116G) and designated the RF mutant. This mutant carried a cDNA copy of ICP0, and therefore, we included as the appropriate control the parent of the RF mutant virus [HSV-1(vCPc0)].
FIG. 1.

Sequences of wild-type and mutant ICP0 genes in this study. The RF domain is highlighted in blue and the CoREST homology site in green, while the CoREST binding site is in red. The key mutants, RF mutant (C116G/C156A), R8507 (D671A/E673A), R8508 (R693A/R695A), R8509 (R703A/R704A), and R8510 (H714A), are shown in brown.
The results of the assays showed that the parent virus and the R8508, R8509, and R8510 mutants yielded approximately equal titers on both cell lines (Table 1). The titers of the RF and R8507 mutants in Vero cells were approximately 100-fold lower than in U2OS cells. From these and other results (not shown), we concluded that the phenotypes of the R8508, R8509, and R8510 mutants were similar to those of the wild-type virus. In all subsequent studies, the R8508 mutant was selected as the representative of this group.
TABLE 1.
Summary of viruses used in the study
| Virus | Titer on Vero | Titer on U2OS | PML degradationa | ICP0 translocationa | CoREST bindinga |
|---|---|---|---|---|---|
| HSV-1(vCPc0) | 1.1 × 1010 | 5.3 × 109 | + | + | + |
| RF mutant | 2.7 × 107 | 2.8 × 109 | − | − | + |
| R8507 | 5.2 × 107 | 1.0 × 109 | +/delayed | +/delayed | − |
| R8508 | 7.6 × 109 | 3.0 × 109 | + | + | + |
| R8509 | 7.5 × 109 | 3.0 × 109 | NA | NA | NA |
| R8510 | 7.7 × 109 | 3.6 × 109 | NA | NA | NA |
+, present; −, absent; NA, not applicable.
We should note at this point that since all of the studies described below involve comparisons of RF and R8507 mutant viruses, we have included in the comparisons the R8708 mutant, representative of amino acid substitutions in HSV-1(F), which exhibited the phenotype of a wild-type virus, and HSV-1(vCPc0), the parent of the RF mutant virus, exhibiting the properties of the wild-type virus.
In the second series of experiments, we compared the replication of these viruses in Vero cells. The cells were infected with 0.1 PFU/cell, and replicate cultures of the infected cells were harvested at 3, 7, 24, and 48 h after infection. The yields of virus harvested at these time points in U2OS cells are shown in Fig. 2. The results show that the initiation of accumulation of progeny virus in cells infected with the RF or R8507 mutant was delayed several hours beyond the time of onset of accumulation of wild-type or R8508 mutant virus. At the completion of the replication cycle of the wild-type virus (24 h), those of the RF mutant and the R8507 mutant lagged behind that of the wild-type virus. However, both viruses continued to accumulate in infected cells, and at least the R8507 mutant attained wild-type yields at 48 h after infection. The pattern of accumulation of the R8508 mutant virus was indistinguishable from that of wild-type virus.
FIG. 2.
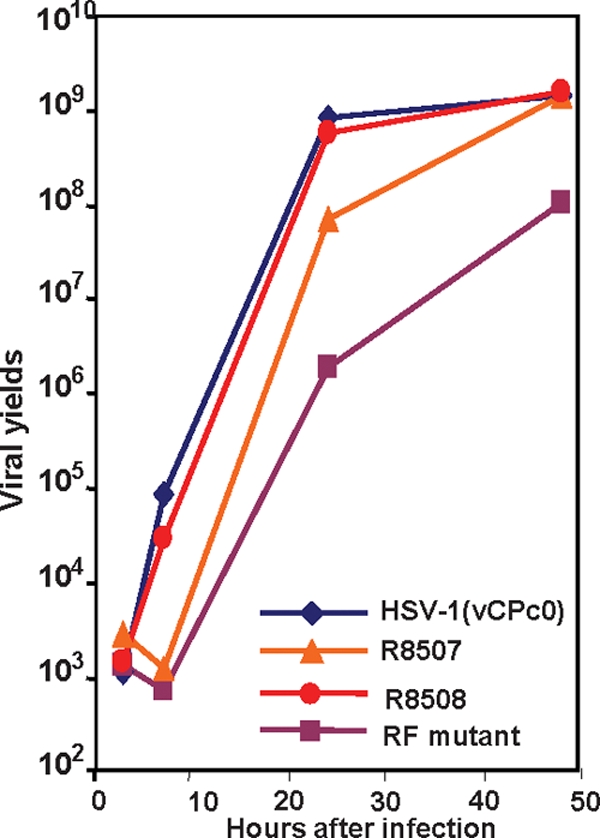
One-step growth curves of the mutants studied. Vero cells were infected with HSV-1(vCPc0), R8507, R8508, and the RF mutant at 0.1 PFU/cell. At 3, 7, 24, and 48 h postinfection, the infected cells were washed and harvested. The titers of the progeny viruses were determined on U2OS cells and are plotted on a logarithmic scale.
We conclude that the growth characteristics of the R8507 mutant carrying amino acid substitutions in the CoREST binding site are similar to those of the RF mutant. Both mutants showed delayed replication in Vero cells.
The ICP0 of the R8507 mutant fails to bind CoREST in pull-down assays.
In this series of experiments, HeLa cells were mock infected or exposed to 10 PFU of HSV-1(vCPc0), RF, R8507, or R8508 virus per cell. The cells were harvested 7 h after infection. Aliquots of the cleared lysates were reacted with ICP0 antibody. The antigen-antibody mixtures were harvested, solubilized, subjected to electrophoresis on denaturing gels and probed with antibody to ICP0 or CoREST. The results were reproducible, and two experiments are shown in Fig. 3. In brief, ICP0 from lysates of the R8507 mutant either failed to pull down CoREST or pulled down less than all other ICP0 mutants tested. The RF mutant and the R8508 mutant ICP0 pulled down reduced amounts of CoREST relative to those of the HSV-1(vCPc0) mutant. The results suggest that even though the properties of the R8508 mutant were, as described below, similar to those of the wild-type virus, its interactions with CoREST were partially impaired.
FIG. 3.

Disruption of CoREST-ICP0 interaction by the D671A/E673A mutation. HeLa cells were mock infected or exposed to 10 PFU of HSV-1(vCPc0), the RF mutant, R8507, or R8508 per cell for 7 h. Immunoprecipitation (IP) was carried out as described in Materials and Methods. The immunoprecipitates were immunoblotted with CoREST or ICP0 antibody. The input was 5% of the total lysates used for immunoprecipitation.
Effects of parent and mutant viruses on PML.
Replicate cultures of HeLa cells were harvested 17 h after infection with 5 PFU of HSV-1(vCPc0), RF, R8507, and R8508 mutant viruses per cell. The cells were solubilized, subjected to electrophoresis on denaturing gels, and probed with antibodies to PML, ICP0, and ICP27. The results (Fig. 4A) showed that PML was degraded in cells infected with the HSV-1(vCPc0) and R8508 mutant viruses. PML was not degraded in cells infected with the RF mutant and was largely, but not completely, degraded in cells infected with the R8507 mutant. The accumulation levels of ICP0 and ICP27 were indistinguishable in cells infected with these viruses.
FIG. 4.
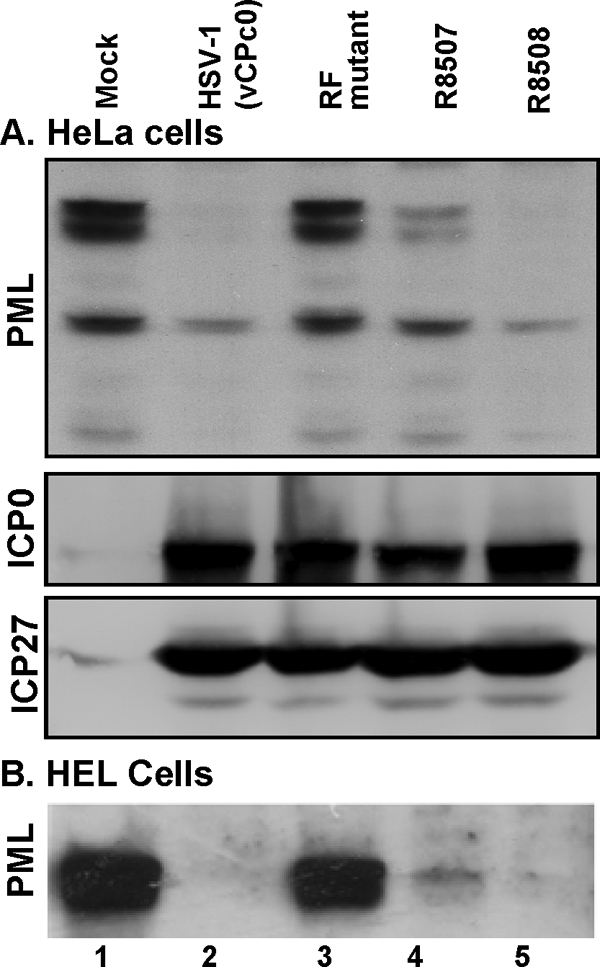
Delay of PML degradation in R8507-infected cells. HeLa cells (A) or HEL cells (B) were mock infected or infected as described above with HSV-1(vCc0), the RF mutant, R8507, or R8508. The cells were harvested 17 h after infection or mock infection. Total cell lysates were electrophoretically separated and immunoblotted for PML, ICP0, or ICP27.
The status of PML in cells infected with HSV-1(vCPc0) and its stabilization in cells infected with the RF mutant were as expected. The degradation of PML in cells infected with the R8508 mutant was consistent with its wild-type phenotype. The partial degradation of PML in cells infected with the R8507 mutant suggests that the rate of degradation of PML in cells infected with this mutant is lower than that of wild-type viruses.
The delay of PML degradation in R8507-infected cells was also observed in HEL cells, as shown in Fig. 4B.
The dispersal of ND10 and translocation of ICP0 are delayed in cells infected with the RF and R8507 mutant viruses.
HEL fibroblasts grown in four-well slides were mock infected or exposed to 10 PFU of HSV-1(vCPc0), RF, R8507, or R8508 mutant virus per cell. At 5 h after infection, the cells were reacted with polyclonal antibody to ICP0 and monoclonal antibody to PML. In a parallel experiment, at 7 h after infection, the cells were reacted with antibodies to Daxx and ICP0. Representative images are shown in Fig. 5A to L and AA to LL, respectively. The results were as follows. (i) In mock-infected cells, PML was entirely contained in punctate nuclear structures known as ND10 (Fig. 5A and C). Daxx (Fig. 5AA and CC), by contrast, was partially contained in punctate structures and partially dispersed in the nucleus. (ii) In cells infected with HSV-1(vCPc0), PML was no longer detectable at 5 h after infection and ICP0 was largely in the cytoplasm, although in some cells it was still retained in the nucleus (Fig. 5D to F). At 7 h after infection, Daxx was largely released from ND10s, reflecting the dispersal of ND10s after the degradation of PML. ICP0 was largely in the cytoplasm (Fig. 5DD to FF). (iii) In cells infected with the RF mutant, ICP0 and PML colocalized in punctate structures at 5 h after infection (Fig. 5G to I). At 7 h, ICP0 increased in amount and in many cells began to fill the nucleus (Fig. 5GG to II). The distribution of Daxx did not significantly change relative to that in mock-infected cells. In the few infected cells where ICP0 filled the nucleus, Daxx was released from the ND10s (Fig. 5GG). (iv). In cells infected with the R8507 mutant, at 5 h after infection, ICP0 was contained in the nucleus or in both the nucleus and the cytoplasm (Fig. 5J to L). PML was barely detectable by immunofluorescence, consistent with the results shown in Fig. 4. The distribution of ICP0 did not appear to change much at 7 h after infection. In these cells, Daxx was dispersed in the nucleus (Fig. 5JJ to LL). (v) The rate of degradation of PML determined from visual analyses of the images shown in Fig. 5 is consistent with the amount of PML detected at 17 h after infection, shown in Fig. 4. To obtain a more accurate determination of the distribution of ICP0, we took serial photographs of adjacent fields of HEL cells 9 h after infection. Earlier studies had shown that at 9 h after infection with wild-type viruses, ICP0 was found almost exclusively in the cytoplasm (8, 14, 20). The distribution of ICP0, whether it was contained entirely in the nucleus, in both the nucleus and the cytoplasm, or totally exported to the cytoplasm, was then determined by cell counts. The results shown in Fig. 6 indicate that, as expected, ICP0 was totally exported to the cytoplasm in at least 90% of cells in cultures infected with HSV-1(vCPc0) or the R8508 mutant. In cells infected with the RF mutant, ICP0 was retained in the nucleus, and only a very small fraction of the total infected cells contained ICP0 in both the cytoplasm and the nucleus. In cells infected with the R8507 mutant, ICP0 was largely in both the cytoplasm and the nucleus or entirely in the nucleus.
FIG. 5.

PML and Daxx distribution in cells infected with the mutants. HEL cells were infected with HSV-1(vCPc0), the RF mutant, or R8507 for 5 or 7 h, as indicated. An immunofluorescence assay was carried out as described in Materials and Methods. Images of cells reacted with antibody to PML and ICP0 (A to L) or Daxx and ICP0 (AA to LL) were collected with a Zeiss confocal microscope (100× objective).
FIG. 6.

ICP0 distribution in R8507-infected cells. HEL cells were fixed 9 h after infection with HSV-1(vCPc0), the RF mutant, R8507, or R8508 and reacted with antibody to PML and ICP0 antibodies. Photographs were taken of adjacent fields and tabulated on the basis of localization of ICP0 in infected cells. At least 200 infected cells were tabulated for each virus.
RF and R8507 mutants are unable to overcome the inhibition of viral replication by IFN-α or IFN-γ.
In another series of experiments, replicate cultures of Vero cells were mock treated or exposed to 1,000 units of IFN-α or IFN-γ per ml (Fig. 7). After 24 h, the cells were mock infected or exposed to 0.1 PFU of HSV-1(vCPc0), RF, R8507, or R8508 mutant virus per cell. The cells were harvested at 3, 7, 24, or 48 h after infection, and titers were determined on U2OS cells. As expected, HSV-1(vCPc0) or R8508 mutant virus was unaffected by either IFN-α or IFN-γ. Also as predicted, the onset of accumulation of the R8507 and RF mutants was delayed, and while the R8507 mutant attained the same yields at 48 h as HSV-1(vCPc0), the yield of RF was reduced >10-fold. Both the RF and R8507 mutants were inhibited by the interferons. The effect on the RF mutant was appreciably greater than that on the R8507 mutant.
FIG. 7.

Defective response of R8507 to interferon treatment. (A) Time line of interferon treatment and infection. (B) Growth curves. Vero cells were treated with 1,000 U of IFN-α or IFN-γ per ml for 24 h and then exposed to 0.1 PFU of HSV-1(vCPc0), RF, R8507, or R8508 mutant virus. At 3, 7, 24, and 48 h after infection, the infected cells were harvested. The titers of the progeny viruses were determined on U2OS cells.
DISCUSSION
ICP0 performs multiple functions, but at least two appear to be specifically related to the phenotype of ΔICP0 mutants. The first involves precluding the inhibition of viral replication by interferon (12, 24). The second involves blocking the silencing of viral DNA after α-gene expression by host proteins (7, 27, 29). The first function appears to take place in or in the immediate vicinity of ND10 nuclear bodies. Numerous reports have shown that transfected DNA, as well as HSV-1 DNA, on entry into the nucleus accumulates in ND10 nuclear bodies (6, 13, 22, 33, 35). The sequence of events reported in numerous studies and also illustrated in this report is that ICP0 is translocated to the ND10 structures but does not colocalize with PML (8, 19, 21) and that PML, along with other ND10 proteins, is degraded (3, 8). ICP0 then becomes dispersed throughout the nucleus, and once viral-DNA synthesis begins, it is transported to the cytoplasm (14, 20). Since ICP0 acts as a ubiquitin ligase for the degradation of PML and SP100 (1, 8, 11), this activity most likely takes place in the ND10 structures. The full range of objectives attained by the degradation of the PML and SP100 components of ND10 is unknown, but studies of PML+/+ and PML−/− murine cells indicate that destruction of PML may render cells nonsusceptible to exogenous interferon (2).
The sum of evidence obtained to date indicates that in the absence of ICP0, post-α genes are silenced but may be partially rescued by inhibitors of HDACs (29). ICP0 blocks the silencing by displacing HDAC1/2 from the CoREST-REST complex (7, 9). Both HDACs and the CoREST-REST complex are then transported to the cytoplasm (7). Evidence favoring a role for CoREST in blocking post-α gene silencing is based on the observation that a mutant virus in which the ICP0 genes were replaced by a truncated CoREST bereft of the ability to bind HDAC1 enabled the virus to replicate to wild-type levels in Vero cells and to at least 10-fold higher titers than the ΔICP0 mutant in other cell lines (9). Inasmuch as CoREST is dispersed throughout the nucleus, the question arose as to whether the two functions of ICP0 are concurrent or are independent of each other. In this report, we compared two mutants carrying amino acid substitutions in the RF domain and in the domain of ICP0 that is required to pull down CoREST. We selected as a representative of a wild-type virus the parent virus of the RF mutant, since both carry a cDNA copy of wild-type ICP0, and R8508, a mutant that carries two amino acid substitutions in the binding domain of ICP0 for CoREST but which in all respects tested in the studies reported here exhibits a wild-type phenotype. The results and their significance may be summarized as follows. (i) The onset of accumulation of both RF and R8507 mutant viruses is delayed by several hours relative to the wild-type viruses used in this study. Both viruses continue to replicate long after the accumulation of progeny in cells infected with wild-type viruses ceases. We have no evidence that the accumulation of RF virus in Vero cells (Fig. 2) levels off after 48 h. The results shown in Table 1 and Fig. 2 indicate that the R8507 virus can replicate, albeit with some delay, in both Vero and U20S cells to the same levels as the wild-type parent but that the ability to form a plaque is impaired, possibly because as the virus spreads from cell to cell at each step, the replication is significantly delayed. (ii) Only a fraction of the CoREST present in HeLa cells was pulled down by ICP0 encoded by the wild-type surrogate HSV-1(vCPc0). This is not surprising, inasmuch as CoREST is abundant, is dispersed throughout the cell, and may form different functional aggregations. This observation is consistent with the observation that late in infection only a fraction of CoREST is exported from the nucleus to the cytoplasm (7). In contrast, the ICP0 encoded by the R8507 mutant was impaired in its ability to pull down CoREST, consistent with the mutation in the binding site for CoREST. It is noteworthy, however, that the ICP0s encoded by both the RF and the R8508 mutants pulled down reduced amounts of CoREST. The implication of these findings is twofold. In the case of the R8508 mutant, the mutation reduced the ability of ICP0 to bind CoREST, but the residual affinity of ICP0 was sufficient to enable the virus to replicate as a wild-type virus. The reduced capacity of the RF mutant to bind CoREST is less clear; it may reflect changes in the secondary or tertiary structure of ICP0. It is noteworthy that the dimerization domain of ICP0 was mapped between residues 617 and 711 (4), that is, roughly between the sequence homologous to CoREST and the binding site of CoREST (Fig. 1). An alternative hypothesis is that undegraded components of the ND10 structure block the interaction of ICP0 with the CoREST/REST complex. (iii) PML was degraded in both the HeLa and HEL cell lines infected with wild-type virus and was spared in cells infected with the RF mutant. The degradation of PML was delayed but was almost complete in HEL cells 17 h after infection with the R8507 mutant (Fig. 4A and 5). A similar delay was noted in HeLa cells (Fig. 4B). (iv) The ICP0s of both mutant viruses are retained in ND10 structures longer than those of wild-type viruses. At 9 h after infection, at the time the ICP0 of wild-type virus is fully exported, that of the RF virus is retained in the nucleus and that of R8507 is partially exported to the cytoplasm but has not yet vacated the nucleus (Fig. 6). (v) Pretreatment of Vero cells with IFN-α or IFN-γ reduced the yields of the RF and R8507 mutants but had no effect on the replication of wild-type viruses. The experimental design of these studies was similar to that employed in the studies of PML+/− and PML−/− cells (2). Since in those studies the resistance to interferons was linked to the presence of PML, it is tempting to conclude that the sensitivity of the mutant viruses to interferons reflects the delayed degradation of (R8507) or failure to degrade (RF mutant) PML.
It is convenient to view the results of this report in the context of a model of the early events. In brief, several studies have shown that on entry into the nucleus, ND10 structures form or aggregate around the DNA (13, 22). The key events taking place at this time are degradation of PML by the ubiquitin ligase expressed by ICP0 and activation of the transcription of β genes. From the point of view of viral replication, the key event is the activation of transcription of β and γ genes. In ICP0 mutant virus-infected cells, activation of β genes, albeit inefficiently, can be achieved by inhibitors of HDACs (27, 29). As noted above, replacement of ICP0 by a gene encoding a truncated CoREST lacking the first 145 residues and incapable of binding HDAC1 is a more efficient way of activating the HSV-1 gene cascade (9). Activation of the cascade is also effected at high multiplicities of infection. We conclude from these considerations that degradation of PML and activation of the transcription of β and γ genes are independent events. The studies presented in this report indicate that in the context of intact ICP0 encoding a mutation in RF or in the CoREST binding site, the two events are interdependent. One explanation of the interdependence is that the biochemical processes at the two ends of ICP0, in the RF domain and at the C terminus, have evolved to proceed in tandem and that interference with one process affects the other. Consistent with this view is the fact that both functions are expressed by the same protein early in infection. The failure to degrade or the delay in the degradation of PML by the mutations in RF and in the CoREST binding site may be sufficient to enable the expression of the antiviral state and to result in diminished virus yields.
It is noteworthy that the truncated CoREST protein that compensates entirely or in part in a cell-dependent manner for the absence of ICP0 targets the HDAC1- or HDAC2 CoREST/REST complex and not the known constituents of ND10 structures (9). One implication of this observation in light of current results is that the truncated CoREST enables the ΔICP0 mutant virus to bypass the block imposed by the intact PML.
Acknowledgments
These studies were aided by National Cancer Institute grant CA78766.
Footnotes
Published ahead of print on 22 October 2008.
REFERENCES
- 1.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chee, A. V., P. Lopez, P. P. Pandolfi, and B. Roizman. 2003. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J. Virol. 777101-7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chelbi-Alix, M. K., and H. de The. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18935-941. [DOI] [PubMed] [Google Scholar]
- 4.Ciufo, D. M., M. A. Mullen, and G. S. Hayward. 1994. Identification of a dimerization domain in the C-terminal segment of the IE110 transactivator protein from herpes simplex virus. J. Virol. 683267-3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Everett, R. D., W. C. Earnshaw, J. Findlay, and P. Lomonte. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 181526-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Everett, R. D., and J. Murray. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 795078-5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu, H., Y. Liang, G. Mandel, and B. Roizman. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. USA 1027571-7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu, H., and B. Roizman. 2003. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 1008963-8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu, H., and B. Roizman. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. USA 10417134-17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hagglund, R., and B. Roizman. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 782169-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagglund, R., C. Van Sant, P. Lopez, and B. Roizman. 2002. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA 99631-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harle, P., B. Sainz, Jr., D. J. Carr, and W. P. Halford. 2002. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology 293295-304. [DOI] [PubMed] [Google Scholar]
- 13.Ishov, A. M., and G. G. Maul. 1996. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 134815-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawaguchi, Y., R. Bruni, and B. Roizman. 1997. Interaction of herpes simplex virus 1 α regulatory protein ICP0 with elongation factor 1δ: ICP0 affects translational machinery. J. Virol. 711019-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawaguchi, Y., C. Van Sant, and B. Roizman. 1997. Herpes simplex virus 1 α regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 717328-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee, M. G., C. Wynder, N. Cooch, and R. Shiekhattar. 2005. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437432-435. [DOI] [PubMed] [Google Scholar]
- 17.Lium, E. K., and S. Silverstein. 1997. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential α27 gene. J. Virol. 718602-8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lomonte, P., K. F. Sullivan, and R. D. Everett. 2001. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 2765829-5835. [DOI] [PubMed] [Google Scholar]
- 19.Lopez, P., R. J. Jacob, and B. Roizman. 2002. Overexpression of promyelocytic leukemia protein precludes the dispersal of ND10 structures and has no effect on accumulation of infectious herpes simplex virus 1 or its proteins. J. Virol. 769355-9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez, P., C. Van Sant, and B. Roizman. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol. 753832-3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maul, G. G., H. H. Guldner, and J. G. Spivack. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 742679-2690. [DOI] [PubMed] [Google Scholar]
- 22.Maul, G. G., A. M. Ishov, and R. D. Everett. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 21767-75. [DOI] [PubMed] [Google Scholar]
- 23.Maul, G. G., D. Negorev, P. Bell, and A. M. Ishov. 2000. Properties and assembly mechanisms of ND10, PML bodies, or PODs. J. Struct. Biol. 129278-287. [DOI] [PubMed] [Google Scholar]
- 24.Mossman, K. L., H. A. Saffran, and J. R. Smiley. 2000. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 742052-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Negorev, D., and G. G. Maul. 2001. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene 207234-7242. [DOI] [PubMed] [Google Scholar]
- 26.Parkinson, J., S. P. Lees-Miller, and R. D. Everett. 1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73650-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poon, A. P., Y. Liang, and B. Roizman. 2003. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. 0. J. Virol. 7712671-12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poon, A. P., S. J. Silverstein, and B. Roizman. 2002. An early regulatory function required in a cell type-dependent manner is expressed by the genomic but not the cDNA copy of the herpes simplex virus 1 gene encoding infected cell protein 0. J. Virol. 769744-9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poon, A. P. W., H. Gu, and B. Roizman. 2006. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. USA 1039993-9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roizman, B., D. M. Knipe, and R. J. Whitley. 2007. Herpes simplex viruses, p. 2501-2601. In D. M. Knipe, P. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, New York, NY.
- 31.Sacks, W. R., and P. A. Schaffer. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 61829-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi, Y., F. Lan, C. Matson, P. Mulligan, J. R. Whetstine, P. A. Cole, R. A. Casero, and Y. Shi. 2004. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119941-953. [DOI] [PubMed] [Google Scholar]
- 33.Sourvinos, G., and R. D. Everett. 2002. Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J. 214989-4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 672571-2585. [DOI] [PubMed] [Google Scholar]
- 35.Tsukamoto, T., N. Hashiguchi, S. M. Janicki, T. Tumbar, A. S. Belmont, and D. L. Spector. 2000. Visualization of gene activity in living cells. Nat. Cell Biol. 2871-878. [DOI] [PubMed] [Google Scholar]