

Abstract
Studies with herpes simplex virus type 1 (HSV-1) have shown that secondary envelopment and virus release are blocked in mutants deleted for the tegument protein gene UL36 or UL37, leading to the accumulation of DNA-containing capsids in the cytoplasm of infected cells. The failure to assemble infectious virions has meant that the roles of these genes in the initial stages of infection could not be investigated. To circumvent this, cells infected at a low multiplicity were fused to form syncytia, thereby allowing capsids released from infected nuclei access to uninfected nuclei without having to cross a plasma membrane. Visualization of virus DNA replication showed that a UL37-minus mutant was capable of transmitting infection to all the nuclei within a syncytium as efficiently as the wild-type HSV-1 strain 17+ did, whereas infection by UL36-minus mutants failed to spread. Thus, these inner tegument proteins have differing functions, with pUL36 being essential during both the assembly and uptake stages of infection, while pUL37 is needed for the formation of virions but is not required during the initial stages of infection. Analysis of noninfectious enveloped particles (L-particles) further showed that pUL36 and pUL37 are dependent on each other for incorporation into tegument.
Herpesvirus virions have characteristic structures that combine symmetrical and nonsymmetrical components (22, 45, 58). The viral DNA genome is contained within an icosahedral capsid, a protein layer called the tegument surrounds the capsid, and the virion is bounded by a lipid envelope, which contains numerous glycoprotein spikes. Capsids are robust structures that can be readily purified from infected cells and studied in isolation from the other virion components. They are classified according to their internal composition as A-capsids (empty), B-capsids (containing scaffold), and C-capsids (containing DNA) (19, 23, 42, 50). The symmetry and uniformity of the capsid shell make it amenable to structural analysis, and its composition, structure, and morphogenesis have been studied extensively (23, 50, 59). In contrast, the bulk of the tegument appears to be highly variable, with the numbers of some component proteins differing widely among individual virus particles (7, 11). Compositionally, it is the most complex part of the virion, containing more than 15 viral gene products (23, 36, 50), many of which can be deleted without obviously affecting the virion structure. The tegument is usually described as amorphous, and its structure appears not to be correlated to that of the capsid, except at its inner boundary where a pattern of icosahedrally arranged elements interacting with capsid proteins is evident. These connections are distributed across the entire surface of the capsid in simian and human cytomegaloviruses (6, 53) but are confined to the vertices in herpes simplex virus type 1 (HSV-1) (22, 58).
Capsid assembly and DNA packaging take place in the nuclei of infected cells. The DNA-containing capsids (C-capsids) exit the nucleus by traversing the two layers of the nuclear membrane (36). They become enveloped at the inner leaflet but lose this primary envelope by fusion with the outer leaflet, which releases the capsid into the cytoplasm. Two viral gene products (genes UL31 and UL34) are required for this process (40, 44), but they do not form part of the mature virus particle (18). The US3-encoded protein kinase (pUS3), which is a constituent of tegument, is important for efficient nuclear exit (41, 56), but no tegument protein is known to be essential for capsid exit from the nucleus. The sequence and location of tegument assembly are still poorly understood. It has been reported that the HSV-1 tegument protein, pUL48 (VP16), and the HSV-1 and pseudorabies virus (PrV) pUS3 proteins are associated with primary enveloped capsids in the perinuclear space (21, 38, 41). However, the bulk of the tegument appears to be added in the cytoplasm, following which the mature virion is thought to be formed by envelopment at vesicles of the trans-Golgi network.
The products of HSV-1 genes UL36 and UL37 are tegument proteins which are believed to be closely associated with the capsid. The UL36 protein (pUL36) has been proposed as a possible candidate for the tegument protein attached to the vertices of capsids within virions (58), although recently it has been suggested that the proteins encoded by UL17 and UL25 form part of this material (53). There is no direct evidence for the location of the UL37 protein (pUL37) in the tegument, but it is known to interact with pUL36 (25, 37, 55) and has been described as an inner tegument protein. It is not clear where pUL36 and pUL37 become incorporated into virions, although both proteins have been reported as associating with capsids purified from the nuclei of infected cells (5). The putative interaction between pUL36 and the capsid may not entirely account for its presence in the virion tegument, as it is also found in L-particles, which resemble virions in size and shape but lack capsids and therefore consist solely of enveloped tegument (52). L-particles are formed both in productive infections and in circumstances where virion assembly is blocked (43). Although pUL36 and pUL37 are components of L-particles (34, 52), studies with PrV mutants have shown that either one is dispensable for L-particle production (17, 26).
Mutational analysis has established that UL36 is essential for virus replication in both HSV-1 (3, 14) and PrV (17). However, although UL37 is essential in HSV-1 (13), deletion of this gene in PrV results in impairment but not abolition of growth (26). The phenotypes of HSV-1 mutants deleted for UL36 and UL37 are similar, and in both cases C-capsids accumulate in the cytoplasm of infected cells. This implies that the proteins are not required for capsid assembly, DNA packaging, or exit from the nucleus but are important for the further addition of tegument and envelope.
As well as being important in virus assembly and egress, UL36 plays a role during the initial phase of infection. Thus, infection of cells at nonpermissive temperatures with an HSV-1 temperature-sensitive mutant of UL36 (tsB7) prevents release of the viral DNA from capsids and causes them to accumulate in the vicinity of nuclear pores (3). No temperature-sensitive mutant has been reported for UL37, and its importance in initiation of infection is unknown. However, both pUL36 and pUL37 remain associated with capsids during transport to the nucleus (20, 29), indicating a possible role for pUL37 at this stage of infection.
In this paper we use deletion mutants with deletions of the UL36 and UL37 genes to study their roles during viral infection. Our studies confirm that neither protein is needed for formation of cytoplasmic C-capsids or for bulk tegument assembly, as evidenced by L-particle formation, making it likely that they act as the link between these two virion compartments. In addition, by examining the spread of infection within syncytia, we show that capsids lacking pUL36 are unable to transmit infection between nuclei, whereas initiating a new infectious cycle does not require pUL37.
MATERIALS AND METHODS
Cell culture.
Human fetal foreskin fibroblasts (HFFF2; European Collection of Cell Cultures) were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Gibco). Baby hamster kidney cells (BHK 21 clone 13; ATCC) were grown in Glasgow modified Eagle medium supplemented with 10% newborn bovine serum (Gibco) and tryptose phosphate broth. Rabbit skin (RS) cells (2) were grown in DMEM supplemented with 10% fetal bovine serum and 1% nonessential amino acids (Gibco).
Cell lines expressing HSV-1 genes.
All complementing cell lines were derived from RS cells. Plasmids expressing the appropriate HSV-1 genes (see below) were mixed with pSV2Neo (Clontech) or with pC1-Neo (Promega) and cotransfected by calcium phosphate precipitation (51) into 35-mm dishes of RS cells. Transfected cells were incubated for 24 h at 37°C before subculture into 24-well plates. Cells were overlaid with medium containing 1 mg/ml G418 (Gibco). The selection medium was replaced every 3 days, until colony growth was evident. The drug-resistant colonies were then selected, expanded, and tested for their ability to support growth of the mutant viruses.
(i) UL19.
UL19(1) cells were provided by V. Preston (MRC Virology Unit). Briefly, the open reading frame (ORF) of UL19 (residues 40,528 to 36,353 [32]) was isolated from pJN6 (39) as a BglII fragment and cloned into the BamHI site of pApV (27). G418-resistant cell lines produced as described above were tested for their ability to support growth of the UL19 deletion mutant K5ΔZ (12), and one line was selected for further use (V. Preston, personal communication).
(ii) UL36.
The expression plasmid pApV (27) was modified using the double-stranded oligonucleotide pApVBstBI generated from the complementary sequences 5′-CTAGAACGGATCCGTCGACTTCGAAC and 5′-GATCGTTCGAAGTCGACGGATCCGTT. pApVBstBI was inserted between the unique XbaI and BamHI sites to produce pApVB. This creates a new BamHI site and introduces a BstBI site between the HSV-1 ICP6 (UL39) promoter and simian virus 40 polyadenylation sequence. The C-terminal ∼8,000 nucleotides of the UL36 ORF were isolated from cosmid cos14 (9) as a BamHI fragment (see Fig. S1 in the supplemental material) and cloned into BamHI-digested pApVB to form pApVB36C. pApVB36C was digested with BstBI and religated to give plasmid pApVB36CBs, which has only 32 bp of HSV-1 sequence downstream of the UL36 ORF. The missing N-terminal region of UL36 was generated by PCR using primers UL36_HA_Nterm and UL36CTBamHIF (see Fig. S1 in the supplemental material) and cloned as an XbaI/BamHI fragment into XbaI/BamHI-digested pApVB36CBs to produce pHAUL36. This contains the full-length UL36 ORF (residues 80,540 to 71,016), fused to an influenza virus hemagglutinin (HA) epitope tag inserted at the N terminus. Cell lines generated using pHAUL36 and pSV2Neo (see above) were tested for their ability to complement the growth of the temperature-sensitive mutant tsB7 and the deletion mutant KΔUL36 (14). One line (HAUL36-1) was selected and used for all subsequent experiments.
(iii) UL37.
Cell lines expressing UL37 were produced by two methods. Initially, the green fluorescent protein (GFP) vector, GFPemd (Packard), was modified to introduce SpeI sites at the N and C termini of the GFP ORF. The resulting SpeI fragment was cloned into the unique SpeI site located adjacent to the 3′ end of the UL37 ORF (see Fig. S1 in the supplemental material) in plasmid pGX336, which contains the BamHI O fragment (residues 79,441 to 86,980) cloned into pUC118. The resulting plasmid, pGX336GFP, which expresses a pUL37-GFP fusion protein was cotransfected with pSV2neo into RS cells. G418-resistant clones were screened for GFP expression following infection with wild-type (WT) HSV-1. One clone [1.40(2)] was selected and used in the production of the UL37 deletion mutant FRΔUL37 (see below). The plasmid, pGX336GFP, used to produce cell line 1.40(2) contains the UL37 ORF flanked by extensive HSV-1 sequences. To reduce the possibility that recombination with these sequences would rescue the FRΔUL37 mutation (see below), a second pUL37-expressing cell line was produced. The UL37 ORF (residues 84,171 to 80,707) was isolated from pUL373 (33) by BamHI digestion and cloned into BamHI-digested pApV to form pApV-UL373. pApV-UL373 was cotransfected into RS cells with pC1-Neo. G418-resistant clones were screened for their ability to complement the growth of FRΔUL37. One clone (80C02) was selected and used for all subsequent experiments.
Virus mutants.
K5ΔZ (minus UL19) and KΔUL36 were provided by P. Desai (12, 14).
(i) UL36.
Because the existing mutant, KΔUL36, contained only a partial deletion of the UL36 ORF, a second, complete deletion mutant was made using the HSV-1 bacterial artificial chromosome (BAC) pBAC SR27. In pBAC SR27 (supplied by C. Cunningham [MRC Virology Unit]), loxP recombination sites flank the bacterial maintenance sequences and a Cre recombinase gene, which are inserted between genes US1 and US2. The Cre recombinase is expressed following transfection into mammalian cells and results in the loss of the bacterial sequences (48). The UL36 ORF was removed from pBAC SR27 using a variation on the Lambda RED recombination system (Genebridges, GmbH). PCR was carried out on the supplied rpsL-neo template using primers UL36rm_loxFAS_F and UL36rm_loxFAS_R (see Fig. S1 in the supplemental material). The resulting PCR product contains the neomycin cassette flanked by nested FAS sites and sequences from upstream and downstream of the UL36 ORF. FAS sites are variant loxP sequences that will not undergo heterologous recombination with the archetypal loxP sites present in pBAC SR27 (47). The PCR fragment was recombined into pBAC SR27, and bacterial colonies were selected for neomycin resistance. The resulting BAC (pBAC SR27Δ36-1) has the UL36 ORF replaced by the neomycin cassette. Following transfection into HAUL36-1 cells, Cre recombinase is expressed, which excises both the FAS-flanked neomycin cassette and the loxP-flanked BAC maintenance sequences. Individual virus plaques were picked, and their ability to grow on HAUL36-1 and control RS cells was examined. Deletion of the UL36 ORF was confirmed by PCR and DNA sequencing, and one isolate (ARΔUL36) was selected for further use.
(ii) UL37.
pGX336 was digested with ClaI and HpaI, which flank the UL37 ORF (see Fig. S1 in the supplemental material). The ClaI site was rendered blunt ended by treatment with T4 polymerase, and the plasmid was recircularized by ligation to generate pGX336-37minus. The red fluorescent protein (RFP) ORF from pDsRed-Monomer-N1 (Clontech) was isolated as an NheI/XbaI fragment and ligated into SpeI-digested pGX336-37minus to generate pFRΔUL37. This removes the UL37 ORF apart from the three C-terminal amino acids and places the RFP ORF under the control of the UL37 promoter. pFRΔUL37 was cotransfected with HSV-1 strain 17+ virion DNA into 1.40(2) cells (see above). Red fluorescent plaques were selected and screened for their differential ability to grow on 1.40(2) cells and parental RS cells. One isolate (FRΔUL37) was selected and grown for further use on 80C02 cells.
Rescuants.
The NheI fragment (residues 68,662 to 85,304) containing the UL36 and UL37 sequences was cloned into the XbaI site of pUC19 to generate pUCNhe4. Noncomplementing RS cells transfected with pUCNhe4 were superinfected with 1 PFU of ARΔUL36 or FRΔUL37 per cell. Progeny virus was screened for the ability to grow on RS cells, and one virus from each rescue, designated ARΔUL36R and FRΔUL37R, was selected and grown for further use.
Cell fusion.
HFFF2 cells were seeded onto 22-mm square cover glasses in 35-mm culture dishes at 5 × 105 cells per dish and incubated at 37°C overnight. Duplicate plates of cells were infected for 1 h at 37°C with 0.01 PFU of the appropriate virus per cell. One plate from each pair was then overlaid with 2 ml of DMEM supplemented with 10% pooled human serum (Cellect). The second plate was washed twice in serum-free DMEM and then overlaid with 1 ml of 50% polyethylene glycol 1500 (PEG1500) in 75 mM HEPES (pH 8) (Roche) for 1 min at 37°C (28). The PEG solution was removed, and the cells were rinsed three times in DMEM containing 15% dimethyl sulfoxide and three times in DMEM and then overlaid with DMEM containing 10% pooled human serum. Both plates were maintained at 37°C for a further 23 h.
Preparation of fluorescent DNA probe.
A pseudolibrary was produced by SacII digestion of a bacmid containing the entire HSV-1 genome and cloning 200- to 500-bp gel-purified fragments into pEGFP-C1 (Clontech). DNA from this pseudolibrary was used as a template for a PCR using Cy3-conjugated pEGFP-C1-specific primers (MWG). PCR products of suitable size (200 to 500 bp) were gel purified and used as a probe for fluorescent in situ hybridization (FISH).
FISH.
Cells grown on cover glasses as described above were rinsed twice in phosphate-buffered saline (PBS) and fixed in 95% ethanol and 5% acetic acid at −20°C for 5 min. After fixation, the cells were air dried, rehydrated in PBS, and stored at 4°C until required. To carry out FISH (16), the cover glasses were incubated in hybridization buffer (50% formamide, 10% dextran sulfate, and 4× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]) for 30 min at 37°C and then in probe solution (1 μl HSV-1 probe, 0.5 μl salmon testis DNA [10 mg/ml], 8.5 μl hybridization buffer) for 2 min at 95°C, before they were placed, cell side down, onto glass slides and incubated overnight at 37°C in a humidified hybridization chamber (Camlabs). They were washed twice at 65°C and once at room temperature with 2× SSC and overlaid with PBS before processing for immunofluorescence staining. If cytoplasmic labeling was required, they were then incubated for 30 min in PBS containing 0.5 μg/ml CellMask deep red (Invitrogen) and washed three times in PBS. All cover glasses were mounted in 2.5% 1,4-diazabicyclo[2.2.2]octane (Sigma) in Mowiol (Harco) containing 1 μg/ml 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma). Samples were examined using a Zeiss LSM 510 meta confocal microscope.
Antibodies.
The following antibodies were used: mouse monoclonal antibodies (MAbs) E12-E3 raised against the N-terminal 1 to 287 amino acids of pUL36 (1), DM165 against VP5 (31), VP16 (1-21) against pUL48 (Santa Cruz Biotechnology), MCA406 against VP22a (AbD Serotec), and DM1A against α-tubulin (Sigma); rabbit polyclonal antibody M780 against pUL37 (46); AGV031 against pUL49 (15); Alexa Fluor 488-conjugated goat anti-mouse antibody (Molecular Probes); and horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit antibodies (Sigma).
Capsid and L-particle preparation.
Ten 850-cm2 roller bottles of BHK cells were infected at 5 PFU/cell with the appropriate virus. Three hours after infection, the cells were washed with 50 ml of PBS twice to remove unbound virus, then overlaid with 40 ml of fresh Glasgow modified Eagle medium supplemented with 10% newborn bovine serum and tryptose phosphate broth, and incubated for a further 21 h at 37°C. The culture medium was removed and retained for isolation of extracellular particles. Infected cells were removed, and nuclear and cytoplasmic fractions were separated by overlaying the monolayers with 25 ml of ice-cold PBS containing 1% IGEPAL [(octylphenoxy)polyethoxyethanol] CA-630 (Sigma) and protease inhibitors (complete, EDTA-free; Roche). Nuclei were harvested by centrifugation at 3,000 rpm for 10 min and resuspended in 30 ml of NTE buffer (0.5 M NaCl, 0.02 M Tris [ph 7.4], 0.01 M EDTA) containing 1% IGEPAL. Both the nuclear and cytoplasmic fractions were disrupted with a Branson sonifier 350 and clarified by centrifugation at 3,000 rpm for 10 min. The supernatants were transferred to fresh tubes, and the capsids were pelleted through 40% (wt/vol) sucrose cushions before being separated on 20 to 50% (wt/vol) sucrose gradients by centrifugation for 1 h at 25,000 rpm on an AH629 rotor (Sorvall).
Extracellular particles were purified from the culture medium on 5 to 15% (wt/vol) Ficoll gradients as described previously (52).
Western blot analysis.
Protein samples separated by electrophoresis on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels (SDS-polyacrylamide gel electrophoresis [SDS-PAGE]) were transferred electrophoretically to Hybond ECL membrane (Amersham). Blots were blocked for 30 min with 5% Marvel (Premium Brands) milk powder in 20 mM Tris (pH 8), 0.15 M NaCl, 1% Tween 20 (TBS-Tween) and incubated overnight at 4°C with appropriate antibodies diluted in 1% Marvel milk powder in TBS-Tween. Immunodetection was by enhanced chemiluminescence (ECL; Amersham). Before the blots were reprobed, the bound antibodies were stripped by incubating the membrane at 50°C in 62.5 mM Tris (pH 7), 2% SDS, and 100 mM β-mercaptoethanol for 30 min and washed two times in TBS-Tween.
Electron microscopy.
Thirty-five-millimeter dishes of cells were fixed with 2.5% glutaraldehyde and 1% osmium tetroxide. Fixed cells were harvested and pelleted through 1% SeaPlaque agarose (Flowgen). The cell pellets were dehydrated through a graded alcohol series and embedded in Epon 812 resin. Thin sections were cut and examined in a JEOL 1200 EX II electron microscope.
RESULTS
Characterization of UL36 and UL37 mutants.
The virus mutants studied in the following experiments were made using a variety of techniques, were generated from different parental strains, and include both complete (FRΔUL37 and ARΔUL36 [strain 17]) and partial (K5ΔZ and KΔUL36 [strain KOS]) gene deletions. Therefore, to compare their phenotypes, the growth characteristics and particle assembly pathways of the mutants were examined. Titration of virus stocks showed that the WT HSV-1 and the rescuants FRΔUL37R and ARΔUL36R grew equally well on complementing and noncomplementing cells and confirmed that all mutations were lethal for virus growth, with reductions in titer of >105 in each case (Table 1). Since each of the complementing cell lines was generated to contain only the relevant HSV-1 ORF, their ability to complement growth of the mutant viruses implies that no secondary mutations were contributing to the growth defects. This was confirmed by single-step growth analysis on complementing cells, which showed that all the mutant viruses exhibited similar growth patterns and grew to titers equivalent to that of wild-type HSV-1 (Fig. 1).
TABLE 1.
Growth of mutant viruses on complementing and noncomplementing cells
| Virus | Growth of virus (titer) on:
|
|
|---|---|---|
| Noncomplementing cells | Complementing cells | |
| WT HSV-1 | 1.8 × 1010 | 1.2 × 1010 |
| K5ΔZ | <104a | 1.6 × 109 |
| FRΔUL37 | <104a | 1.9 × 109 |
| FRΔUL37R | 6.2 × 1010 | 9 × 1010 |
| KΔUL36 | <104a | 3.1 × 109 |
| ARΔUL36 | <104a | 2.0 × 109 |
| ARΔUL36R | 2.5 × 1010 | 2.4 × 1010 |
The input virus caused severe cytopathic effects at lower dilutions.
FIG. 1.

Single-step virus growth. Replicate 35-mm dishes of complementing RS cells were infected with 10 PFU/cell of HSV-1 strain 17+ (WT), K5ΔZ, FRΔUL37, KΔUL36, or ARΔUL36 mutant virus. After 1 h at 37°C, the cells were washed at low pH to remove residual input infectivity and overlaid with 2 ml of DMEM, and incubation was continued at 37°C. Wild-type HSV-1 was grown on RS cells in an identical fashion as a control. At 3, 6, 12, and 24 h after infection, the cells were harvested by scraping into the supernatant medium, and the progeny virus was titrated on complementing cells.
To determine the effects of the deletions on virus assembly, infected cells were examined by electron microscopy (Fig. 2). No enveloped virions were seen either on the cell surface or in cytoplasmic vacuoles of cells infected with any of the UL36 and UL37 mutants, while large numbers of nonenveloped C-capsids were present in the cytoplasm, confirming that these viruses are defective in envelopment and virus release. In FRΔUL37-infected cells, the cytoplasmic C-capsids congregated in large aggregates similar to those seen with the independently derived mutant KΔUL37 (13) and their PrV counterpart, PrV-ΔUL37 (26). The tendency of these capsids to aggregate is indicative of the presence of tegument proteins, which are known to cause clumping of de-enveloped virions (35). However, they lack the densely staining tegument found associated with cytoplasmic PrV capsids when secondary envelopment was blocked by mutations in glycoproteins gE/gI and gM (4). Similar aggregations of cytoplasmic C-capsids were present in the KΔUL36-infected cells. In contrast, no aggregates were seen in ARΔUL36-infected cells with individual C-capsids being distributed throughout the cytoplasm, reproducing the pattern seen with the PrV mutant, PrV-ΔUL36F (17). The distribution of capsids in infected cells was quantified by counting the numbers present in electron microscopic images of randomly selected cell sections (Table 2). This confirmed that C-capsids accumulated in the cytoplasm of FRΔUL37-, KΔUL36-, and ARΔUL36-infected cells, with the majority of the FRΔUL37 and KΔUL36 C-capsids occurring in aggregates.
FIG. 2.

Cytoplasmic capsids in infected cells. Replicate monolayers of HFFF2 cells were infected with 5 PFU/cell of HSV-1 strain 17+ (WT) or K5ΔZ, FRΔUL37, KΔUL36, or ARΔUL36 mutant virus. Cells were fixed and prepared for electron microscopy at 24 h postinfection. Both free capsids (black arrowheads) and enveloped virions (white arrowheads) were present in the cytoplasm of WT HSV-1-infected cells. In addition, large numbers of virions were present on the cell surfaces (not shown). KΔUL36 and FRΔUL37 cytoplasmic capsids accumulated in aggregates, while ARΔUL36 capsids were dispersed individually throughout the cytoplasm. No enveloped virions were seen with any of the mutant viruses. Nuclear (nuc) and cytoplasmic (cyt) compartments are labeled. Bar = 1 μm.
TABLE 2.
Nuclear and cytoplasmic distribution of capsids in WT and mutant HSV-1-infected cells
| Virus and capsid type | Nuclear
|
Cytoplasmic
|
|||
|---|---|---|---|---|---|
| No. of capids | % of total nuclear capsids | No. of free capids | No. of aggregated capsids | % of total DNA-containing capsids | |
| WT HSV-1 | |||||
| A | 154 | 17 | 2 | ||
| B | 691 | 76 | 2 | ||
| C | 63 | 7 | 40 | ||
| Virionsa | 217 | ||||
| FRΔUL37 | |||||
| A | 227 | 13 | 2 | 3 | |
| B | 1,380 | 77 | 4 | 21 | |
| C | 177 | 10 | 39 | 425 | 72 |
| KΔUL36 | |||||
| A | 107 | 14 | 17 | 1 | |
| B | 586 | 77 | 10 | 2 | |
| C | 71 | 9 | 149 | 324 | 87 |
| ARΔUL36 | |||||
| A | 161 | 19 | 13 | ||
| B | 544 | 66 | 68 | ||
| C | 123 | 15 | 276 | 69 | |
Includes enveloped cytoplasmic and extracellular capsids.
Sucrose gradient sedimentation was performed to analyze the properties of the mutant capsids. When nuclear extracts were sedimented through sucrose gradients, three bands corresponding to A-, B- and C-capsids were visible in all samples apart from K5ΔZ, which is deleted for the major capsid protein gene (12) and fails to assemble capsids (see Fig. S2A in the supplemental material). When cytoplasmic extracts from the same cells were analyzed, capsid bands were again present in KΔUL36 and ARΔUL36 gradients, but none were seen for WT HSV-1 or FRΔUL37 mutant virus (see Fig. S2B in the supplemental material). To determine the fate of the large number of cytoplasmic capsids present in FRΔUL37-infected cells, nuclear and cytoplasmic fractions from PBS-IGEPAL-treated cells were embedded, sectioned, and examined by electron microscopy (not shown). Very few capsids were found in the cytoplasmic fraction, while large numbers were present in the nuclear fraction, where the expected pattern of A-, B-, and C-capsids was observed inside nuclei. In addition, clusters of C-capsids were present outside the nuclear membrane, indicating that the large aggregations of cytoplasmic capsids seen in Fig. 2 had copelleted with the nuclei.
The differing distributions (aggregated and dispersed, respectively) of KΔUL36 and ARΔUL36 cytoplasmic C-capsids seen in Fig. 2, may reflect differences in the nature of their mutations. Thus, in ARΔUL36, the entire UL36 ORF has been deleted, whereas in KΔUL36 only the central portion has been removed (14), leaving 361 codons at the N terminus (plus a further 42 codons arising from a frameshift) that could encode a truncated form of pUL36. To determine whether this truncated form was expressed, the protein profiles of cells infected with WT HSV-1 or with FRΔUL37, KΔUL36, or ARΔUL36 mutant virus were probed with the pUL36-specific antibody, E12-E3. Western blotting confirmed that pUL36 bands of the expected sizes were present in the WT HSV-1- and FRΔUL37-infected cells, but not in the KΔUL36- or ARΔUL36-infected cells (Fig. 3A). However, the KΔUL36 sample did contain a strong band of the size expected for the residual N-terminal portion of pUL36 present in this mutant (predicted size of 43 kDa). Capsids were prepared from the cytoplasm of KΔUL36-infected cells and purified on sucrose gradients. Western blotting of gradient fractions showed that the 43-kDa protein fragment cosedimented with the capsid bands, confirming its association with KΔUL36 capsids (Fig. 3B). No signal was detected when these blots were probed with antibodies against pUL37 or the outer tegument protein, pUL48 (not shown).
FIG. 3.
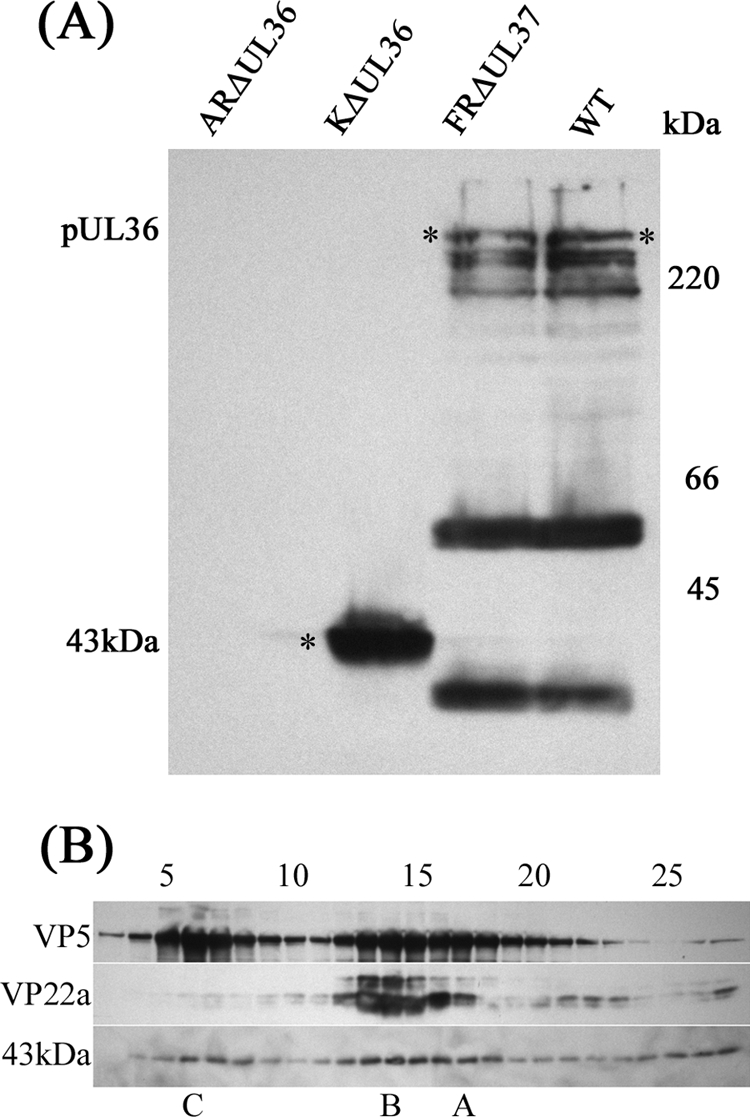
KΔUL36 protein expression. (A) BHK cells were harvested 24 h after infection with 5 PFU/cell of HSV-1 strain 17+ (WT) or FRΔUL37, KΔUL36, or ARΔUL36 mutant virus. Proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with MAb E12-E3 directed against pUL36. The positions of full-length pUL36 and the 43-kDa N-terminal fragment (*) are indicated to the left of the gel and in the gel, and the positions of the protein size standards (in kilodaltons) are shown to the right of the gel. (B) Cytoplasmic capsids from KΔUL36-infected cells were separated by sucrose gradient sedimentation (see Fig. S2 in the supplemental material). The gradients were collected from the bottom in 30 equal fractions. Gradient fractions 3 to 27 were resolved by SDS-PAGE and analyzed by Western blotting. Blots were probed sequentially with MAb E12-E3 (anti-pUL36), DM165 (anti-VP5; to show the distribution of all capsid types), and MCA406 (anti-VP22a scaffolding protein; to show the location of B-capsids). The positions at which A-, B-, and C-capsids migrated are indicated below the blots. The positions of gradient fractions 5, 10, 15, 20, and 25 are shown above the blots.
Roles of UL36 and UL37 in the initial stages of virus infection.
Since pUL36 and pUL37 are required for assembly of infectious virions, it is not possible to purify virions lacking these proteins in order to examine their behavior during the initial stages of infection. To overcome this limitation, we required a system in which virus could spread without needing to exit from and reenter cells. This is achieved naturally by syncytial viruses, in which the fusion of neighboring cell membranes exposes naïve cells to the contents of an infected cell's cytoplasm. To mimic this situation, we carried out studies in which cells, after infection, were induced to form syncytia by exposure to polyethylene glycol. Typical syncytia under these circumstances contained less than 20 nuclei. Therefore, infection at 0.01 PFU/cell ensured that few syncytia contained more than one initially infected cell. Following infection with the UL36 and UL37 mutants, the nuclear stages of assembly take place as normal, and capsids containing intact viral genomes are released into the cytoplasm. These progeny capsids have access to the other nuclei within the syncytium, and their ability to “infect” them can be determined by looking for replicating viral DNA.
Experiments in BHK and HFFF2 cells gave similar results, and the HFFF2 data are shown here as they produced more clearly delineated syncytia. To examine the spread of infection, monolayers of HFFF2 cells were infected with HSV-1 strain 17+ (WT) or with K5ΔZ, ARΔUL36, KΔUL36, or FRΔUL37 mutant virus (Fig. 4). WT HSV-1 was used as a positive control for spread within syncytia, and K5ΔZ was used as a negative control. After 1 h, the virus inoculum was removed, and the cells were either treated with PEG1500 (Roche) to induce fusion or left untreated. The cells were incubated for a further 23 h to allow time for infection to spread, then they were fixed, and the distribution of viral DNA was analyzed by fluorescent in situ hybridization. Infected nuclei were identified by the presence of fluorescent areas indicating replicating viral DNA. The addition of phosphonoacetic acid (an inhibitor of HSV-1 DNA polymerase) abolished labeling, confirming that the FISH probe was specifically recognizing newly synthesized virus DNA (not shown). Cells were counterstained with DAPI and CellMask deep red to highlight nuclei and cytoplasm, respectively.
FIG. 4.

Spread of virus infection. Replicate monolayers of HFFF2 cells were infected with 0.01 PFU/cell of HSV-1 strain 17+ (WT) or K5ΔZ, FRΔUL37, KΔUL36, or ARΔUL36 mutant virus. (A) Unfused cells; (B) cells after treatment with PEG and dimethyl sulfoxide at 1 h postinfection to induce syncytium formation. The cells were fixed and labeled at 24 h postinfection. Viral DNA was visualized by FISH using Cy3-labeled probe (red), nuclei were stained with DAPI (blue), and cell cytoplasm was stained with CellMask deep red (yellow). Bars, 50 μm in all panels.
Infection of unfused cells with WT virus gave rise to patches of cells with fluorescent nuclei, confirming that the virus was able to spread from an initially infected cell (Fig. 4A). As expected for a spreading infection, the pattern of labeling differed among individual cells, in some cases involving the entire nucleus and in others only localized patches. In contrast, infection of unfused cells with K5ΔZ, KΔUL36, ARΔUL36, or FRΔUL37 mutant virus resulted in only isolated fluorescing cells; the lack of spread to adjacent cells reflecting the inability of these mutants to produce infectious virus particles.
In PEG-treated cells, WT virus had also spread between nuclei, generating labeling patterns within syncytia similar to those seen in unfused cells (Fig. 4B). In contrast, the mutant viruses exhibited a range of labeling patterns. As would be predicted, in K5ΔZ-infected samples, viral DNA was confined to individual nuclei within syncytia, showing that unpackaged viral DNA could not spread between nuclei. In contrast, the FRΔUL37-infected samples contained multiple labeled nuclei. This demonstrates that formation of intact virions is not necessary for the spread of infection within syncytia but that this process can be mediated directly by cytoplasmic C-capsids. In both KΔUL36- and ARΔUL36-infected samples, replicating viral DNA was again confined to individual nuclei, indicating that not all cytoplasmic C-capsids are equally capable of transferring infection between nuclei and suggesting that, unlike pUL37, pUL36 plays an essential role in initiation of infection.
To determine the presence of cytoplasmic labeling, the experiment was repeated but without using CellMask deep red (Fig. 5). Cytoplasmic labeling clearly denoted the positions of extranuclear C-capsids or virions. Thus, fluorescent spots indicative of C-capsids were present in the cytoplasm of the WT HSV-1-, KΔUL36-, ARΔUL36-, and FRΔUL37-infected cells, but none were seen in cells infected with the major capsid protein mutant, K5ΔZ. Furthermore, the cytoplasmic fluorescence patterns confirmed the capsid distributions seen by electron microscopy. Thus, in FRΔUL37-infected cells, much of the labeling was in large irregular patches corresponding to the capsid aggregates shown in Fig. 2, although widely dispersed individual capsids were also evident. A similar pattern was seen in KΔUL36-infected cells, although the labeled patches were generally smaller than in FRΔUL37-infected cells. However, in ARΔUL36-infected cells, all cytoplasmic labeling was in uniformly distributed, discrete spots. That these patterns of cytoplasmic DNA labeling accurately reflected the distribution of cytoplasmic capsids was confirmed by immunofluorescent labeling using an antibody against the major capsid protein (see Fig. S3 in the supplemental material). The failure of KΔUL36 and ARΔUL36 to “infect” naïve nuclei within syncytia despite the evident ability of their capsids to spread throughout the cytoplasm suggests that the defect is not a result of reduced capsid mobility, while the divergent behaviors of the ΔUL36 and ΔUL37 mutants imply different roles for these two inner tegument proteins in capsid transport and DNA release during virus entry.
FIG. 5.

Capsid distribution in infected syncytia. Replicate monolayers of HFFF2 cells were infected and induced to form syncytia as described in the legend to Fig. 4. Viral DNA was visualized by FISH using Cy3-labeled probe (red), and nuclei were stained with DAPI (blue). Bars, 50 μm in all panels.
Role of microtubules in the spread of infection.
To examine the role of microtubules in the spread of virus, cells were treated with nocodazole to depolymerize the microtubule network. Nocodazole was added 3 h after infection, and cells were incubated at 37°C for a further 21 h before being fixed and processed for FISH as before. Disruption of the microtubule network was confirmed by immunofluorescence (Fig. 6).
FIG. 6.

Effect of nocodazole on spread of virus infection within syncytia. Replicate monolayers of HFFF2 cells were infected with HSV-1 strain 17+ (WT) or FRΔUL37 mutant virus and induced to form syncytia as described in the legend to Fig. 4. Nocodazole (0.5 μg/ml) was added at 3 h postinfection, and incubation was continued for a further 21 h. For immunofluorescence, cells were fixed (54), and microtubule proteins were identified with MAb DM1A and Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (green). Viral DNA was visualized by FISH using Cy3-labeled probe (red). Nuclei were stained with DAPI (blue) The top panels show that the fibrillar microtubule network seen in uninfected syncytia [no nocodazole (−noc)] is disrupted by the addition of nocodazole (+noc). The bottom panels show the distribution of viral DNA in nocodazole-treated, WT HSV-1- and FRΔUL37-infected syncytia. Bars, 50 μm in all panels.
Nocodazole treatment had no obvious effect on the overall distribution of cytoplasmic capsids with the patterns of spread and aggregation in wild-type and mutant virus-infected syncytia resembling those seen in Fig. 4 and 5. Furthermore, nocodazole treatment of WT HSV-1-infected syncytia did not prevent the spread of infection but reduced its extent, with approximately half the number of labeled nuclei observed in nocodazole-treated syncytia (Fig. 6 and Table 3). This agrees with previous studies on unfused cells, which showed that disruption of microtubules delayed but did not prevent infection (49). A similar result was obtained with FRΔUL37, with nocodazole inhibiting but not abolishing spread within syncytia (Fig. 6 and Table 3). Unsurprisingly, nocodazole treatment had no effect on ARΔUL36 and KΔUL36 infections, which remained confined to individual nuclei (not shown). The disruption of the microtubule network did have any obvious effect on the gross distribution of capsids in infected syncytia, suggesting that extensive movement through the cytoplasm could occur by alternative mechanisms.
TABLE 3.
Percentage of labeled nuclei in WT and mutant HSV-1-infected syncytiaa
| Virus | % of labeled nuclei
|
|
|---|---|---|
| Not treated with nocodazole | Treated with nocodazole | |
| WT HSV-1 | 98 | 46 |
| FRΔUL37 | 76 | 32 |
| ARΔUL36 | 10 | 13 |
Only syncytia with at least one labeled nucleus were counted.
Incorporation of pUL36 and pUL37 into L-particles.
pUL36 and pUL37 are components of virions and L-particles, and it has been shown for PrV that neither protein is required for L-particle assembly. To analyze tegument assembly in these virus mutants, particles released into infected-cell culture medium were collected and purified by Ficoll gradient sedimentation. All of the mutants gave similar sedimentation profiles with a broad light-scattering band in the position expected for L-particles and a complete absence of the prominent virion band found in WT HSV-1 (see Fig. S4 in the supplemental material). Gradient fractions were resolved by SDS-PAGE, and the distribution of virions and L-particles was determined using an antibody (AGV031) against the abundant tegument protein pUL49 (VP22; Fig. 7A). Interestingly, this appears to show that L-particles produced by the UL36 and UL37 mutants extended higher up the gradients than those of WT or K5ΔZ. The presence of pUL36 (Fig. 7B) and pUL37 (Fig. 7C) was determined by Western blotting with antibodies E12-E3 and M780, respectively. As expected, full-length pUL36 was detected in WT virions and L-particles and in K5ΔZ L-particles, but not in ARΔUL36 or KΔUL36 L-particles. However, the 43-kDa N-terminal fragment (Fig. 7B) of KΔUL36 had a sedimentation profile identical to that of pUL49, indicating that it had been incorporated into L-particles. Interestingly, only traces of pUL36 were detected in FRΔUL37 L-particles despite being present in normal amounts in the infected cells (Fig. 3A). Similarly, pUL37 was also present in WT HSV-1 and K5ΔZ particles and absent from FRΔUL37 L-particles (Fig. 7C). However, it was not detected in either ARΔUL36 or KΔUL36 L-particles, providing further evidence that pUL36 and pUL37 are dependent on one another for incorporation into tegument.
FIG. 7.
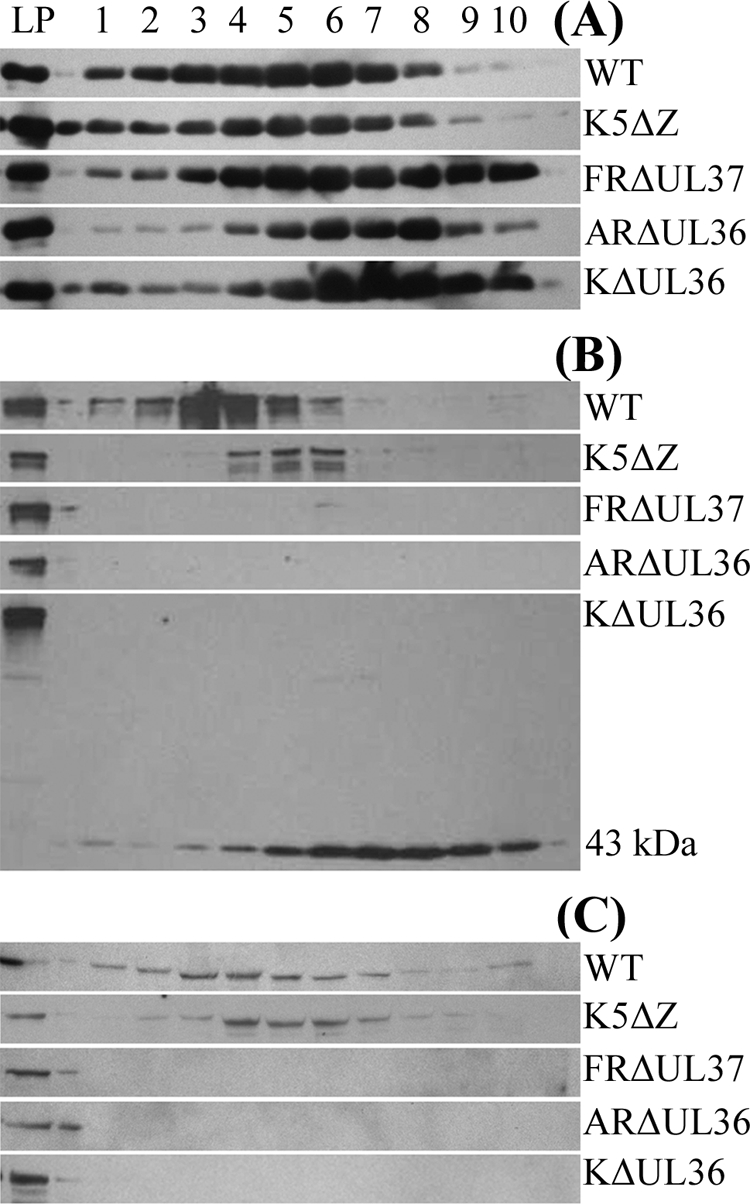
Protein content of virions and L-particles. Extracellular virions and L-particles from cells infected with HSV-1 strain 17+ (WT) and L-particles from cells infected with mutant virus K5ΔZ, FRΔUL37, KΔUL36, or ARΔUL36 were separated by Ficoll gradient sedimentation (see Fig. S4 in the supplemental material). The gradients were collected from the bottom in 10 equal fractions. A control sample of HSV-1 strain 17+ L-particles (LP) was run on each gel. Gradient fractions were resolved by SDS-PAGE and analyzed by Western blotting. Blots were probed sequentially with E12-E3 (anti-pUL36) (B), M780 (anti-pUL37) (C), and AGV031 (anti-pUL49) (A). The position of the 43-kDa N-terminal fragment of pUL36 is indicated in panel B.
DISCUSSION
Previous studies to analyze the properties of HSV-1 and PrV mutants with deletions in the UL36 and UL37 genes have described the effects of the mutations on virion assembly and egress. Thus, UL36 and UL37 in HSV-1 (13, 14) and UL36 in PrV (17, 30) are all essential for assembly of infectious virions, while studies in PrV showed that loss of UL37 reduced but did not abolish virus replication (26, 30). The results presented here using independently derived HSV-1 mutants support those of Desai et al. (13, 14) and confirm the phenotypic divergence between the HSV-1 and PrV UL37 mutants. A common feature of all the UL36 and UL37 deletion mutants described so far is that they produce large numbers of nonenveloped C-capsids when they infect noncomplementing cells. However, there are differing accounts of their effects on the nuclear and cytoplasmic distributions of these capsids. In UL37-minus mutants of both HSV-1 and PrV, the C-capsids form large aggregates in the cytoplasm (13, 26) (Fig. 2 and 5). Desai et al. (13) reported that their HSV-1 mutant, KΔUL37, also accumulated large numbers of capsids around the inner surface of the nuclear membrane, which they interpreted as indicating a block on nuclear exit. We did not see intranuclear accumulations with our independently derived HSV-1 mutant, FRΔUL37, although the total number of nuclear capsids was higher than found with WT HSV-1 or either UL36 mutant (Table 2). Furthermore, the ratio of A-, B-, and C-capsids in the nuclei suggested no specific accumulation of C-capsids. To quantify the effect of their mutation on capsid exit from the nucleus, Desai et al. (13) carried out gradient analysis of lysates prepared from nuclear and cytoplasmic fractions of KΔUL37-infected cells which showed that 75% of the DNA-containing capsids were associated with the nuclear fraction. This contrasted with data from our electron micrographs, which showed that the majority (72%) of the DNA-containing capsids were in the cytoplasm (Table 2). In an attempt to resolve this apparent contradiction, we repeated the separation of FRΔUL37-infected cells into nuclear and cytoplasmic fractions and analyzed their capsid content by electron microscopy. The electron microscopy images revealed that most C-capsids were indeed present in the nuclear pellet but were located outside nuclei. This suggests that the preferential association of C-capsids in the nuclear fraction is misleading and results from the large cytoplasmic capsid aggregates, which are a feature of the UL37 deletion mutants, copelleting with the nuclei. Inhibition of nuclear exit has also been reported for a UL37 mutant of PrV (30), although studies on a second PrV mutant (17, 26) produced no evidence of retention of capsids in the nucleus. The reasons for the differing behaviors of these mutations are unknown, but our studies and those of Klupp et al. (26) suggest that the major effect of deleting UL37 is to block secondary envelopment in the cytoplasm and that any role of pUL37 in nuclear exit is minor.
Differing behaviors have also been described for the UL36 mutants. Luxton et al. (30) reported that deletion of UL36 from PrV caused a dramatic reduction in capsid egress from nuclei. They suggested that the absence of such an effect in the HSV-1 mutant KΔUL36 could be due to the incomplete nature of its deletion. However, as shown here, removal of the entire UL36 ORF did not prevent efficient egress of capsids from the nucleus (Fig. 2 and 5; Table 2), nor was this effect observed with a different UL36 mutant of PrV (17). This phenotype therefore is not typical of other UL36 mutants. Another difference in the behavior of UL36 mutants is in their cytoplasmic distributions. Thus, KΔUL36 showed a tendency for capsids to accumulate in cytoplasmic clusters (14) in contrast to the PrV mutant, PrV-ΔUL36F, where capsids were dispersed evenly throughout the cytoplasm (17). However, the results described here for KΔUL36 and our independent HSV-1 mutant, ARΔUL36, show that this apparent difference does not reflect any biological distinction between HSV-1 and PrV but is most likely a consequence of the incomplete deletion in KΔUL36. Western blotting (Fig. 3) confirmed that the 43-kDa N-terminal truncation product of UL36 copurified with cytoplasmic KΔUL36 capsids, although they did not contain either pUL37 or pUL48. Therefore, it appears that this fragment is sufficient to cause their aggregation. The “stickiness” of UL36 has previously been suggested to account for the formation of large capsid aggregates by the UL37-minus mutants (17). Interestingly, the association of the 43-kDa fragment with capsids implies that capsid binding sequences map in this N-terminal region. Since a binding site for the minor capsid protein, pUL25, has recently been mapped to the C-terminal 62 residues of pUL36 (8), it appears that both N- and C-terminal regions of pUL36 interact with capsids.
Perhaps surprisingly in view of their close association with capsids, both pUL36 and pUL37 are found in L-particles, which lack capsids and consist solely of tegument and envelope. It is likely that this reflects the probable function of these proteins as an interface between the capsid and outer tegument and that interaction with one or more of the outer tegument proteins accounts for their presence in L-particles. Indeed, L-particles from cells infected with a PrV, UL48-minus mutant did not contain either pUL36 or pUL37 (17). In view of this, it is interesting that pUL36 and pUL37 were dependent on one another for incorporation into L-particles (Fig. 7), suggesting that they enter L-particles as a complex. If these two proteins must form a complex before interacting with the outer tegument proteins, it would explain why tegument-like material is not detected around pUL36- or pUL37-minus cytoplasmic capsids (Fig. 2) (26). The occurrence of the 43-kDa N-terminal fragment of pUL36 in KΔUL36 L-particles is intriguing. This fragment does not include the region homologous to the pUL37 binding site in PrV (17), although it does overlap with a pUL48 interaction domain in HSV-1 (37). Its presence demonstrates that, unlike the full-length protein, this region of pUL36 can be incorporated into L-particles in the absence of pUL37.
Although the involvement of UL36 and UL37 in virus egress has been well documented, their role in virus entry is less easy to analyze. Studies with an HSV-1 temperature-sensitive mutant of UL36 (tsB7), which showed that capsids migrated to the vicinity of the nuclear pores but failed to release their DNA into the nucleus, provided the first indication that pUL36 functioned during the initial stages of infection (3). This is supported by recent work which showed that protease inhibitors prevented infection (10, 24) and linked their action to inhibition of cleavage of pUL36 on incoming capsids (24). Because UL36 deletion mutants do not form virions, they have not previously been used to study the role of pUL36 in entry. To circumvent this problem, we examined the behavior of the UL36 deletion mutants in artificially induced syncytia, since in the absence of plasma membranes separating neighboring cells, the virus capsids leaving an infected nucleus can gain direct access to uninfected nuclei without needing to undergo envelopment. Under these conditions, the environment of the capsids present in the cytoplasm of these syncytia was broadly equivalent to that of capsids from infecting virions that had undergone a normal fusion event. The failure of UL36 deletion mutants KΔUL36 and ARΔUL36 to infect new nuclei within syncytia despite having ready access to them confirms that pUL36 is involved in the initiation of infection and further strongly suggests that it must be present on the capsids. Therefore, the inhibition of infection seen with tsB7 or in the presence of protease inhibitors is not due to any failure to remove pUL36 from particles but reflects a positive requirement for pUL36 to ensure correct capsid transport and/or DNA release.
As with UL36, UL37 deletion mutants of HSV-1 fail to produce virions, and although UL37 mutants of PrV are viable, they are severely disabled, and it has not been possible to determine whether the small number of virions they produce have reduced specific infectivity (26, 30). Since no temperature-sensitive mutants in UL37 are available, essentially nothing was known of the role of this protein in virus entry. The observation that cytoplasmic capsids produced by FRΔUL37 were able to infect new nuclei clearly showed that pUL37 does not play an essential role in the initiation of infection and that its primary function is in the virion envelopment/release pathway, where it appears to act together with pUL36 as a template for assembly of outer tegument proteins.
The spread of infection within a syncytium will depend on the ability of the mutant virus particles to reach nuclear pores, to interact appropriately with them, and to release the virus genome into the nucleus. The retention of pUL36 and pUL37 on capsids during transport to the nucleus (29) would allow them to function at any or all of these stages. There is considerable evidence both from studies using an in vitro microtubule model (57) and from fluorescence microscopy of live cells (30) that the inner tegument proteins are important for capsid motility. In particular, studies of cells infected with PrV mutants showed that during virus egress pUL36 was absolutely required for processive capsid transport along microtubules, while pUL37 increased its efficiency (30). Furthermore, it was known that intact microtubules were needed to direct incoming capsids to the nuclear membrane (49), raising the possibility that failure of capsids to reach the nuclear pore could explain the block on infection by the UL36 deletion mutants. However, disrupting microtubules with nocodazole did not completely block HSV-1 infection of individual cells (49) and although the rate of spread was decreased, both WT HSV-1 and FRΔUL37 were able to infect naïve nuclei in nocodazole-treated syncytia. In addition, the widespread distribution of ARΔUL36 and KΔUL36 capsids throughout infected syncytia suggests that the uninfected nuclei were not physically inaccessible to them. Therefore, it is highly unlikely that the restriction of infection seen with the UL36 mutants is due solely to inefficient transport, and an additional role in nuclear pore recognition and/or DNA release seems probable.
In conclusion, we have shown that two inner tegument proteins that remain on capsids during the initial stages of infection have differing roles. One, pUL37, is dispensable during the initial stages of virus infection, while the second, pUL36, plays an essential, but as yet undefined, role in DNA release from the capsid and entry into the nucleus. The syncytial infection model should allow further investigation of the role of UL36 in the initial stages of infection.
Supplementary Material
Acknowledgments
The HSV-1 bacterial artificial chromosome, pBAC SR27, was provided by C. Cunningham and A. J. Davison. Antibodies M780 and AGV031 were provided by F. Jenkins, (University of Pittsburgh) and G. Elliott (Imperial College), respectively. We thank M. McElwee for excellent technical support and V. Preston and D. McGeoch for critically reading the manuscript.
Footnotes
Published ahead of print on 29 October 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Abaitua, F., and P. O'Hare. 2008. Identification of a highly conserved functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1-2 tegument protein. J. Virol. 825234-5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baines, J. D., and B. Roizman. 1991. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 65938-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batterson, W., D. Furlong, and B. Roizman. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 45397-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brack, A. R., J. M. Dijkstra, H. Granzow, B. G. Klupp, and T. C. Mettenleiter. 1999. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J. Virol. 735364-5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bucks, M. A., K. J. O'Regan, M. A. Murphy, J. W. Wills, and R. J. Courtney. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361316-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, D. H., H. Jiang, M. Lee, F. Y. Liu, and Z. H. Zhou. 1999. Three-dimensional visualization of tegument/capsid interactions in the intact human cytomegalovirus. Virology 26010-16. [DOI] [PubMed] [Google Scholar]
- 7.Clarke, R. W., N. Monnier, H. T. Li, D. J. Zhou, H. Browne, and D. Klenerman. 2007. Two-color fluorescence analysis of individual virions determines the distribution of the copy number of proteins in herpes simplex virus particles. Biophys. J. 931329-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coller, K. E., J. I.-H. Lee, A. Ueda, and G. A. Smith. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 8111790-11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham, C., and A. J. Davison. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197116-124. [DOI] [PubMed] [Google Scholar]
- 10.Delboy, M. G., D. G. Roller, and A. V. Nicola. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 823381-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.del Rio, T., T. H. Ch'ng, E. A. Flood, S. P. Gross, and L. W. Enquist. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 793903-3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai, P., N. A. Deluca, J. C. Glorioso, and S. Person. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J. Virol. 671357-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai, P., G. L. Sexton, J. M. McCaffery, and S. Person. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 7510259-10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desai, P. J. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 7411608-11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott, G., D. O'Reilly, and P. O'Hare. 1996. Phosphorylation of the herpes simplex virus type-I tegument protein VP22. Virology 226140-145. [DOI] [PubMed] [Google Scholar]
- 16.Everett, R. D., J. Murray, A. Orr, and C. M. Preston. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 8110991-11004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs, W., B. G. Klupp, H. Granzow, and T. C. Mettenleiter. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 7811879-11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs, W., B. G. Klupp, H. Granzow, N. Osterrieder, and T. C. Mettenleiter. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76364-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson, W., and B. Roizman. 1972. Proteins specified by herpes simplex virus. VIII. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 101044-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 793200-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2004. The pseudorabies virus US3 protein is a component of primary and of mature virions. J. Virol. 781314-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grunewald, K., P. Desai, D. C. Winkler, J. B. Heymann, D. M. Belnap, W. Baumeister, and A. C. Steven. 2003. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 3021396-1398. [DOI] [PubMed] [Google Scholar]
- 23.Homa, F. L., and J. C. Brown. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 7107-122. [DOI] [PubMed] [Google Scholar]
- 24.Jovasevic, V., L. Liang, and B. Roizman. 2008. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 823311-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klupp, B. G., W. Fuchs, H. Granzow, R. Nixdorf, and T. C. Mettenleiter. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 763065-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klupp, B. G., H. Granzow, E. Mundt, and T. C. Mettenleiter. 2001. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 758927-8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamberti, C., and S. K. Weller. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 722463-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis, L., and G. Albrechtbuehler. 1987. Distribution of multiple centrospheres determines migration of BHK syncytia. Cell Motil. Cytoskelet. 7282-290. [DOI] [PubMed] [Google Scholar]
- 29.Luxton, G. W. G., S. Haverlock, K. E. Coller, S. E. Antinone, A. Pincetic, and G. A. Smith. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 1025832-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luxton, G. W. G., J. I. H. Lee, S. Haverlock-Moyns, J. M. Schober, and G. A. Smith. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80201-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McClelland, D. A., J. D. Aitken, D. Bhella, D. McNab, J. Mitchell, S. M. Kelly, N. C. Price, and F. J. Rixon. 2002. pH reduction as a trigger for dissociation of herpes simplex virus type 1 scaffolds. J. Virol. 767407-7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 691531-1574. [DOI] [PubMed] [Google Scholar]
- 33.McLauchlan, J. 1997. The abundance of the herpes simplex virus type 1 UL37 tegument protein in virus particles is closely controlled. J. Gen. Virol. 78189-194. [DOI] [PubMed] [Google Scholar]
- 34.McLauchlan, J., K. Liefkens, and N. D. Stow. 1994. The herpes simplex virus type 1 UL37 gene-product is a component of virus particles. J. Gen. Virol. 752047-2052. [DOI] [PubMed] [Google Scholar]
- 35.McLauchlan, J., and F. J. Rixon. 1992. Characterization of enveloped tegument structures (L-particles) produced by alphaherpesviruses: integrity of the tegument does not depend on the presence of capsid or envelope. J. Gen. Virol. 73269-276. [DOI] [PubMed] [Google Scholar]
- 36.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 761537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mijatov, B., A. L. Cunningham, and R. J. Diefenbach. 2007. Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 36826-31. [DOI] [PubMed] [Google Scholar]
- 38.Naldinho-Souto, R., H. Browne, and T. Minson. 2006. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J. Virol. 802582-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicholson, P., C. Addison, A. M. Cross, J. Kennard, V. G. Preston, and F. J. Rixon. 1994. Localization of the herpes simplex virus type 1 major capsid protein VP5 to the cell nucleus requires the abundant scaffolding protein VP22a. J. Gen. Virol. 751091-1099. [DOI] [PubMed] [Google Scholar]
- 40.Reynolds, A. E., B. J. Ryckman, J. D. Baines, Y. P. Zhou, L. Liang, and R. J. Roller. 2001. UL31 and UL34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 758803-8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 768939-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rixon, F. J. 1993. Structure and assembly of herpesviruses. Semin. Virol. 4135-144. [Google Scholar]
- 43.Rixon, F. J., C. Addison, and J. McLauchlan. 1992. Assembly of enveloped tegument structures (L-particles) can occur independently of virion maturation in herpes-simplex virus type 1-infected cells. J. Gen. Virol. 73277-284. [DOI] [PubMed] [Google Scholar]
- 44.Roller, R. J., Y. P. Zhou, R. Schnetzer, J. Ferguson, and D. DeSalvo. 2000. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 74117-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrag, J. D., B. V. V. Prasad, F. J. Rixon, and W. Chiu. 1989. Three-dimensional structure of the HSV-1 nucleocapsid. Cell 56651-660. [DOI] [PubMed] [Google Scholar]
- 46.Shelton, L. S., A. G. Albright, W. T. Ruyechan, and F. J. Jenkins. 1994. Retention of the herpes simplex virus type 1 (HSV-1) UL37 protein on single-stranded DNA columns requires the HSV-1 ICP8 protein. J. Virol. 68521-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siegel, R. W., R. Jain, and A. Bradbury. 2001. Using an in vivo phagemid system to identify non-compatible loxP sequences. FEBS Lett. 499147-153. [DOI] [PubMed] [Google Scholar]
- 48.Smith, G. A., and L. W. Enquist. 2000. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc. Natl. Acad. Sci. USA 974873-4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sodeik, B., M. W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1361007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steven, A. C., and P. G. Spear. 1997. Herpesvirus capsid assembly and envelopment, p. 312-351. In W. Chiu, R. M. Burnett, and R. Garcea (ed.), Structural biology of viruses. Oxford University Press, Oxford, United Kingdom.
- 51.Stow, N. D., and N. M. Wilkie. 1976. An improved technique for obtaining enhanced infectivity with herpes simplex virus type 1 DNA. J. Gen. Virol. 33447-458. [DOI] [PubMed] [Google Scholar]
- 52.Szilagyi, J. F., and C. Cunningham. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72661-668. [DOI] [PubMed] [Google Scholar]
- 53.Trus, B. L., W. W. Newcomb, N. Q. Cheng, G. Cardone, L. Marekov, F. L. Horna, J. C. Brown, and A. C. Steven. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vielkind, U., and S. H. Swierenga. 1989. A simple fixation procedure for immunofluorescent detection of different cytoskeletal components within the same cell. Histochemistry 9181-88. [DOI] [PubMed] [Google Scholar]
- 55.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 799566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wagenaar, F., J. M. A. Pol, B. Peeters, A. L. J. Gielkens, N. Dewind, and T. G. Kimman. 1995. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 761851-1859. [DOI] [PubMed] [Google Scholar]
- 57.Wolfstein, A., C. H. Nagel, K. Radtke, K. Dohner, V. J. Allan, and B. Sodeik. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7227-237. [DOI] [PubMed] [Google Scholar]
- 58.Zhou, Z. H., D. H. Chen, J. Jakana, F. J. Rixon, and W. Chiu. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 733210-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou, Z. H., M. Dougherty, J. Jakana, J. He, F. J. Rixon, and W. Chiu. 2000. Seeing the herpesvirus capsid at 8.5 Å. Science 288877-880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.