

Abstract
Vesicular stomatitis virus (VSV) matrix protein inhibits nuclear-cytoplasmic mRNA transport. The goal of this work is to determine whether VSV inhibits the nuclear-cytoplasmic transport of heterogeneous ribonucleoproteins (hnRNPs), which are thought to serve as mRNA export factors. Confocal microscopy experiments showed that hnRNPA1, hnRNPK, and hnRNPC1/C2, but not hnRNPB1 or lamin A/C, are relocalized to the cytoplasm during VSV infection. We determined whether protein import is inhibited by VSV by transfecting cells with a plasmid encoding enhanced green fluorescent protein (EGFP) tagged with either the M9 nuclear localization sequence (NLS) or the classical NLS. These experiments revealed that both the M9 NLS and the classical NLS are functional during VSV infection. These data suggest that the inhibition of protein import is not responsible for hnRNP relocalization during VSV infection but that hnRNP export is enhanced. We found that hnRNPA1 relocalization was significantly reduced following the silencing of the mRNA export factor Rae1, indicating that Rae1 is necessary for hnRNP export. In order to determine the role of hnRNPA1 in VSV infection, we silenced hnRNPA1 in HeLa cells and assayed three aspects of the viral life cycle: host protein synthesis shutoff concurrent with the onset of viral protein synthesis, replication by plaque assay, and cell killing. We observed that host shutoff and replication are unaffected by the reduction in hnRNPA1 but that the rate of VSV-induced apoptosis is slower in cells that have reduced hnRNPA1. These data suggest that VSV promotes hnRNPA1 relocalization in a Rae1-dependent manner for apoptotic signaling.
Several RNA viruses inhibit the nuclear-cytoplasmic trafficking of cellular RNA and proteins, despite the fact that viral replication occurs in the cytoplasm of the host cells. The disruption of nuclear-cytoplasmic trafficking by these viruses in the host cell may facilitate viral replication and help subvert the host antiviral response. For example, infection with some picornaviruses disrupts several protein import pathways and triggers the degradation of nuclear pore complex components (24, 25). This causes the accumulation of host nuclear proteins in the cytoplasm, where these proteins may aid in viral replication (23). Viral proteins encoded by henipaviruses and Ebola and severe acute respiratory syndrome viruses recruit host import factors or STAT1 directly to block STAT1 transport into the nucleus, resulting in the suppression of interferon signaling (21, 52-54).
Vesicular stomatitis virus (VSV) inhibits host gene expression at multiple steps, including transcription (2, 6, 63), translation (14, 15, 17), and mRNA export (19, 26, 61). The inhibition of host gene expression is due to the activity of the VSV matrix (M) protein (36). M protein also plays a major role in virus assembly. The function of M protein in virus assembly is genetically separable from its function in inhibiting host gene expression (7). M protein lacks any known enzymatic activity and may inhibit host gene expression by binding directly to host factors and inhibiting their function. Recently, it was shown that M protein binds host factor Rae1, which may lead to the inhibition of mRNA export in infected cells (19). Rae1 is involved in the regulation of mitotic spindle formation and the cell cycle (4, 9, 58, 66) and has been implicated in mRNA transport from the nucleus (8, 11, 41, 51). However, Rae1 is not essential for mRNA transport in higher eukaryotic cells (4, 58), leaving open the question of its role in VSV infection. It is unknown whether Rae1 plays a role in protein export or import.
Whether VSV infection induces defects in the nuclear-cytoplasmic trafficking of proteins is somewhat ambiguous in the literature. Some reports have shown that M protein inhibits the import of proteins and ribonucleoproteins (RNPs) into the nucleus (45), while others have shown that protein import was minimally affected by VSV infection (5, 19). These studies have primarily addressed the classical import pathway. Thus, it is unknown whether the M9 import pathway is altered by VSV infection or whether VSV affects the export of proteins from the nucleus to the cytoplasm. The goal of the experiments reported here was to determine whether VSV alters nucleocytoplasmic trafficking by analyzing cellular heterogeneous nuclear RNPs (hnRNPs) that are transported between the nucleus and the cytoplasm by several different pathways.
In human cells, hnRNPs are abundant nuclear proteins that have RNA binding abilities. A number of functions have been demonstrated for hnRNPs, including splicing, 3′ end processing and mRNA transport, and translation (31). Different hnRNPs have distinct signals which enable them to move between the nuclear and cytoplasmic compartments. hnRNPA1 contains the M9 nuclear localization sequence (NLS) and is imported by the M9/transportin pathway (20, 27, 49). hnRNPA1 rapidly shuttles between the nucleus and the cytoplasm (47) and is likely involved in mRNA export (27), although the receptor for hnRNPA1 export is unknown. Another hnRNP which is predominately localized to the nucleus, hnRNPC1/C2, utilizes a different pathway for its import and does not shuttle (42). hnRNPC1/C2 contains a classical NLS which binds importin α and allows the import of cargo proteins by the importin β receptor. A third hnRNP, hnRNPK, uses a pathway distinct from hnRNPA1 and hnRNPC1/C2 (39).
In this study, we determined whether VSV alters protein trafficking in the host cell by examining the localization of several cellular hnRNPs following VSV infection. We found that VSV infection results in the relocalization of hnRNPA1, hnRNPC1/C2, and hnRNPK, but not hnRNPB1 or lamin A/C, from the nucleus to the cytoplasm. The ability of hnRNPA1 to relocalize to the cytoplasm correlated with the ability of M protein to shut off host gene expression. However, the classical and M9 protein import pathways do not appear to be disrupted as a result of VSV infection. This argues either that these factors are actively retained in the cytoplasm or that nuclear export is increased in contrast to the reported effect on mRNA transport. The silencing of host factor Rae1 expression showed that Rae1 was required for the relocalization of hnRNPA1 during VSV infection. We hypothesized that the relocalization of hnRNPs is important in the virus life cycle and thus examined a possible role for hnRNPA1 by silencing its expression. While there was little if any difference in the viral gene expression or yield of progeny virus in cells containing decreased hnRNPA1 levels, these cells entered apoptosis more slowly following VSV infection than cells containing normal levels of hnRNPA1, indicating that the relocalization of hnRNPA1 likely plays a role in apoptotic signaling in VSV-infected cells.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 7% fetal bovine serum (FBS) and 2 mM glutamine. The cells were grown to 70 to 80% confluence before virus infection. Wild-type VSV derived from the Indiana serotype Orsay (wto) strain (64) was used to infect cells, as shown in Fig. 1. The recombinant viruses rwt and rM51R-M have been described previously (30).
FIG. 1.

Confocal microscopy analysis of cellular hnRNPs during VSV infection. HeLa cells were infected with the wto strain; then fixed, permeabilized, and processed for confocal microscopy using primary antibodies to VSV G protein and various nuclear factors; and then stained with TOPRO-iodide or DAPI as indicated to label the DNA in the nuclei. (A) hnRNPA1; (B) hnRNPC1/C2; (C) hnRNPK; (D) hnRNPB1; (E) lamin A/C. The cells shown in all panels except panel C were fixed 6 h postinfection; the cells shown in panel C were fixed 7 h postinfection.
Indirect immunofluorescence.
Cells were plated onto six-well plates containing sterile, poly(l)-lysine treated coverslips. Following infection or transfection, the cells were washed briefly with phosphate-buffered saline (PBS) at room temperature and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. The cells were then permeabilized with 0.2% Triton X-100 for 5 min and rinsed twice with Tris-buffered saline containing 0.5 mg/ml bovine serum albumin and 0.1 mg/ml glycine (1× TBS-BG). Antibodies for hnRNPA1 (sc32301; 1:500), hnRNPC1/C2 (sc32308; 1:750), hnRNP A2/B1 (sc 32316; 1:500), hnRNPK (sc 28380; 1:750) (all from Santa Cruz Biotechnology), and VSV G-tag (1:250 to 1:500) (11) were mixed in 1× TBS-BG and added to the cells for 2 h at room temperature. For lamin A/C, cells were permeabilized in methanol at −20°C for 5 min. Primary antibodies for lamin A/C (sc-7292; 1:100) were incubated in 1% fish skin gelatin with 0.05% Triton X-100 in PBS as described previously (25). Following three to five washes with 1× TBS-BG, the cells were incubated with secondary anti-mouse and anti-rabbit AlexaFluor 488-nm and 568-nm antibodies (Invitrogen Molecular Probes) for 2 h at room temperature. The cells were washed with 1× TBS-BG as above, and to detect DNA, the cells were incubated in 1× TBS-BG containing TOPRO-iodide at a dilution of 1:2,000 (Invitrogen) for 5 min at room temperature and then washed three times with 1× TBS-BG. Coverslips were then inverted onto glass slides containing mounting medium for analysis by microscopy. If DAPI (4′,6-diamidino-2-phenylindole) was used to detect the DNA, the mounting medium contained 10 μg/ml DAPI.
Microscopy.
For confocal microscopy, a Zeiss LSM 510 confocal laser scanning device fitted to an Axioplan 2 microscope was used. Images were taken with a 40× (hnRNPK) or 63× lens, using a 3× digital magnification. The Zeiss LSM510 software was used to determine the mean pixel intensity of hnRNPA1 in the cytoplasm and nucleus using the histogram function. The localization of hnRNPC1/C2 and hnRNPB1 (Fig. 1), as well as the enhanced green fluorescent protein (EGFP) (see Fig. 2 and 5) images were generated with the fluorescent microscope. Images were acquired as 8-bit TIFF files with a monochrome Retiga EX 1350 digital camera (QImaging Corp.) attached to a Nikon Eclipse TE300 inverted microscope. The resulting images were imported into Adobe Photoshop, and the individual images were pseudocolored in their respective RGB channels.
FIG. 2.

Fluorescence microscopy analysis of the import of EGFP containing M9 or classical NLS during VSV infection. HeLa cells were transiently transfected with EGFP plasmids containing various NLSs, and at 48 h posttransfection, the cells were infected with rwt virus for 6 h. Following infection, cells were fixed and labeled with DAPI to indicate the DNA (nucleus). (A) EGFP-M9 contains the wild type M9 NLS; (B) EGFP-M9 MT contains a mutation in the M9 NLS which abrogates the retention of the protein to the nucleus; (C) EGFP-TAG NLS contains the NLS from the simian virus 40 large T antigen; (D) EGFP lacking any NLS, which localizes to both the cytoplasm and the nucleus.
FIG. 5.

Immunofluorescence analysis of hnRNPA1 localization in Rae1-silenced cells treated with the transcriptional inhibitor actinomycin D. HeLa cells were transfected with siRNAs directed against a nontargeting siRNA (A) or Rae1 siRNA (B). At 72 h posttransfection, cells were mock treated or treated for 4 h with actinomycin D and processed for confocal microscopy using antibody against hnRNPA1 and TOPRO-iodide. (A) Confocal microscopy images of cells transfected with nontargeting siRNAs. (B) Images of Rae1-silenced cells.
siRNA transfections.
For Rae1, cells were transfected with small interfering RNA (siRNA) (Dharmacon catalog number D-011482-02) at a final concentration of 5 nmol using the HiPerfect reagent according to the manufacturer's instructions (Qiagen Corporation). For hnRNPA1, HeLa cells were transfected with Dharmacon catalog number J-008221-11 siRNA at a final concentration of 33 nmol per reaction using TransIT siQuest reagent according to the manufacturer's instructions (Mirus Bio Corporation). The medium on the cells was changed to DMEM (Gibco) containing 7% FBS at 24 h after transfection, and the cells were split at 48 h after transfection to obtain cells that were 60 to 80% confluent at 72 h after transfection. The lysates were harvested at 72 h after transfection to verify silencing by immunoblotting. The nontargeting siRNA used was Dharmacon catalog number D-001210-01-05.
Immunoblotting.
The lysates were prepared by removing medium from the cells, washing once with PBS, and adding cold radioimmunoprecipitation assay (Ripa) buffer containing 1 mM each benzamidine and phenylmethylsulfonyl fluoride. The six-well plates were rocked for 5 min at 4°C, and the cells were scraped and transferred to a 1.5-ml microcentrifuge tube. The cells were spun in Ripa buffer at 14,000 × g for 10 min at 4°C. Following centrifugation, the supernatants were removed and placed into a new tube. These lysates were assayed for their protein concentrations using the DC protein assay (Bio-Rad), and equal amounts of lysate (10 to 30 μg) were loaded on a 10% Bis-Tris NuPage gel (Invitrogen). The proteins were transferred to polyvinylidene difluoride using the Invitrogen transfer buffer. The membranes were blocked with 3% milk in 1× TBS-0.1% Tween 20 overnight and probed using antibodies to Rae1 (provided by Jan van Deursen, Mayo Clinic), hnRNPA1 (sc32301; Santa Cruz Biotechnology), or actin (Sigma A5441) diluted in a 1% milk solution in 1× TBS-0.1% Tween. The blots were next washed five times in 1× TBS-0.1% Tween and incubated with ECL anti-rabbit immunoglobulin G secondary antibody linked to horseradish peroxidase (Amersham) at 1:20,000 in a 1% milk solution in 1× TBS-0.1% Tween. The blots were washed five times as described above, and protein was detected using the SuperSignal West Pico substrate (Pierce).
EGFP transfections.
pEGFP-M9, pEGFPM9-MT, pEGFPTAG, and pEGFPantiTAG (24) DNAs (1 μg each) were transfected into HeLa cells in six-well dishes using Effectene (Qiagen) according to the manufacturer's instructions. The cells were approximately 40 to 60% confluent at the time of transfection, and the transfection mixtures were added to cells in 1.6 ml DMEM containing 7% FBS and 2 mM glutamine. At 24 h posttransfection, the medium was removed, the cells were washed gently with DMEM, and fresh medium was added. At 48 h posttransfection, the cells were infected with rwt VSV at a multiplicity of infection (MOI) of 10 PFU/cell. At 6 h postinfection, the cells were washed with PBS and fixed in 4% paraformaldehyde for 20 min. The cells were next washed three times with PBS containing 1% BSA, 1 drop of ProLong Gold antifade reagent with DAPI (Invitrogen Molecular Probes) was added to each well, and coverslips were added.
Cotransfection of M and EGFP mRNAs.
The templates for mRNAs for EGFP or M protein were generated by linearizing plasmid pSD4EGFP or pSDM (65) with SalI, followed by phenol chloroform extraction and ethanol precipitation. mRNA was transcribed in vitro using the mMessage Machine SP6 kit (Ambion) according to the manufacturer's instructions, and the RNA was precipitated with lithium chloride. The Mirus TransIT mRNA reagent was used to transfect 4 μg GFP RNA and 10, 30, 100, or 300 ng M mRNA as well as various amounts of yeast tRNA to bring the RNA level to a total of 750 ng into HeLa cells in six-well dishes containing 2.5 ml DMEM with 7% FBS. The cells were 40% to 60% confluent at the time of transfection.
Metabolic labeling.
Metabolic labeling was performed as described previously (15) by infecting with VSV at an MOI of 10 PFU/cell and pulsing for 15 min with 35S-methionine at either 4 or 8 h postinfection.
Growth curve analysis.
The cells were plated to a density of 70 to 80% and infected with rwt virus at an MOI of 0.01 PFU/cell. At 1 h postinfection, virus was removed from the cells, the cells were washed with DMEM, and fresh medium was added. At the indicated postinfection time points in Fig. 7B, 100-μl aliquots of medium were removed from the cells and frozen at −80°C. The yield of virus was determined by plaque assay on BHK cells.
FIG. 7.

Role of hnRNPA1 in VSV replication. (A) Western blot analysis showing hnRNPA1-silenced cells (A1), cells transfected with a nontargeting siRNA (NT), or mock transfected with no siRNA (−). (B) Plaque assay of VSV growth in untreated cells (diamonds), cells transfected with nontargeting siRNA (siNT [squares]), or cells transfected with hnRNPA1 siRNA (triangles). Plaque assays were performed in duplicate at least three times. (C) Analysis of viral and host protein synthesis. HeLa cells were treated with siRNA directed against hnRNPA1 or control siRNA. At 72 h posttransfection, cells were infected with VSV at an MOI of 10 and then labeled with 35S-methionine at 4 or 8 h posttransfection, harvested, and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and phosphorimaging. A representative image from three independent experiments is shown.
Time lapse microscopy.
At 48 h postsilencing, the cells were split into 24-well dishes for time lapse microscopy. One day later, the cells were infected at an MOI of 10 PFU/cell, placed in an environmental chamber attached to a Zeiss Axiovert S200 microscope, and imaged every 15 min for 24 h as described previously (22). Still-frame grabs of QuickTime movie files were imported into Adobe Photoshop, resized to 5 in. by 2 in. and set at 300 dpi. The images were flattened and changed to grayscale mode and saved as uncompressed TIFF files.
Caspase assays.
HeLa cells were silenced as above, and at 48 h postsilencing, the cells were split into 96-well dishes to obtain approximately 20,000 cells per well. At 72 h postsilencing, the cells were mock infected, infected with rwt virus at an MOI of 10 PFU/ml, or treated with 0.5 μM staurosporine. The cells were harvested by centrifugation at 500 × g for 5 min, and the medium was removed. The cells were resuspended in 50 μl lysis buffer containing 50 mM HEPES (pH 7.4), 150 mM NaCl, 0.1% (wt/vol) each CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate) and NP-40, 292 mM sucrose, 2 mM EDTA, 10 mM dithiothreitol, and 1 mM each phenylmethylsulfonyl fluoride and benzamidine, transferred to a 96-well dish, and rocked gently at 4°C for 5 min. Two aliquots of 5 μl from each well were removed to assay protein concentration in duplicate using the Bio-Rad protein assay. Immediately prior to assaying the caspase activity, 50 μl of activation buffer (100 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 0.2 mM EDTA, and 20% glycerol) containing Ac-DEVD-AFC fluorogenic substrate (BioMol) and 1 mM dithiothreitol were added to the remaining 40 μl of lysed cells. Fluorescence was measured every 3 min using a POLARstar Omega fluorescence microplate reader for 1 h (BMG Labtech), and the rate per minute of DEVDase activity was calculated using Omega data analysis software version 1.00, Build 1.0.1.2 (BMG Labtech).
RESULTS
We sought to determine whether VSV alters protein trafficking in the host cell by examining the net subcellular distribution of several cellular hnRNPs. HeLa cells were infected with the wto strain of VSV and processed for fluorescence or confocal microscopy using antibodies against hnRNPA1, hnRNPC1/C2, hnRNPK, and hnRNPB1. Lamin A/C was analyzed as a control for a nonshuttling nuclear protein. The cells were also labeled with antibody to VSV G protein, which identifies the intracellular membranes in the cytoplasm of infected cells, and with TOPRO-iodide or DAPI, which labels DNA to define the nuclear compartment. Representative images from these experiments are shown in Fig. 1. The mock-infected control samples are shown in the top row for each protein, and the VSV-infected cells are shown in the bottom row. In the control cells, the signal for all of the hnRNPs was coincident with the DNA staining, showing that these proteins were largely in the nucleus. In the infected cells, hnRNPA1, -C1/C2, and -K were redistributed to the cytoplasm (Fig. 1A to C). However, hnRNPB1 remained nuclear in VSV-infected cells (Fig. 1D). There were no alterations to the nuclear lamin in infected cells (Fig. 1E). This result indicates that the structure of the nuclear envelope was not altered at this relatively early time (6 to 7 h postinfection), which is prior to the onset of virus-induced apoptosis.
The relocalization of nuclear proteins to the cytoplasm could be due to either increased export from or decreased import into the nucleus. To determine the effect of VSV infection on nuclear import, we used EGFP linked to the M9 NLS, which is used by hnRNPA1 for nuclear import, or EGFP tagged with the classical NLS, which is used by hnRNPC1/C2. These EGFP proteins are small enough in size (∼30 kDa) to allow diffusion across the nuclear membrane independent of the time of protein synthesis. Thus, their localization reflects the steady state between import and export. HeLa cells were transfected with plasmids encoding the tagged EGFPs, and at 48 h posttransfection, the cells were infected with rwt virus. At 6 h postinfection, the cells were fixed, labeled with DAPI, and imaged using fluorescence microscopy. As shown in Fig. 2A, the EGFP-M9 protein was predominately nuclear, in both mock-infected and virus-infected cells. As a control, the cells were also transfected with a plasmid containing a mutation in the M9 sequence which renders the NLS nonfunctional (24). The fluorescent signal for both mock- and virus-infected cells expressing this mutant, EGFP-M9 MT (Fig. 2B), occurred throughout the cell, similarly to the distribution of EGFP without an NLS (Fig. 2D) and in contrast to the nuclear signal of cells expressing EGFP-M9 (Fig. 2A). As shown in Fig. 2C, the cells were transfected with a plasmid encoding a classical NLS-containing reporter, EGFP-TAG, containing an NLS from the large T antigen of simian virus 40 (24). As expected, EGFP-TAG NLS fluorescence was coincidental with DAPI staining of the nucleus in mock-infected cells (Fig. 2C). VSV infection did not alter the localization of EGFP-TAG (Fig. 2C). We conclude from these results that VSV infection does not alter the M9/transportin import pathway or the classical import pathway, used by hnRNPA1 and hnRNPC1/C2, respectively.
VSV infection results in the inhibition of host transcription due to the activity of the viral matrix (M) protein (6). The inhibition of cellular transcription with actinomycin D has been shown to result in the relocalization of hnRNPA1 to the cytoplasm (48). To determine whether the relocalization of hnRNPA1 is correlated with the ability of VSV to shut off host gene expression, cells were infected with the rwt and M protein mutant (rM51R-M) viruses. These viruses are isogenic except for a point mutation in the M protein of the rM51R-M virus which renders this virus defective in the inhibition of host transcription (7). At 6 h postinfection, the cells were analyzed by confocal microscopy in order to visualize the subcellular localization of hnRNPA1. hnRNPA1 was in the nucleus of mock-treated cells, while the majority of hnRNPA1 in cells infected with rwt virus was in the cytoplasm (Fig. 3). However, infection with rM51R-M virus did not induce the relocalization of hnRNPA1 to the cytoplasm, suggesting that the inhibition of host gene expression by VSV is correlated with alterations in hnRNPA1 distribution. This idea was tested by treating cells infected with rM51R-M virus with pharmacologic inhibitors of host transcription and analyzing hnRNPA1 relocalization. Cells infected with rM51R-M virus were treated with the inhibitors 5,6-dichloro-1-β-d-ribobenzimidazole or actinomycin D at 2 h postinfection, and the cells were processed for microscopy at 6 h postinfection. The presence of either inhibitor resulted in the increased relocalization of hnRNPA1 to the cytoplasm relative to the rM51R-M virus-infected cells in the absence of inhibitor. As a control, the cells were treated with inhibitor alone in the absence of virus infection (bottom panel), and most of the hnRNPA1 appeared in the cytoplasm, in agreement with previous results (47).
FIG. 3.
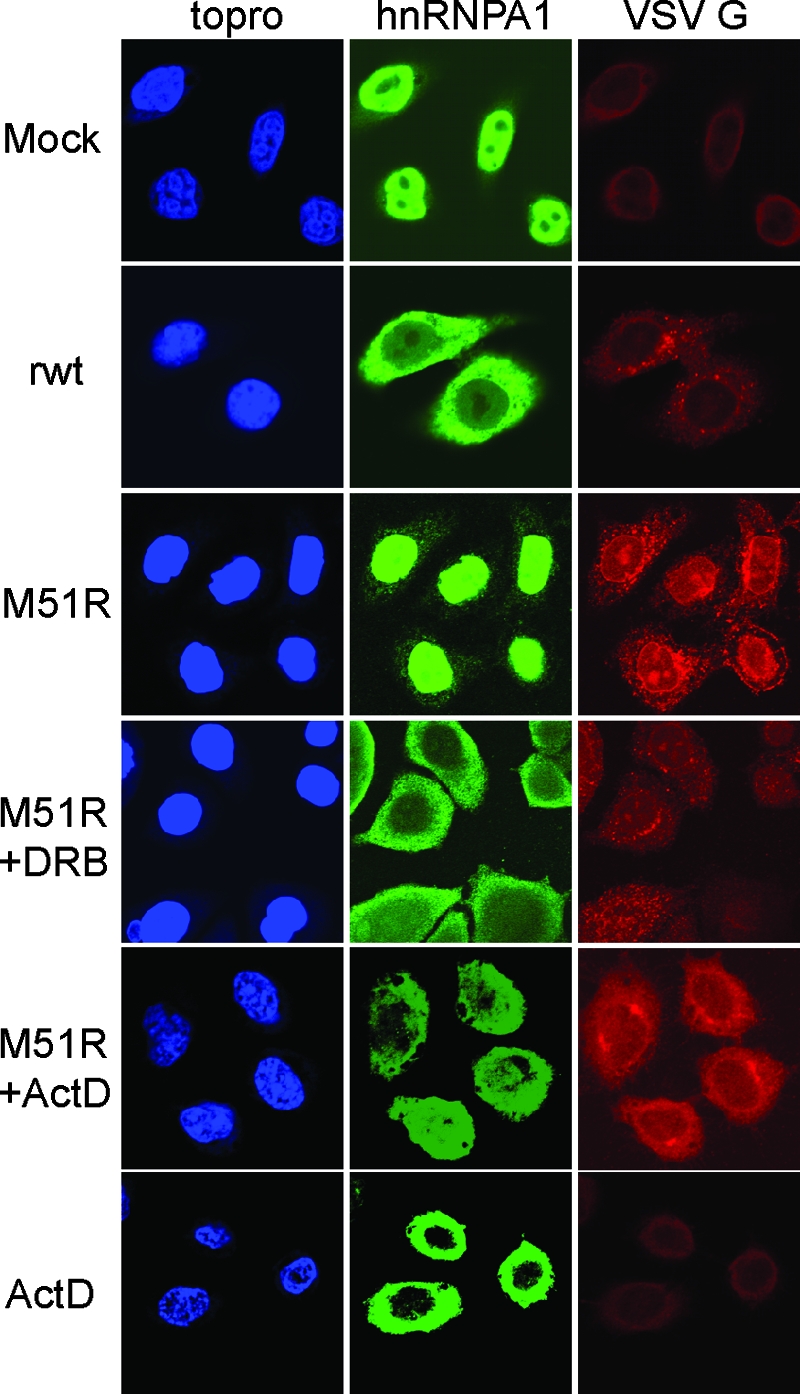
Confocal microscopy analysis of hnRNPA1 relocalization during infection with rwt and M protein mutant VSV. HeLa cells were mock infected or infected with either rwt or rM51R-M virus for 6 h, as indicated on the left. In the bottom three panels, cells were treated with the transcription inhibitors 5,6-dichloro-1-β-d-ribobenzimidazole (DRB) or actinomycin D (ActD) beginning at 2 h postinfection.
We next sought to determine the importance of nuclear export in controlling hnRNP relocalization following virus infection. VSV infection has been reported to inhibit mRNA export by binding the cellular factor Rae1 (19), which has been implicated in RNA transport (8, 11, 41, 51) and as a cell cycle checkpoint regulator (4, 51, 61). Thus, we determined whether Rae1 contributes to hnRNPA1 relocalization during VSV infection. The approach was to transfect cells with siRNAs directed against Rae1, infect the silenced cells, and analyze them by confocal microscopy (Fig. 4A). As a control, cells were transfected with a nontargeting siRNA, whose sequence is scrambled and does not match any sequence in the human genome. Figure 4B shows that Rae1 protein levels were silenced to approximately 10% of the normal levels. As reported previously (9), the silencing of Rae1 expression was not lethal to the cells, which continued to grow and divide during the 72-h time course of the experiment. Interestingly, the VSV infection of cells with reduced Rae1 did not result in the substantial relocalization of hnRNPA1 to the cytoplasm (Fig. 4A, top panel). Although a small fraction of hnRNPA1 was cytoplasmic in Rae1-silenced cells, the majority of hnRNPA1 appeared to be nuclear. Similarly to the mock-infected controls above, in the mock-infected cells in which Rae1 was silenced, hnRNPA1 appeared coincident with the nuclear staining (Fig. 4A, second panel). For the cells which were transfected with the nontargeting siRNA, hnRNPA1 was relocalized to the cytoplasm following VSV infection, as seen in the VSV-infected cells which were mock transfected (Fig. 4A, bottom panels). The signal for hnRNPA1 in the nucleus and in the cytoplasm was quantified for cells in three or more fields in multiple experiments to determine the ratio of cytoplasmic to nuclear hnRNPA1 for each sample. The ratios of cytoplasmic to nuclear signals were then expressed relative to the ratio observed in VSV-infected but nontransfected control cells (Fig. 4A, bottom panel) and are shown in Fig. 4C. Mock-infected cells transfected with Rae1 siRNA had approximately 14% of the relative movement of the control. Similarly, the reduction of Rae1 prevented hnRNPA1 relocalization to the cytoplasm in VSV-infected cells, as indicated by a decrease in movement to 16% of the control. The controls for siRNA transfections had high relative movement (118% of the mock-transfected cells) of hnRNPA1 following infection with VSV. These results indicate that hnRNPA1 relocalization during VSV infection is dependent on the host factor Rae1.
FIG. 4.

hnRNPA1 relocalization during VSV infection following silencing of the host factor Rae1. HeLa cells were transfected with siRNAs directed against Rae1, a nontargeting siRNA (NT), or mock transfected. At 72 h posttransfection, cells were mock infected or infected with the wto strain at an MOI of 10 and processed as described in the legend to Fig. 1. (A) Confocal microscopy images of cells labeled with TOPRO-iodide and antibodies against hnRNPA1 or VSV G protein. (B) Western blot analysis of silenced cells. (C) Quantification of the cytoplasmic-to-nuclear ratio of the hnRNPA1 signal in cells from multiple confocal analysis experiments. The positive control, set at 100%, is the ratio obtained from nontransfected HeLa cells infected with VSV. Error bars correspond to the average standard deviation from the results of three experiments.
The relocalization of hnRNPA1 following VSV infection correlated with the inhibition of gene expression by VSV and was dependent on the host factor Rae1. These results led us to ask whether Rae1 is required for hnRNPA1 relocalization due to transcription inhibition by actinomycin D. To test this possibility, HeLa cells were transfected with nontargeting siRNA or Rae1 siRNA, treated with the transcriptional inhibitor actinomycin D for 4 h, and analyzed by confocal microscopy. As expected, the cells that received nontargeting siRNAs showed the nuclear localization of hnRNPA1 (Fig. 5A, top panel), and actinomycin D treatment caused the relocalization of hnRNPA1 to the cytoplasm (Fig. 5A, bottom panel). Similarly, Rae1-silenced cells (Fig. 5B) showed the cytoplasmic localization of hnRNPA1 following treatment with actinomycin D. These results show that despite reduced levels of Rae1, the inhibition of transcription by pharmacologic inhibitors results in the relocalization of hnRNPA1 to the cytoplasm.
Because Rae1 interacts with the VSV M protein, we tested whether M protein alone would be capable of inducing hnRNPA1 relocalization in the absence of viral infection. HeLa cells were transfected with mRNAs encoding wild-type M protein, and the localization of hnRNPA1 at 18 h posttransfection was examined. Because M protein inhibits host transcription, cells were transfected with M mRNA to avoid the pitfall that M protein inhibits its own expression from plasmid DNA (6, 7). For these experiments, a cotransfected marker is required in order to label equivalently cells transfected with mRNA for wto or mutant M protein or the negative control mRNA. Thus, cells were cotransfected with EGFP mRNA in order to label the transfected cells (Fig. 6). As controls, the cells were mock transfected or transfected with GFP mRNA only (top panels). For all concentrations of M mRNA tested, hnRNPA1 was detected primarily in the nucleus of transfected cells, comparable to the control cells. This result indicates that the inhibition of gene expression by M protein in the absence of other viral components is not sufficient to induce the relocalization of hnRNPA1. One possible explanation for this result is that an additional signal produced during VSV infection is required, although it is also likely that higher concentrations of M protein are necessary to induce relocalization than are achieved in the transfection experiment.
FIG. 6.

Effects of M protein expressed in the absence of other viral components on hnRNPA1 localization. HeLa cells were transfected with mRNA encoding M protein or actin, along with EGFP mRNA as a control for transfection. At 18 h posttransfection, cells were fixed, incubated with antibodies against hnRNPA1, and stained with DAPI.
The finding that hnRNPA1 relocalized to the cytoplasm during VSV infection led us to determine whether there is a role for hnRNPA1 in the viral life cycle. hnRNPA1 expression was silenced in HeLa cells, and replication as well as the rates of viral and host protein synthesis were measured in infected cells. siRNA directed against hnRNPA1 dramatically reduced hnRNPA1 expression relative to the control cells which were transfected with a scrambled, nontargeting siRNA mixture, or cells that received no siRNA, as determined by Western blot analysis (Fig. 7A). The quantification of blots from multiple experiments indicated that hnRNPA1 was reduced to an average of 15% of normal levels. To assay whether hnRNPA1 is required for a productive virus infection, hnRNPA1-silenced cells were infected with rwt virus at an MOI of 0.01, and the virus titer was determined at various times postinfection by plaque assay in BHK cells (Fig. 7B). No difference was observed between viral titers in the cells transfected with siRNA directed against hnRNPA1 compared with the control cells (Fig. 7B). To assay the rates of viral and host protein synthesis, silenced cells were infected under single-cycle conditions (MOI of 10) and pulse-labeled with 35S-methionine at 4 and 8 h postinfection. The cell lysates were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and phosphorimaging (Fig. 7C). The hnRNPA1-silenced cells showed similar levels of viral protein synthesis (distinct dark bands) relative to the controls. Compared to that of the uninfected cells, host protein synthesis was reduced significantly in VSV-infected cells, regardless of siRNA transfection. Thus, the silencing of hnRNPA1 did not affect the ability of VSV to shut off host protein synthesis.
In performing these experiments, we noted that the hnRNPA1-silenced cells appeared to show less cytopathic effect following VSV infection. Representative images from 16 h postinfection are shown in Fig. 8A. The cytopathic effects of VSV infection are due to the rapid induction of apoptosis. To determine the time course of the induction of apoptosis, time lapse microscopy analysis was performed on hnRNPA1-silenced cells. Cells transfected with hnRNPA1 siRNA appeared to retain their flattened morphology at later times postinfection. In HeLa cells infected with VSV, entry into apoptosis is unambiguous as seen by the dramatic onset of membrane blebbing and cell rounding (30). The rate of entry into apoptosis was quantified by scoring cells for membrane blebbing and cell rounding at 2-h intervals following infection (Fig. 8B). hnRNPA1-silenced cells entered apoptosis more slowly following VSV infection than cells treated with control siRNA: the average time for 50% of the cells to enter apoptosis was 18 h for hnRNPA1-silenced cells compared with 13 h for nontargeting cells. The majority of the cells entered apoptosis by 24 h postinfection, and all cells died by 36 to 48 h postinfection (not shown). To confirm this result, we performed caspase 3 activity assays in hnRNPA1-silenced HeLa cells at 8, 12, and 16 h postinfection. We observed that hnRNPA1-silenced cells had lower caspase 3 activity at 8 h postinfection (Fig. 8C). The calculated P values relative to the hnRNPA1-silenced cells in these experiments using a pairwise t test with two-tailed distribution were 1.03 × 10−4 for cells that received nontargeting siRNA and 2.56 × 10−2 for the nontransfected control cells. At the later times postinfection (Fig. 8D and E), the difference in caspase 3 activity between hnRNPA1-silenced cells and the control cells was not statistically significant. In summary, even though virus replication and host shutoff were unaffected by the reduction in hnRNPA1, the rate of VSV-induced apoptosis was slower in cells with reduced hnRNPA1.
FIG. 8.

VSV-induced apoptosis in hnRNPA1-silenced cells. (A) Phase-contrast images of cells transfected with siRNAs and infected with VSV or mock infected at an MOI of 10 for 16 h. A1, hnRNPA1-silenced cells; NT, nontargeting; +, presence; −, absence. (B) Cells transfected with siRNA directed against hnRNPA1 (diamonds) or nontargeting siRNA (squares) were infected as described for panel A and were analyzed by time lapse microscopy. The numbers of cells entering apoptosis as a percentage of total cells were determined every 2 h and plotted against time postinfection. Three fields of cells were analyzed from each of at least two independent experiments, and the error bars represent the average standard deviations. (C) Caspase 3 activity assays in cells transfected with siRNA directed against hnRNPA1 (siA1), nontargeting siRNA (siNT), or untreated cells (No Rx). Cells were mock infected (white bars), infected with rwt virus at an MOI of 10 (black bars), or treated with staurosporine (gray bars) for 8 h. For each experiment, the relative caspase activity was calculated as a percentage of the fluorescence units min−1 μg−1 protein relative to the values obtained in cells which received nontargeting siRNA and were infected with rwt virus. The values represent an average of two to four experiments, with three replicates per experiment. The error bars indicate average standard deviations. Caspase activity assay results are shown for 12 h (D) and 16 h (E) postinfection or posttreatment as described for panel C.
DISCUSSION
The data presented here show that the nuclear-cytoplasmic trafficking of several cellular hnRNPs, including hnRNPA1, hnRNPC1/C2, and hnRNPK, is disrupted following VSV infection. In contrast to the reported disruption of host mRNA export during VSV infection, these nuclear proteins are relocalized to the cytoplasm (Fig. 1). This relocalization is not due to virus-induced defects in protein import by two major pathways (Fig. 2). Instead, this relocalization correlates with the ability of VSV to shut off host gene expression (Fig. 3) and appears to be similar to the relocalization induced by pharmacologic inhibitors of cellular transcription (18, 48). However, the relocalization of hnRNPA1 in infected cells requires Rae1, while relocalization induced by the pharmacologic inhibition of host transcription does not depend on Rae1 expression.
Several viruses which undergo a primarily cytoplasmic life cycle in host cells induce defects in nuclear-cytoplasmic trafficking, such as the relocalization of hnRNPs to the cytoplasm. However, the mechanisms are distinct from those described here for VSV. Following poliovirus infection, several host nuclear proteins, including hnRNPs, La, and nucleolin, relocalize to the cytoplasm where they associate with viral mRNA or proteins (24, 25, 37, 38, 62). The mechanism for this relocalization involves the inactivation of several protein import pathways and the degradation of nuclear pore complex proteins Nup98, Nup153, and p62 (24, 25, 44). The degradation of nuclear pore components during poliovirus infection is likely due to the viral protease 2A (44). Cardioviruses, which are in a different picornaviral genus, encode an L protein that alters the nuclear-cytoplasmic transport of host proteins both into and out of the nucleus (16, 35) by binding directly to Ran-GTPase (50). In contrast, VSV does not encode a protease, and whether any nuclear pore components are targeted for degradation following VSV infection is unknown. Unlike the picornaviruses, VSV infection does not appear to induce defects in protein import in HeLa cells (Fig. 2). Our result differs slightly from previous results obtained in Xenopus oocytes in which the import of classical NLS-containing proteins is slowed but not entirely abrogated by M protein (45). However, these experiments also revealed that the transport receptors are not inactivated, and the results of subsequent experiments with mammalian cells are similar to our results that protein import is not substantially affected by VSV (5, 19). Thus, the relocalization of hnRNPs following VSV infection may be explained by the enhanced export of certain mRNPs or enhanced retention in the cytoplasm.
Because hnRNPs have been implicated as mRNA export factors (27, 43), the relocalization of hnRNPs in VSV-infected cells likely involves the export of a subset of host mRNAs. For example, hnRNPA1 is bound to RNAs during their processing in the nucleus and is also bound to some mRNAs in the cytoplasm (47). hnRNPA1-containing mRNPs are thought to chaperone mRNAs through the nuclear pore complex, undergoing dynamic changes in structure and composition during transport (31, 40, 60). Like hnRNPA1, hnRNPK is a shuttling protein, which may also be linked to mRNA export (39). Nonshuttling proteins, such as hnRNPC1/C2 may also play a role in mRNA export, because their removal from mRNPs might be necessary before mRNA is exported (42, 43). The export of hnRNPs together with mRNAs would not be expected if the primary effect of VSV M protein was to block mRNA export, as has been proposed (19). However, if M protein inhibits host transcription prior to inhibiting mRNA export, then the depletion of newly synthesized mRNAs in the nucleus would lead to hnRNP relocalization, as occurs following treatment with pharmacological inhibitors of transcription (48) (Fig. 3). Alternatively, hnRNPs may be relocalized to the cytoplasm of VSV-infected cells independently of mRNAs.
The relocalization of hnRNPA1 to the cytoplasm in VSV-infected cells was dependent on the expression of host factor Rae1. This result was unexpected, as the interaction between VSV M protein and Rae1 has been proposed to result in mRNA export inhibition (19). However, the depletion of Rae1 from higher eukaryotic cells does not inhibit mRNA export (4, 58), which suggests that the M protein-mediated inhibition of Rae1 function alone is not sufficient to prevent mRNA export. In fact, our data suggest that Rae1 is still functional in protein export in VSV-infected cells. The M51R M protein mutant virus is defective at inhibiting host transcription (2, 3, 6) and does not cause the relocalization of hnRNPA1 to the cytoplasm (Fig. 3). Unlike the wild-type M protein (19), the M51R mutant does not form a complex with Rae1 (K. Rajani, E. Kneller, D. Lyles, unpublished results). Thus, the binding of M protein to Rae1 appears to be required for hnRNP relocalization. In infected cells, the M protein-Rae1 complex may sequester other factors to alter hnRNPA1 localization, inhibit host gene expression, or both. Along these lines, the M protein-Rae1 complex may play a larger role in disrupting host nuclear function by acting as a signal that couples the inhibition of mRNA export, the inhibition of transcription, and the relocalization of hnRNPs. Coupling between transcription and mRNA export in mammalian cells has recently been shown, as transcriptional inactivation by different inhibitors resulted in the inhibition of mRNA export (59). Future studies will determine the role that the M protein-Rae1 complex might play in the VSV-induced inhibition of host nuclear function.
Cellular hnRNPs have been implicated in the life cycles of several viruses. For poliovirus, the relocalization of hnRNPC1/C2 (24) enables hnRNPC1 to associate with viral proteins to promote replication (12). Following infection with the coronavirus mouse hepatitis virus (MHV), hnRNPA1 and hnRNPA2/B1 relocalize to the cytoplasm and bind to the leader region of MHV RNA (34, 57), which has been proposed to regulate viral replication (56). The knockdown of hnRNPA1 has been shown to inhibit hepatitis C virus replication (28). In contrast to MHV and hepatitis C virus, the replication of human T-lymphotropic virus (HTLV-1) is increased in cells lacking hnRNPA1 (32). Thus, hnRNPs can have either positive or negative effects on virus replication. It has been suggested in the literature that hnRNPA1 might play a role in the cytoplasm of VSV-infected cells. A protein of 38 kDa likely to be hnRNPA1 is cross-linked to viral and cellular mRNA in the cytoplasm following VSV infection (1, 18). Recently, it was observed that hnRNPA1 can associate in vitro with VSV G and M mRNAs in complexes containing other cellular factors which might modulate translation in VSV-infected cells (46). However, we found that silencing hnRNPA1 expression did not affect virus growth or protein synthesis, or the shutoff of host translation (Fig. 7). This result does not rule out a role for cellular hnRNPs in the control of VSV translation, since some other hnRNPs may functionally substitute for hnRNPA1 in its absence. Likewise, MHV can replicate in cells lacking hnRNPA1, and hnRNPA2/B1 can serve in its place (55, 57). However, if there is another hnRNP involved for VSV, it is likely not hnRNPA2/B1, as its localization was unchanged following VSV infection (Fig. 1).
We found that VSV-infected cells with reduced hnRNPA1 levels became apoptotic more slowly than control cells (Fig. 8). This would suggest that hnRNPA1 plays a role in promoting apoptosis in response to viral infection. This effect could be due to changes in hnRNPA1-dependent gene expression that would normally promote the induction of apoptosis. For example, changes in the mRNP protein composition of cellular mRNAs in infected cells might enable the preferential translation of specific cellular mRNAs in the induction of apoptosis. hnRNPA1 is recruited to and acts upon the internal ribosome entry sites in mRNAs for factors that regulate apoptosis, including those that encode for XIAP, apaf-1, and FGF-2 (10, 13, 33). Our results suggest a model in which hnRNPA1 relocalization promotes apoptotic signaling as a host response following VSV infection, rather than as a means to promote viral replication. The observation that the relocalization of hnRNPA1 is dependent on Rae1 suggests that Rae1 might have two possible functions in the VSV life cycle. Rae1 in a complex with M protein inhibits mRNA transport; in addition, Rae1 in the context of a viral infection promotes the export of hnRNPA1 to the cytoplasm. Future studies will address the role of Rae1 in inhibiting host gene expression, which is correlated with the induction of apoptosis by M protein (29). These studies will help us distinguish whether the cytoplasmic localization or the protein level of hnRNPA1 in a virus-infected cell is crucial to apoptotic signaling.
Acknowledgments
We thank Karishma Rajani, Griffith Parks, and Maryam Ahmed for critical reading of the manuscript. We also thank Ken Grant at the Wake Forest University School of Medicine Microscopy facility for assistance with confocal imaging, Kurt Gustin (University of Arizona College of Medicine, Phoenix) for the EGFP plasmids tagged with M9 and classical NLSs, and Jan van Deursen (Mayo Clinic, Rochester) for the Rae1 antibody. We are also grateful to Shelby Puckett for assistance with plaque assays; John Wilkinson, Zachary Cary, Peter Antinozzi, and Alicia Pearce for advice in setting up the apoptosis assays; John Johnson for suggestions for improved EGFP and DAPI imaging; and Will Safrit in the Creative Communications department at the Wake Forest University School of Medicine for help with movie files.
This research was supported by Public Health Service grants AI35892 and AI52304 (to D.S.L.) and AI064606 (to J.H.C.) from the National Institute for Allergy and Infectious Diseases. The Microscopy Core Laboratory is supported in part by the core grant (CA12197) for the Comprehensive Cancer Center of Wake Forest University from the National Cancer Institute.
Footnotes
Published ahead of print on 12 November 2008.
REFERENCES
- 1.Adam, S. A., Y. D. Choi, and G. Dreyfuss. 1986. Interaction of mRNA with proteins in vesicular stomatitis virus-infected cells. J. Virol. 57614-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed, M., and D. S. Lyles. 1998. Effect of vesicular stomatitis virus matrix protein on transcription directed by host RNA polymerases I, II, and III. J. Virol. 728413-8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed, M., M. O. McKenzie, S. Puckett, M. Hojnacki, L. Poliquin, and D. S. Lyles. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol. 774646-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babu, J. R., K. B. Jeganathan, D. J. Baker, X. Wu, N. Kang-Decker, and J. M. van Deursen. 2003. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 160341-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belov, G. A., A. G. Evstafieva, Y. P. Rubtsov, O. V. Mikitas, A. B. Vartapetian, and V. I. Agol. 2000. Early alteration of nucleocytoplasmic traffic induced by some RNA viruses. Virology 275244-248. [DOI] [PubMed] [Google Scholar]
- 6.Black, B. L., and D. S. Lyles. 1992. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J. Virol. 664058-4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Black, B. L., R. B. Rhodes, M. McKenzie, and D. S. Lyles. 1993. The role of vesicular stomatitis virus matrix protein in inhibition of host-directed gene expression is genetically separable from its function in virus assembly. J. Virol. 674814-4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blevins, M. B., A. M. Smith, E. M. Phillips, and M. A. Powers. 2003. Complex formation among the RNA export proteins Nup98, Rae1/Gle2, and TAP. J. Biol. Chem. 27820979-20988. [DOI] [PubMed] [Google Scholar]
- 9.Blower, M. D., M. Nachury, R. Heald, and K. Weis. 2005. A Rae1-containing ribonucleoprotein complex is required for mitotic spindle assembly. Cell 121223-234. [DOI] [PubMed] [Google Scholar]
- 10.Bonnal, S., F. Pileur, C. Orsini, F. Parker, F. Pujol, A. C. Prats, and S. Vagner. 2005. Heterogeneous nuclear ribonucleoprotein A1 is a novel internal ribosome entry site trans-acting factor that modulates alternative initiation of translation of the fibroblast growth factor 2 mRNA. J. Biol. Chem. 2804144-4153. [DOI] [PubMed] [Google Scholar]
- 11.Brown, J. A., A. Bharathi, A. Ghosh, W. Whalen, E. Fitzgerald, and R. Dhar. 1995. A mutation in the Schizosaccharomyces pombe rae1 gene causes defects in poly(A)+ RNA export and in the cytoskeleton. J. Biol. Chem. 2707411-7419. [DOI] [PubMed] [Google Scholar]
- 12.Brunner, J., J. H. C. Nguyen, H. H. Roehl, T. V. Ho, K. M. Swiderek, and B. L. Semler. 2005. Functional interaction of heterogeneous nuclear ribonucleoprotein C with poliovirus RNA synthesis initiation complexes. J. Virol. 793254-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cammas, A., F. Pileur, S. Bonnal, S. M. Lewis, N. Leveque, M. Holcik, and S. Vagner. 2007. Cytoplasmic relocalization of heterogeneous nuclear ribonucleoprotein A1 controls translation initiation of specific mRNAs. Mol. Biol. Cell 185048-5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connor, J. H., and D. S. Lyles. 2005. Inhibition of host and viral translation during vesicular stomatitis virus infection. eIF2 is responsible for the inhibition of viral but not host translation. J. Biol. Chem. 28013512-13519. [DOI] [PubMed] [Google Scholar]
- 15.Connor, J. H., and D. S. Lyles. 2002. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 7610177-10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delhaye, S., V. van Pesch, and T. Michiels. 2004. The leader protein of Theiler's virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 784357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dratewka-Kos, E., I. Kiss, J. Lucas-Lenard, H. B. Mehta, C. L. Woodley, and A. J. Wahba. 1984. Catalytic utilization of eIF-2 and mRNA binding proteins are limiting in lysates from vesicular stomatitis virus infected L cells. Biochemistry 236184-6190. [DOI] [PubMed] [Google Scholar]
- 18.Dreyfuss, G., S. A. Adam, and Y. D. Choi. 1984. Physical change in cytoplasmic messenger ribonucleoproteins in cells treated with inhibitors of mRNA transcription. Mol. Cell. Biol. 4415-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faria, P. A., P. Chakraborty, A. Levay, G. N. Barber, H. J. Ezelle, J. Enninga, C. Arana, J. van Deursen, and B. M. A. Fontoura. 2005. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol. Cell 1793-102. [DOI] [PubMed] [Google Scholar]
- 20.Fridell, R., R. Truant, L. Thorne, R. Benson, and B. Cullen. 1997. Nuclear import of hnRNP A1 is mediated by a novel cellular cofactor related to karyopherin-beta. J. Cell Sci. 1101325-1331. [DOI] [PubMed] [Google Scholar]
- 21.Frieman, M., B. Yount, M. Heise, S. A. Kopecky-Bromberg, P. Palese, and R. S. Baric. 2007. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 819812-9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaddy, D. F., and D. S. Lyles. 2007. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J. Virol. 812792-2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gustin, K. E. 2003. Inhibition of nucleo-cytoplasmic trafficking by RNA viruses: targeting the nuclear pore complex. Virus Res. 9535-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gustin, K. E., and P. Sarnow. 2001. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 20240-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gustin, K. E., and P. Sarnow. 2002. Inhibition of nuclear import and alteration of nuclear pore complex composition by rhinovirus. J. Virol. 768787-8796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Her, L.-S., E. Lund, and J. E. Dahlberg. 1997. Inhibition of Ran guanosine triphosphatase-dependent nuclear transport by the matrix protein of vesicular stomatitis virus. Science 2761845-1848. [DOI] [PubMed] [Google Scholar]
- 27.Izaurralde, E., A. Jarmolowski, C. Beisel, I. W. Mattaj, G. Dreyfuss, and U. Fischer. 1997. A role for the M9 transport signal of hnRNPA1 in mRNA nuclear export. J. Cell Biol. 13727-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim, C. S., S. K. Seol, O.-K. Song, J. H. Park, and S. K. Jang. 2007. An RNA-binding protein, hnRNP A1, and a scaffold protein, septin 6, facilitate hepatitis C virus replication. J. Virol. 813852-3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kopecky, S. A., and D. S. Lyles. 2003. Contrasting effects of matrix protein on apoptosis in HeLa and BHK cells infected with vesicular stomatitis virus are due to inhibition of host gene expression. J. Virol. 774658-4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopecky, S. A., M. C. Willingham, and D. S. Lyles. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol. 7512169-12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krecic, A. M., and M. S. Swanson. 1999. hnRNP complexes: composition, structure, and function. Curr. Opin. Cell Biol. 11363-371. [DOI] [PubMed] [Google Scholar]
- 32.Kress, E., H. H. Baydoun, F. Bex, L. Gazzolo, and M. Duc Dodon. 2005. Critical role of hnRNP A1 in HTLV-1 replication in human transformed T lymphocytes. Retrovirology 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis, S. M., A. Veyrier, N. Hosszu Ungureanu, S. Bonnal, S. Vagner, and M. Holcik. 2007. Subcellular relocalization of a trans-acting factor regulates XIAP IRES-dependent translation. Mol. Biol. Cell 181302-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li, H. P., X. Zhang, R. Duncan, L. Comai, and M. M. C. Lai. 1997. Heterogeneous nuclear ribonucleoprotein A1 binds to the transcription-regulatory region of mouse hepatitis virus RNA. Proc. Natl. Acad. Sci. USA 19979544-9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lidsky, P. V., S. Hato, M. V. Bardina, A. G. Aminev, A. C. Palmenberg, E. V. Sheval, V. Y. Polyakov, F. J. van Kuppeveld, and V. I. Agol. 2006. Nucleocytoplasmic traffic disorder induced by cardioviruses. J. Virol. 802705-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyles, D. S. 2000. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol. Mol. Biol. Rev. 64709-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McBride, A. E., A. Schlegel, and K. Kirkegaard. 1996. Human protein Sam68 relocalization and interaction with poliovirus RNA polymerase in infected cells. Proc. Natl. Acad. Sci. USA 932296-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meerovitch, K., J. Pelletier, and N. Sonenberg. 1989. A cellular protein that binds to the 5′-noncoding region of poliovirus RNA: implications for internal translation initiation. Genes Dev. 31026-1034. [DOI] [PubMed] [Google Scholar]
- 39.Michael, W. M., P. S. Eder, and G. Dreyfuss. 1997. The K nuclear shuttling domain: a novel signal for nuclear import and nuclear export in the hnRNPK protein. EMBO J. 163587-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mili, S., H. J. Shu, Y. Zhao, and S. Pinol-Roma. 2001. Distinct RNP complexes of shuttling hnRNP proteins with pre-mRNA and mRNA: candidate intermediates in formation and export of mRNA. Mol. Cell. Biol. 217307-7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy, R., J. L. Watkin, and S. R. Wente. 1996. GLE2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol. Biol. Cell 71921-1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakielny, S., and G. Dreyfuss. 1996. The hnRNP C proteins contain a nuclear retention sequence that can override nuclear export signals. J. Cell Biol. 1341365-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakielny, S., and G. Dreyfuss. 1999. Transport of proteins and RNAs in and out of the nucleus. Cell 99677-690. [DOI] [PubMed] [Google Scholar]
- 44.Park, N., P. Katikaneni, T. Skern, and K. E. Gustin. 2008. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 821647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petersen, J. M., L.-S. Her, and J. E. Dahlberg. 2001. Multiple vesiculoviral matrix proteins inhibit both nuclear export and import. Proc. Natl. Acad. Sci. USA 988590-8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfeifer, I., R. Elsby, M. Fernandez, P. A. Faria, D. R. Nussenzveig, I. S. Lossos, B. M. A. Fontoura, W. D. Martin, and G. N. Barber. 2008. NFAR-1 and -2 modulate translation and are required for efficient host defense. Proc. Natl. Acad. Sci. USA 1054173-4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pinol-Roma, S., and G. Dreyfuss. 1992. Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature 355730-732. [DOI] [PubMed] [Google Scholar]
- 48.Pinol-Roma, S., and G. Dreyfuss. 1991. Transcription-dependent and transcription-independent nuclear transport of hnRNP proteins. Science 253312-314. [DOI] [PubMed] [Google Scholar]
- 49.Pollard, V. W., W. M. Michael, S. Nakielny, M. C. Siomi, F. Wang, and G. Dreyfuss. 1996. A novel receptor-mediated nuclear protein import pathway. Cell 86985-994. [DOI] [PubMed] [Google Scholar]
- 50.Porter, F. W., Y. A. Bochkov, A. J. Albee, C. Wiese, and A. C. Palmenberg. 2006. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 10312417-12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pritchard, C. E. J., M. Fornerod, L. H. Kasper, and J. M. A. van Deursen. 1999. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J. Cell Biol. 145237-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reid, S. P., L. W. Leung, A. L. Hartman, O. Martinez, M. L. Shaw, C. Carbonnelle, V. E. Volchkov, S. T. Nichol, and C. F. Basler. 2006. Ebola virus VP24 binds karyopherin α1 and blocks STAT1 nuclear accumulation. J. Virol. 805156-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez, J. J., J.-P. Parisien, and C. M. Horvath. 2002. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 7611476-11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez, J. J., L.-F. Wang, and C. M. Horvath. 2003. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J. Virol. 7711842-11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen, X., and P. S. Masters. 2001. Evaluation of the role of heterogeneous nuclear ribonucleoprotein as a host factor in murine coronavirus discontinuous transcription and genome replication. Proc. Natl. Acad. Sci. USA 982717-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi, S. T., P. Huang, H.-P. Li, and M. M. C. Lai. 2000. Heterogeneous nuclear ribonucleoprotein A1 regulates RNA synthesis of a cytoplasmic virus. EMBO J. 194701-4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi, S. T., G.-Y. Yu, and M. M. C. Lai. 2003. Multiple type A/B heterogeneous nuclear ribonucleoproteins (hnRNPs) can replace hnRNP A1 in mouse hepatitis virus RNA synthesis. J. Virol. 7710584-10593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sitterlin, D. 2004. Characterization of the Drosophila Rae1 protein as a G1 phase regulator of the cell cycle. Gene 326107-116. [DOI] [PubMed] [Google Scholar]
- 59.Tokunaga, K., T. Shibuya, Y. Ishihama, H. Tadakuma, M. Ide, M. Yoshida, T. Funatsu, Y. Ohshima, and T. Tani. 2006. Nucleocytoplasmic transport of fluorescent mRNA in living mammalian cells: nuclear mRNA export is coupled to ongoing gene transcription. Genes Cells 11305-317. [DOI] [PubMed] [Google Scholar]
- 60.Visa, N., A. T. Alzhanova-Ericsson, X. Sun, E. Kiseleva, B. Bjorkroth, T. Wurtz, and B. Daneholt. 1996. A pre-mRNA-binding protein accompanies the RNA from the gene through the nuclear pores and into polysomes. Cell 84253-264. [DOI] [PubMed] [Google Scholar]
- 61.von Kobbe, C., J. M. A. van Deursen, J. P. Rodrigues, D. Sitterlin, A. Bachi, X. Wu, M. Wilm, M. Carmo-Fonseca, and E. Izaurralde. 2000. Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol. Cell 61243-1252. [DOI] [PubMed] [Google Scholar]
- 62.Waggoner, S., and P. Sarnow. 1998. Viral ribonucleoprotein complex formation and nucleolar-cytoplasmic relocalization of nucleolin in poliovirus-infected cells. J. Virol. 726699-6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weck, P., and R. R. Wagner. 1978. Inhibition of RNA synthesis in mouse myeloma cells infected with vesicular stomatitis virus. J. Virol. 25770-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whelan, S. P. J., L. A. Ball, J. N. Barr, and G. T. W. Wertz. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. USA 928388-8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Whitlow, Z. W., J. H. Connor, and D. S. Lyles. 2008. New mRNAs are preferentially translated during vesicular stomatitis virus infection. J. Virol. 822286-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wong, R. W., G. Blobel, and E. Coutavas. 15 December 2006. Rae1 interaction with NuMA is required for bipolar spindle formation. Proc. Natl. Acad. Sci. USA 10319783-19787. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]