

Abstract
Human endogenous retrovirus K (HERV-K) is the most intact retrovirus in the human genome. However, no single HERV-K provirus in the human genome today appears to be infectious. Since the Gag protein is the central component for the production of retrovirus particles, we investigated the abilities of Gag from two HERV-K proviruses to support production of virus-like particles and viral infectivity. HERV-K113 has full-length open reading frames for all viral proteins, while HERV-K101 has a full-length gag open reading frame and is expressed in human male germ cell tumors. The Gag of HERV-K101 allowed production of viral particles and infectivity, although at lower levels than observed with a consensus sequence Gag. Thus, including HERV-K109, at least two HERV-K proviruses in human genome today have functional Gag proteins. In contrast, HERV-K113 Gag supported only very low levels of particle production, and no infectivity was detectable due to a single amino acid substitution (I516M) near the extreme C terminus of the CA protein within Gag. The sequence of this portion of HERV-K CA showed similarities to that of human immunodeficiency virus type 1 and other primate immunodeficiency viruses. The extreme C terminus of CA may be a general determinant of retrovirus particle production. In addition, precise mapping of the defects in HERV-K proviruses as was done here identifies the key polymorphisms that need to be analyzed to assess the possible existence of infectious HERV-K alleles within the human population.
Approximately 8% of the human genome comprises endogenous retroviruses (ERVs) (33, 59). These viruses infect germ lineage cells and thereby enter the genome of the host species. Thus, endogenous proviruses (the integrated form of retroviral DNA) are transmitted from parents to offspring in genomic DNA. If ERV genomes are intact, viral particles may be generated that can reinfect the germ line and form proviruses at new positions in the host genome. However, ERVs are subject to the same mutagenic processes over evolutionary time as any cellular gene. In the absence of selective pressure on the host to maintain intact viral genomes, endogenous retroviral proviruses accrue mutations over evolutionary time that inactivate viral infectivity. Most of the ERVs in the human genome have converted to solo long terminal repeats (solo LTRs), which are the product of homologous recombination between LTRs at the ends of the complete viral genome. Other types of mutations, such as nucleotide substitutions, insertions, and deletions, can also affect ERV proviruses, and many of the retroviral proviruses in the human genome have been inactivated by such mutations, which created premature stop codons or frameshifts in viral open reading frames (ORFs). The vast majority of the ERVs present in humans today (and perhaps all of them) have incurred mutations that inactivated viral infectivity.
One provirus that exists in the genome of approximately 20% of humans, human ERV K113 (HERV-K113, referred to here as K113), has full-length ORFs for all viral proteins (8, 63). However, this provirus does not appear to be infectious, as the pol and env genes of K113 do not support infectivity (9, 19, 20). K113 belongs to a subset of HERV-K called HML-2 (43). Since the human and chimpanzee lineages diverged about 6 million years ago (52), the only proviruses that entered the genome of the human lineage belong to this subgroup, although other members of this subgroup entered the germ line prior to the divergence of the human and chimpanzee lineages (8, 27, 44, 63). The human-specific proviruses of this subgroup are the most intact retroviruses in the human genome. Infectious HERV-K particles have been generated using two different approaches based on their DNA sequences. HERV-KCON (K-CON) was constructed based on the consensus sequence of human-specific HERV-K proviruses (34). Infectious HERV-K particles were also generated by combining pieces from three separate proviruses, HERV-K109 (K109) gag-pro, HERV-K115 pol, and HERV-K108 (K108) env (20). Thus, it may be that no single provirus is infectious, but recombination and/or genetic complementation among multiple genomic proviruses may be required to produce infectious HERV-K particles. This raises the questions of whether multiple functional HERV-K components exist in the human genome today and how close these components are to being able to form a functional viral genome that might be capable of reinfecting human cells.
To begin addressing these issues, we examined two of the full-length HERV-K gag genes that exist in the human genome today. Like all retroviruses, HERV-K contains the four genes necessary for viral replication: gag, pro, pol, and env. The human-specific HERV-K proviruses exist in two forms, type I and type II (38, 39). The type II proviruses contain gag, pro, pol, and env plus an accessory gene, rec, that encodes a protein (Rec) that functions in nuclear export of unspliced viral RNA in a manner analogous to that of human immunodeficiency virus type 1 (HIV-1) Rev (12, 40, 41, 65, 66). In type I proviruses, the pol and env genes are fused in frame by a 292-bp deletion that includes the first coding exon of rec, and the viruses encode an additional protein called Np9 (5, 48). The gag genes are relevant to whether HERV-K components in the human genome today might form an infectious virus, as the Gag protein is sufficient to produce virus-like particles (VLPs) in the absence of other viral proteins (4). Formation of such particles is an essential step for subsequent viral replication. Therefore, we decided to investigate whether Gag proteins from K113 and a second provirus, HERV-K101 (K101), are active in functional assays.
MATERIALS AND METHODS
Plasmid engineering and cells.
Plasmids used in this study were generated by standard cloning techniques and PCR amplification. In every instance where PCR amplification was used, the stretches of the resulting plasmids that were obtained by PCR amplification were completely sequenced to confirm the absence of PCR-induced mutations. Plasmids for expression of Gag proteins from various HERV-K proviruses were derived from the vector pCRU5-X-HA (Fig. 1A). pCRU5-X-HA was derived from an expression plasmid for an infectious molecular clone of Jaagsiekte sheep retrovirus (JSRV) clone 21 (49). Initial studies using other expression plasmids had failed to yield detectable gag RNA. In pCRU5-X-HA, gag, pro, and pol have been deleted and replaced by a convenient multiple cloning site between the major splice donor and the env splice acceptor of JSRV (49). Plasmids pCRU5-K101, pCRU5-K108, pCRU5-K113, and pCRU5-CON were generated by PCR amplification of the gag genes from cognate proviruses. K108 and K113 gag genes were amplified from bacterial artificial chromosome (BAC) templates RP11-169N2 and RP11-398B1, respectively. BACs were obtained from BacPac Resources. K101 gag was amplified from the plasmid pcGU1 (13). The K-CON gag was amplified from the plasmid template CHKCG (34). The resulting 2.0-kb amplicons were digested with the restriction enzymes EcoRI and HindIII and cloned into the pCRU5 plasmid at the corresponding sites.
FIG. 1.

Structures of the HERV-K Gag plasmids and positions of the individual HERV-K Gag mutations. (A) pCRU5-X-HA contained JSRV sequences, including the viral LTRs and env gene. The U3 region of the 5′ LTR was replaced with the CMV IE promoter to drive expression in mammalian cells. pCRU5-HERV-K gag was generated by cloning a HERV-K gag cDNA isolated from MGCT cells (13) into pCRU5-X-HA using the EcoRI (R) and HindIII (H) sites. The cloned HERV-K gag was sequenced and identified as K101 gag. K-CON, K108, and K113 gag genes were also cloned into this vector at the same positions. XbaI (X) and Nru I (N) sites were used to delete almost all of JSRV env from upstream of the start codon, up to and including the aspartate residue at position 577 (D577), to create pCRU5-K101 Δenv. pCRU5-K101-FS env was engineered by introducing a frameshift mutation (*) at the XmaI (M) site located at the alanine residue at position 207 (A207) in JSRV env. (B) Amino acid sequences of K101, K108, and K113 Gag proteins are aligned with the K-CON Gag sequence. The amino acid changes relative to the consensus Gag are indicated wherever one of the genomic proviral Gag proteins differs. The positions of the individual differences are also indicated. The corresponding amino acids for each position are also shown for K-CON. Amino acid variants that are shared among multiple HERV-K proviruses (shared polymorphisms) are underlined. The remaining amino acids are found only in the provirus indicated and not in any other HERV-K proviruses in the human genome. The bracket at the bottom shows the position of CA in Gag based on similarity to MPMV CA. (C) The infectious CHKCG plasmid (34) is composed of the HERV-K consensus sequence with the U3 region of the 5′ LTR replaced by the CMV IE promoter to drive expression in mammalian cells. The env gene was disrupted with a CMV IE-GFP cassette, resulting in a nonfunctional env. The SalI (S) and the PshAI (P) restriction sites used to clone K101 and K113 gag into CHKCG are indicated.
Recombinant plasmids were engineered between pairs of viruses by digesting pCRU5-K101 or pCRU5-K113 with either EcoRI plus EcoRV, EcoRV plus PstI, or PstI plus HindIII to divide gag into three fragments. These fragments were then ligated into the corresponding plasmid digested with the same enzymes to generate recombinant Gag expression plasmids.
CHKCG is a plasmid that contains a gag-pro-pol segment of HERV-K based on the consensus sequence (K-CON) of human-specific HERV-K proviruses (34). To engineer variants containing the gag genes of specific proviruses (CHKCG-K101 and CHKCG-K113), pBluescript II SK(+) was digested with SalI and XhoI and religated to mutate the SalI site, creating pBlue-ΔSal. CHKCG was then digested with ClaI and XbaI, and the resulting 6.5-kb fragment was gel purified and cloned into pBlue-ΔSal, creating pBlue-ΔSal-CHKCG C/X. K101 and K113 gag genes were PCR amplified using the cosmid c48A12 and BAC RP11-398B1, respectively, as templates. Each 2.2-kb amplicon was cloned into the plasmid pCR2.1-TOPO (Invitrogen). Following sequencing, each pCR2.1-TOPO clone was digested with SalI and PshAI, and the 1.9-kb fragments were gel purified and subcloned into the compatible sites in pBlue-ΔSal-CHKCG C/X. Finally, each pBlue-ΔSal-CHKCG C/X-subcloned gag was digested with ClaI and XbaI and cloned into CHKCG digested with the same enzymes.
A mutant of CHKCG containing a substitution of a methionine codon for an isoleucine codon at Gag amino acid position 516 (K-CON I516M) was generated by first digesting another plasmid that contains the consensus HERV-K gag-pro-pol, pCRVI/Gag-Pro-Pol (34). This plasmid contains two BssHII restriction sites and was first modified by digesting it with BssHII to delete the small BssHII fragment and religating it to make the BssHII site unique. The Gag I516M mutation was introduced by standard PCR mutagenesis techniques. The resulting 2.2-kb mutagenized PCR product was digested with BssHII and PshAI and cloned into the compatible sites in pCRVI/Gag-Pro-Pol, creating pCRVI/I516M. Finally, the 4.9-kb EcoRV-XbaI fragment of pCRVI/I516M was isolated and cloned into the corresponding sites in CHKCG. The resulting plasmid is identical to CHKCG except for the single base pair change encoding methionine instead of isoleucine, a change of ATC to ATG.
pcDNA3.1/VSV-G was generated by PCR amplification of the vesicular stomatitis virus glycoprotein (VSV-G) from the pVSV-G vector (Stratagene). The resulting 1.5-kb PCR product was cloned into the pCR2.1-TOPO vector (Invitrogen). After sequencing, VSV-G was cloned out of pCR2.1-TOPO using the restriction enzyme EcoRI and cloned into the compatible sites in pcDNA3.1/Zeo (Invitrogen).
pCRU5-K101/Δenv was created by digesting pCRU5-K101 with XbaI and NruI, followed by filling in the cohesive ends with the Klenow fragment of DNA polymerase I and religating, resulting in the deletion of the JSRV env initiation codon up to and including an aspartate codon encoding Env amino acid 577.
pCRU5-K101/FS env was generated by digesting pCRU5-K101 with XmaI, treating the ends with Klenow fragment, and religating, resulting in a 4-bp frameshift mutation immediately following an alanine codon at the location corresponding to Env amino acid 207.
HEK-293T (293T) cells were grown at 37°C, 5% CO2, and 95% humidity in Dulbecco's modified Eagle medium with 5% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. HeLa and Cos cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum at 37°C, 5% CO2, and 95% humidity.
Production of HERV-K CA antibodies.
A polyclonal Gag antiserum was produced by immunizing rabbits with a recombinant protein (expressed in bacteria) of the major capsid protein (CA) of K101 (Proteintech).
Viral particle production assays.
For each pCRU5-HERV-K gag expression vector, 2 μg or 10 μg was transfected into 293T cells using Opti-Mem and Lipofectamine 2000 (Invitrogen), and particle production was assessed by Western blotting. Culture supernatants were collected at various times, passed through 0.22-μm-pore-size filters, and centrifuged at 14,000 × g for 60 min at 4°C. Pelleted particles were resuspended with 50 μl of lysis buffer containing 200 mM Tris, 2.5% sodium dodecyl sulfate (SDS), 22.5% glycerol, 0.6 mg/ml bromphenol blue, and 100 mM dithiothreitol. Cell lysates were prepared 48 h posttransfection by lysing the cells in 100 μl of the same lysis buffer. All samples were incubated at 100°C for 5 min and then separated by SDS-polyacrylamide gel electorphoresis using NuPAGE Novex bis-Tris gels (Invitrogen). Proteins were then transferred to nitrocellulose membranes and incubated in blocking buffer overnight at 4°C. The membranes were then incubated with the rabbit antiserum directed against HERV-K Gag for 1 h at room temperature. After washing with a phosphate-buffered saline-0.1% Tween-20 solution, the membranes were incubated with a goat anti-rabbit immunoglobuline G (IgG) antibody conjugated to horseradish peroxidase (HRP) for 1 h at room temperature. After washing, the blots were visualized using SuperSignal West Pico chemiluminescent substrate (Pierce). Western blot analyses were also performed in parallel with infectivity experiments using the CHKCG vectors.
Confocal microscopy.
Experiments were performed in HeLa, Cos, and 293T cells grown in two-well chambered glass slides and transfected with HERV-K Gag plasmids using Lipofectamine (Invitrogen) according to the manufacturer's instructions. Cells were fixed 24 h posttransfection with methanol for 5 min at −20°C and processed essentially as already described (7, 47). HERV-K Gag was detected using the polyclonal rabbit antibody described above. Goat anti-rabbit IgG conjugated with Alexa Fluor 488 (Molecular Probes) was used as the secondary antibody. Slides were mounted with medium containing DAPI (4′,6-diamidino-2-phenylindole; Vectashield; Vector Laboratories) and analyzed using a Leica TCS SP2 confocal microscope. In experiments to assess protein distribution, the pattern of Gag staining was recorded as accumulating primarily (i) at the plasma membrane, (ii) in the cytoplasm, or (iii) at both the cytoplasm and the plasma membrane when accumulation near the plasma membrane was clear. For each viral Gag tested, at least 100 Gag-positive cells in random fields were counted and scored in two independent experiments in HeLa, Cos, and 293T cells.
Infectivity experiments.
293T cells were seeded in six-well plates at 5.0 × 105 cells per well 16 h prior to transfection. Plasmids were transfected as indicated in the figure legends in the following amounts based on the results of Lee and Bieniasz (34): 1.3 μg of CHKCG, CHKCG-K101, CHKCG-K113, or CHKCG-I516M, 0.5 μg of pCR3.1/Rec, and 0.2 μg of pcDNA3.1/VSV-G. At 24 h posttransfection, the medium was replaced with medium containing 5 mM sodium butyrate and incubated overnight. Producer cell supernatants were harvested and passed through 0.22-μm filters. 293T target cells were seeded in six-well plates at 1.0 × 105 cell per well and incubated overnight. Then, 1 ml of freshly filtered supernatant was applied to each well of target cells with 5 μg/ml Polybrene and incubated for 24 h. Green fluorescent protein (GFP)-positive foci were counted 48 to 72 h postinfection to quantify infectious events.
Generation of phylogenetic trees.
Trees were generated using full-length CA amino acid sequences or the C-terminal 22 amino acids. Sequences were aligned using Muscle (22) with parameters set to defaults. Trees were generated using the maximum parsimony method in PAUP, version 4B10 (61). Bootstrapping support values were derived from 10,000 replications.
RESULTS
VLP production by HERV-K gag genes.
To begin assessing how many functional HERV-K gag genes exist in the human genome, those from three proviruses, K101, K108, and K113, were tested for the ability to generate VLPs by cloning them into an expression vector (Fig. 1A). The proviruses used in this study belong to a set of full-length proviruses within the HML-2 subgroup of HERV-K (43) and have very similar sequences, as exemplified by the viral Gag proteins (Fig. 1B). Nonetheless, each provirus has various nucleotide substitutions within its genome (Fig. 1B). Some of these mutations are shared among multiple HERV-K proviruses in the human genome and are referred to here as shared polymorphisms. Others are unique to individual proviruses and represent mutations that occurred either (i) during the transcription or reverse transcription events of the replication cycle that generated a particular provirus, (ii) in the most immediate progenitor proviruses (and such proviruses for various possible reasons are not in the human genome today), or (iii) over evolutionary time subsequent to the formation of the cognate proviruses.
We cloned specific HERV-K gag genes into a JSRV-based expression vector in order to test their ability to generate VLPs (Fig. 1A). In this vector, the U3 region of the JSRV 5′ LTR was replaced by the cytomegalovirus immediate early (CMV IE) promoter to drive transcription in mammalian cells. The gag genes of K101, K108, K113, and the consensus Gag K-CON were inserted into this plasmid. The K101 Gag was derived from a plasmid containing a HERV-K gag cDNA from RNA expressed in a human male germ cell tumor (MGCT) (13). MGCTs frequently produce high levels of HERV-K particles (14, 17, 37, 38). Since the specific individual provirus(es) encoding these particles had not been identified, we sequenced this HERV-K gag gene and identified it based on unique mutations as K101, a human-specific type I provirus located on chromosome 22 (8). Thus, K101 is responsible for at least part of the production of the viral particles observed in human MGCTs. Each pCRU5-HERV-K gag vector was transfected into HEK-293T (293T) cells and assayed for cellular gag expression and viral particle release into the culture supernatants by Western blotting using a rabbit polyclonal anti-HERV-K CA serum. All the viral gag genes yielded a 75-kDa Gag precursor protein that accumulated within cells (Fig. 2A). However, the levels of VLPs released from the cells varied widely. K-CON, K101, and K108 Gag proteins were able to undergo particle assembly and release from the transfected cells. Although K113 Gag protein was expressed in the transfected cells, only very low levels of VLPs were detected in the culture supernatants, suggesting a defect in K113 particle assembly and/or release (Fig. 2A).
FIG. 2.

Expression of HERV-K Gag proteins and viral particle production. (A) Western blot analysis of 293T cells transfected with 2 μg of pCRU5 vector containing K-CON, K101, K108, or K113 was performed on both culture supernatants and whole-cell lysates (Cells) 36 h posttransfection. Proteins were separated by SDS-polyacrylamide gel electrophoresis, and the approximately 75-kDa unprocessed Gag product was visualized using a rabbit antiserum directed against HERV-K Gag, a secondary anti-rabbit-IgG-HRP antibody, and electrochemiluminescence. (B) Western blot analysis of 293T cells transfected with 10 μg of pCRU5 containing K101, K108, or K113 gag was performed on both cultured supernatants and whole-cell lysates (Cells) 48 h posttransfection. (C) Western blot analysis of 293T cells transfected with 2 μg of pCRU5 containing K-CON, K101, K108, or K113 gag, incubated at either 28°C or 37°C, was carried out 48 h posttransfection. (D) Western blot analysis of 293T cells transfected with 2 μg of pCRU5 containing K-CON, K101/Δenv, or K101/FS env was carried out 24 h and 48 h posttransfection.
Recently, it was reported that K113 Gag is capable of producing VLPs in a baculovirus vector system using Sf9 cells (15). As baculovirus vectors drive extremely high levels of protein expression, we tested whether increasing the amount of pCRU5-K113 vector DNA allowed VLP production. Transfection of 10 μg of pCRU5-K113 DNA did allow some production of particles at a lower level than observed with either K101 or K108 (Fig. 2B). As insect cells are generally grown at 28°C, we also tested the possibility that the mutations unique to K113 blocked VLP production specifically at higher temperatures, but we found K113 Gag to be defective in particle production at 28°C (Fig. 2C). K101 and K108 Gag proteins each yielded VLPs at 28°C (Fig. 2C). Thus, it appears that K113 Gag is essentially defective in VLP production, although when expressed at very high levels, it is able to release some particles into the supernatant.
Confocal microscopy experiments were used to address the defect in K113 Gag further (Fig. 3). HERV-K was reported to assemble at the cell membrane, unlike other betaretroviruses, including Mason-Pfizer monkey virus (MPMV) and JSRV, which assemble in the cytoplasm in the pericentrosomal region (20, 34, 47, 57). HeLa, Cos, and 293T cells expressing K101 or K-CON generally exhibited Gag staining that was diffuse or dispersed in the cytoplasm, with some cytoplasmic puncta visible, and a minority of the cells showed Gag puncta at the plasma membrane. With K113, a higher percentage of cells showed accumulation of viral Gag protein at the plasma membrane. These results are consistent with the interpretation that K113 Gag can traffic to the cell membrane but then accumulates there and is not able to exit the cell.
FIG. 3.

K113 Gag proteins accumulate at the plasma membrane. (A) Representative example of staining pattern of cells expressing K-CON and K-113 Gag. Cells were fixed at 24 h posttransfection and processed as described in Materials and Methods. HERV-K Gag is in green, while nuclei (DAPI) are in blue. Gag is visible as dispersed dots in the cytoplasm (Cyt) or in the cytoplasm and the plasma membrane (Cyt+PM) of transfected cells. The majority of cells expressing K113 show accumulation of Gag staining at the plasma membrane. Panels a to f show representative examples of experiments performed in Cos cells, while panels g and h show 293T cells. Bars, 10 μm. (B) Quantitation of Gag staining pattern in confocal microscopy of HeLa, Cos and 293T cells expressing K-CON, K-101, and K-113 Gag at 24 h posttransfection. The graph represents the number of cells in which Gag proteins display a staining dispersed in the cytoplasm (Cyt) versus cells showing both cytoplasmic and intense plasma membrane staining (PM+Cyt), as shown in panel A. Approximately 100 cells were randomly chosen in each experiment. Data are averages from two independent experiments.
The pCRU5 vector used in these experiments contained a functional JSRV env gene. We tested whether this was important for the production of HERV-K VLPs. First, almost all of JSRV env, including the methionine initiation codon, was deleted in pCRU5-K101, creating pCRU5-K101/Δ env (Fig. 1A). We were unable to detect expression of K101 Gag within cells following transfection of this construct (Fig. 2D). This suggested that JSRV env sequences might be important for Gag production. To test whether the viral Env protein was important for HERV-K Gag expression, another construct was generated with a 4-bp frameshifting insertion 207 amino acids downstream of the start codon of JSRV env (Fig. 1A). Transfection of the plasmid containing this mutation resulted in levels of HERV-K Gag expression and VLP detection in the supernatants similar to that of K101 gag in pCRU5 with the intact env gene (Fig. 2D). Thus, full-length Env protein is not necessary for HERV-K Gag particle assembly and cellular egress in these assays; rather, another element that does not require full-length Env protein expression is present in the JSRV vector. In another study, we discovered that the leader sequence of the JSRV Env (positioned in the N-terminal 80 amino acid residues of Env) acts as a Rev-like protein facilitating mRNA export by interacting with a RNA response element situated in the 3′ end of the JSRV env (M. Caporale, F. Arnaud, M. Mura, C. Murgia, and M. Palmarini, unpublished data). Thus, Gag expression in the context of the pCRU5 constructs is dependent on expression of the JSRV leader sequence.
K101 and K113 gag genes in infectivity assays.
The ability of the K101 and K113 gag genes to support viral replication was tested using a recently generated plasmid that contained the complete genome of consensus sequence HERV-K, except that the U3 of the 5′ LTR was replaced by the CMV IE enhancer-promoter, and a GFP expression cassette was inserted into env, disrupting this ORF (34) (Fig. 1C). The activity of the HERV-K U3 enhancer-promoter is very low in many human cell lines, and the use of the CMV element allows expression in many cell types (53). Transient cotransfection of this K-CON gag-pro-pol plasmid plus two additional plasmids, one encoding VSV-G as a viral envelope protein and the other encoding HERV-K Rec protein to promote nuclear export of the unspliced gag-pro-pol RNA, yields virus that infects 293T cells (34).
The K101 gag gene could support viral replication in place of the consensus sequence of K-CON, although to lower levels (Fig. 4A). The level of infectivity seen with K-CON in different experiments was in the range of 1 × 102 to 4 × 102 infectious units/ml, which is similar to that reported previously (20, 34). In contrast, no infections were observed with K113, which was consistent with the lack of VLP release observed in Fig. 2. Western blot analysis of the cells and particles in the culture supernatants from the same experiments indicated that the K101 gag yielded lower levels of virus particles than K-CON gag (Fig. 4B). The viral Gag protein was processed by the viral protease to a CA protein of approximately 30 kDa in K-CON and K101. No mature CA was detected in lysates of cells expressing K113. These results are consistent with the decreased levels of virus particle production at least in part accounting for the lower infectious activities of the two gag genes from proviruses in the human genome.
FIG. 4.

Infectivity and virus production by K101, K108, and K113 Gag proteins. (A) Infectious titers of HERV-K particles produced by transfection of 293T cells with Rec, VSV-G, and the indicated CHKCG-gag vectors. Producer cell supernatants were used to infect 293T target cells 48 h posttransfection, and GFP foci were counted 72 h postinfection and expressed as infectious units per milliliter (IU/ml). The data are means plus standard deviations (n = 3 [K101 and K113] or 2 [K-CON]). (B) Western blot analysis was performed on the producer cells used for panel A. Supernatants were collected, and whole-cell lysates were prepared from the producer cells at the time of target cell infection (48 h posttransfection). The approximately 75-kDa Gag precursor and 30-kDa processed Gag (CA) proteins are indicated. Only the processed form of Gag was detected in the culture supernatants.
The K113 Gag defect is due principally to a single amino acid substitution.
K113 gag encodes seven amino acid differences compared to the K-CON sequence (Fig. 1). The two closest to the N terminus, a lysine at position 118 and a serine at position 129, are shared with other human-specific HERV-Ks, including K101, and were thus considered highly unlikely to account for the K113 Gag defect. There are five additional amino acid substitutions that are unique to K113 (Fig. 5). Gag chimeras between K101 and K113 were constructed to identify which determined the K113 Gag defect (Fig. 5). By using convenient restriction sites in the pCRU5-HERV-K gag vectors, the K113 gag gene was divided into three sections, and six recombinants were generated (Fig. 5A). Those that contained the C-terminal third of gag from K113 were defective for VLP production, while those with that region from K101 generated particles (Fig. 5B). The only mutation in K113 Gag in this segment was a substitution of methionine for isoleucine at Gag amino acid 516 (I516M). Thus, this single amino acid substitution in K113 Gag was responsible for the defect in VLP production.
FIG. 5.

Genetic mapping of the K113 defect in viral particle production. (A) K101 and K113 Gag proteins are depicted to indicate the amino acid residues at which they differ from the K-CON Gag sequence. Underlined amino acids represent shared polymorphisms among HERV-K Gag proteins. The unique mutations in K113 Gag are indicated by shaded boxes. The restriction sites for EcoRI (R), EcoRV (V), PstI (P), and HindIII (H) were used to generate the recombinants between K101 gag and K113 gag that are shown. The bracket at the bottom shows the position of CA in Gag based on similarity to MPMV CA. (B) Western blot analysis of 293T cells transfected with 2 μg of K101, K113, or the indicated recombinant gag pCRU5 vectors was performed on both cultured supernatants and whole-cell lysates (Cells) 24 h posttransfection. The approximately 75-kDa Gag unprocessed HERV-K Gag product was visualized using a rabbit antiserum directed against HERV-K Gag, a secondary anti-rabbit-IgG-HRP antibody, and enhanced chemiluminescence.
The importance of the I516M substitution was also tested in infectivity assays. A single nucleotide was altered in the K-CON consensus HERV-K gag-pro-pol plasmid to place this mutation into an otherwise isogenic background. No infections were detected with this single amino acid change (Fig. 6A). Western blotting confirmed the lack of viral particles release by K-CON I516M Gag and the failure of this Gag precursor to be processed into mature CA (Fig. 6B). Moreover, when mixed with an equal amount of the infectious K-CON plasmid, I516M caused a modest reduction in the production of virus particles and reduced activity by about 70% in the infectivity assay (Fig. 6).
FIG. 6.

Infectivity and particle production of HERV-K viruses containing the K113 Gag I516M mutation. (A) Infectious titers of HERV-K particles produced by transfection of 293T cells with Rec, VSV-G, and the indicated CHKCG vectors. Producer cell supernatants were used to infect 293T target cells 48 h posttransfection, and GFP foci were counted 72 h postinfection and expressed as infectious units per milliliter (IU/ml). The data are means plus standard deviations (n = 4). (B) Western blot analysis of the producer cells used for panel A. Supernatants were collected, and whole-cell lysates were prepared at the time of target cell infection (48 h posttransfection). The approximately 75-kDa Gag precursor and 30-kDa processed CA proteins are indicated. Only the processed form was detected in the culture supernatants.
DISCUSSION
The Gag protein is the key structural component responsible for the production of retroviral particles. The data presented here show that one of the HERV-K proviruses in the human genome, K101, contains a gag gene that can support viral particle production and infectivity. The gag gene of K109 is also active (19). Thus, at least two HERV-K gag genes in the human genome today might be capable of contributing to virus particle production and replication. HERV-K particles are frequently detectable in human MGCTs (14, 17, 38). The results obtained here with a cDNA clone from a MGCT show that K101 can contribute to production of these particles. Others have shown that K101 and K108 are transcribed in MGCTs (23, 54), and RNAs from these proviruses are packaged into particles produced in MGCTs (54). HERV-K particle production has also been reported to occur in normal human placenta (21, 29, 58), and K101, K108, and K109 are candidates for encoding those particles.
Mutations in HERV-K proviruses.
The gag genes of K101 and K113 yielded about fourfold less infectious virus than the consensus sequence gag gene. K113 gag did not yield infectious particles. Thus, each of the gag genes tested had mutations that reduced the Gag function to a lesser or greater extent. These mutations could have occurred over evolutionary time after the formation of the cognate provirus. Alternatively, they could have occurred during the viral replicative cycle that generated a particular provirus, or they could have occurred in immediate predecessors of the provirus that for various possible reasons are not in the human genome today.
Some of the mutations are shared polymorphisms that are present in multiple proviruses. Positions 118 and 129 of the gag genes are such mutations (Fig. 1B). K101 and K113 share a lysine and serine, respectively, at these positions (here termed the KS shared polymorphism type), as do additional HERV-K proviruses in the human genome. Two of the proviruses, one on chromosome 10 and another on chromosome 12, are also present at the orthologous positions in the genomes of chimpanzees and gorillas. Thus, these amino acids were present in Gag proteins of ancestral HERV-K that was active prior to the divergence of the gorilla and human-chimpanzee lineages. K108 Gag has an asparagine and a glycine at these positions (here called the NG shared polymorphism type). These two amino acids are also encoded by other human-specific HERV-K proviruses and were chosen for the generation of the consensus HERV-K sequence in K-CON (Fig. 1B) (34). They are best explained as shared derived differences among a subset of the human-specific HERV-K proviruses, and thus, there was a common ancestral origin (identity by descent) for this portion of gag in those proviruses.
Each provirus also contains unique mutations within the gag genes, five in K113 and three each in K101 (Fig. 1B). The genetic analyses performed here showed that only a subset of these may account for the defects in VLP production and infectivity. The unique I516M change in K113 was the key mutation in this provirus. In contrast, both the ancestral KS and the newer NG shared polymorphism types supported VLP production and viral infectivity in K101 and K-CON, respectively. This is consistent with the interpretation that the shared polymorphisms were more likely to have survived selective pressure for viral fitness in the past.
Implications for examining HERV-K in the human population.
It remains uncertain whether infectious HERV-K proviruses exist in the human population today (10). If the unique mutations occurred over evolutionary time subsequent to the formation of the proviruses in the genome of the human lineage, then they might not be uniform in the human population. For a population with the size and generation time of ancestral humans, two alleles with approximately neutral fitness could have persisted in the genome for only roughly one million years before one allele would have become fixed due to genetic drift (18, 50, 55, 62). The I516M mutation that inactivated K113 gag was a single nucleotide substitution, ATC to ATG. If that change occurred during the most recent one million years or so, it is possible that some humans do not have it. If the total number of mutations that inactivates any specific provirus is actually low, then it is possible that some individuals have proviruses that lack any inactivating mutations. As DNA sequencing technology rapidly progresses, the complete genome sequences of many individual humans will become available. Mapping the defects in HERV-K proviruses as was done here will allow identification of the key polymorphisms that need to be analyzed in genetically diverse humans in the search for an infectious ERV within the human population.
Effect of K113 gag on the consensus gag.
At least under certain circumstances, ERVs with defects caused by mutation can act to block infection by an exogenous virus, and this may be an advantage that ERVs might confer on a host species (28). The murine Fv1 gene is the classic example of this, where the gag remnant of an ancient provirus can inhibit infection by gammaretroviruses (11, 36). Another example is endogenous betaretroviruses of sheep (endogenous JSRVs), which can interfere with Gag trafficking and viral exit of the related exogenous JSRV by the expression of a transdominant Gag. Endogenous JSRVs with dominant negative properties have been positively selected in the domestic sheep and act as proper restriction factors (6, 7, 46). The partial reduction in production of infectious HERV-K particles caused by coexpression of K113 Gag is consistent with the possibility that such mutations in endogenous viruses might play a role in reducing the level of virus infection in a host.
Importance of the extreme C terminus of CA to HERV-K and its similarity to HIV-1.
All orthoretrovirus Gag proteins are cleaved by the viral protease into the mature components, including MA, CA, and NC. The CA protein is a crucial determinant of assembly of retroviral particles (16, 32, 51, 56, 60). Alignment of the amino acids around the position of the K113 I516M mutation with the Gag sequences of other betaretroviruses showed that this mutation is very close to the carboxyl terminus of the CA protein (Fig. 7A). The position of the cleavage generating the C terminus of MPMV was mapped to the C-terminal side of the alanine residue that is homologous to the alanine at position 527 in HERV-K Gag (26) and is shown in Fig. 7A. The cleavages to generate the C termini of HIV-1 CA (Fig. 7A) and other retroviruses likewise occur close to this position. The I516M mutation occurred 11 amino acids upstream of HERV-K Gag alanine 527 (Fig. 7A).
FIG. 7.
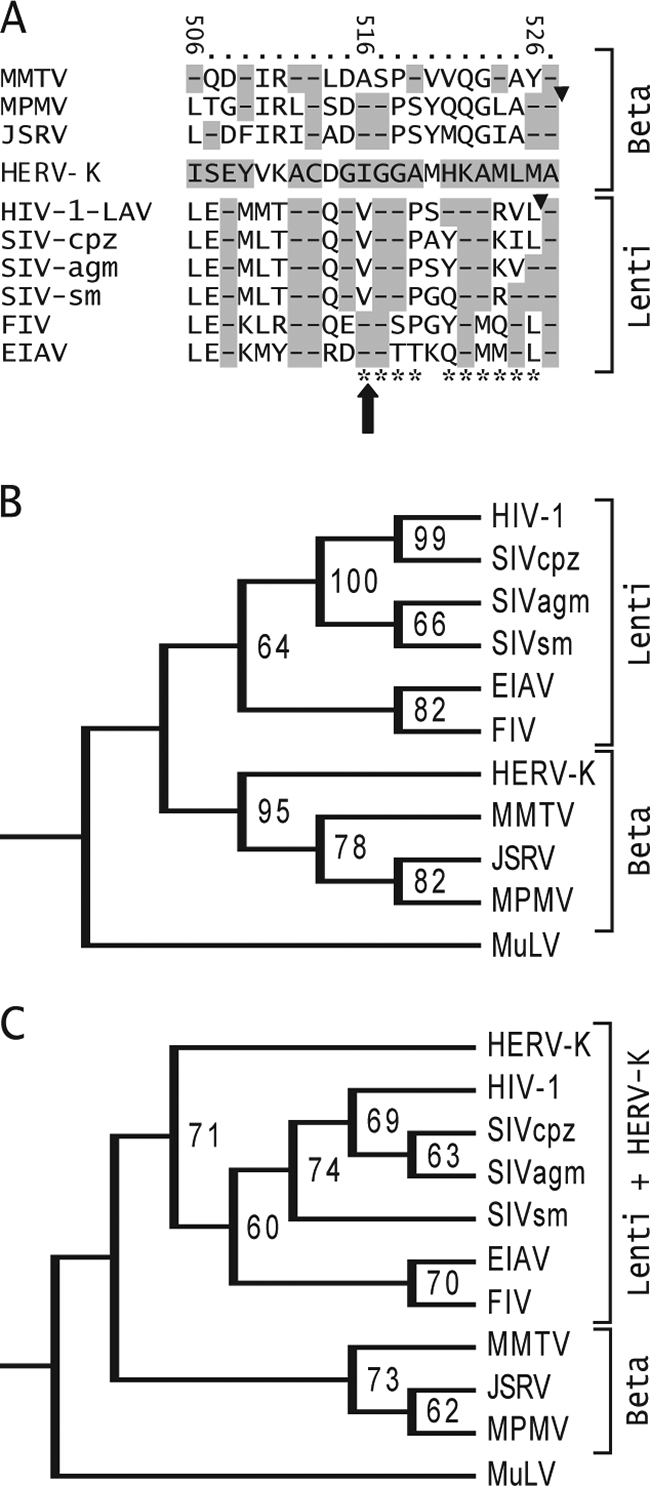
Similarity between the extreme C termini of HERV-K and other retroviruses. (A) Alignment of HERV-K Gag amino acids 506 to 527 with the corresponding Gag sequences of other retroviruses. The amino acids of HERV-K Gag are indicated, and the numbers at the top refer to position within HERV-K Gag. Other betaretroviruses are shown above HERV-K, and lentiviruses are shown below it. The large arrow shows the position of the I516M mutation unique to K113. Triangles show the positions of the protease cleavage sites that generate the C termini of MPMV and HIV-1 CA. Highlighted dashes indicate amino acid identity. The HIV-1 Gag amino acid positions identified by Melamed et al. to be highly detrimental to HIV-1 Gag particle production and infectivity are indicated by asterisks at the bottom (45). Retroviruses included in the alignment are as follows (GenBank accession numbers indicated): mouse mammary tumor virus (MMTV, AAF31467), MPMV (P07567), JSRV (CAA01899), HIV-1 LAV (AAB59747), simian immunodeficiency virus (SIV) from chimpanzee (SIV-cpz, AA013959), SIV from African green monkey (SIV-agm, BAF32563), SIV from sooty mangabey (SIV-sm, AAC68655), feline immunodeficiency virus (FIV, CAA48157), and equine anemia infectious virus (EIAV, AAA43011). (B) A phylogenetic tree of retrovirus CA amino acids sequences was generated using maximum parsimony in PAUP, version 4B10. Bootstrapping values were derived from 10,000 replications. The tree was rooted using the gammaretrovirus Moloney murine leukemia virus (AAB59942) as the outlier. (C) Tree generated using maximum parsimony on the amino acids at the extreme C terminus of CA. The sequences shown in panel A were used, along with the corresponding amino acids of Moloney murine leukemia virus (AAB59942) as the outlier.
Interestingly, the extreme C terminus of HIV-1 CA was shown to be very important for viral particle production and infectivity. Mutation of almost any of the amino acids at the extreme C terminus of HIV-1 CA substantially reduced viral particle release from cells and infectivity (45). These amino acids were among the sequences present within a minimal Gag construct of HIV-1 that was sufficient for assembly and release of Gag particles from cells (2). Amino acids in a segment extending from the extreme C terminus of HIV-1 CA to the immediate downstream SP1 peptide were crucial for viral particle maturation (1, 24, 35, 45). Amino acids spanning CA to SP1 also appear to be the target for a potent inhibitor of HIV-1 particle maturation (3, 30, 42, 67-70). The spacer region immediately downstream of the CA C terminus is also crucial for formation of bovine immunodeficiency virus and Rous sarcoma virus particles (25, 31). Alignment of the amino acid sequences of HIV-1 Gag and those of other lentiviruses with that of HERV-K Gag showed considerable identity and similarity in this region of CA (Fig. 7A). Isoleucine 516 of HERV-K corresponds to a valine residue in HIV-1 and other primate immunodeficiency viruses (Fig. 7A). Mutation of this valine to alanine reduced HIV-1 particle formation and infectivity about 30-fold (45). The I516M in HERV-K yielded equivalent effects. Although the precise structure of the extreme C terminus of HIV-1 CA and the role of this domain in particle assembly and maturation are unclear, it is hypothesized to function as a linker between the rest of CA and the immediate downstream SP1 region (64). The latter is hypothesized to be structured within a concentric layer beneath the rest of CA in immature particles and then lost as particles mature (64). The equivalent region is also important for assembly of RSV (31). The large reduction in particle release and Gag precursor processing caused by mutation of the extreme C terminus of HERV-K CA as well as HIV-1 CA suggests that this segment might play a general role in retroviral particle formation.
Phylogenetic comparison of the full-length HERV-K CA protein to those of other betaretroviruses and lentiviruses showed that it was more similar to other betaretroviruses, as expected (Fig. 7B). Curiously, inspection of the sequences at the extreme C terminus (Fig. 7A) showed positions where HERV-K had identity to lentiviruses but not betaretroviruses (HERV-K Gag amino acids E508, G515, G518, K522, A523, and L525), including three (G515, G518, and A523) where HERV-K matched all four primate immunodeficiency viruses analyzed and not the other lentiviruses or betaretroviruses. Comparison of the extreme C-terminal sequences by the maximum parsimony method supported the similarity of HERV-K to lentiviruses (Fig. 7C). This similarity to primate lentiviruses may be just coincidental, although it can be speculated that it might not be. If not, one possible explanation might be convergent evolution of CA termini of HERV-K and the primate immunodeficiency viruses (but not MPMV), and a less likely possibility is an ancient recombination event in this portion of Gag between an ancestor of the primate immunodeficiency viruses and an ancestor of the HERV-Ks.
Acknowledgments
This work was supported by NIH grant CA044822 and a program grant from the Welcome Trust. D.H. was supported by NIH training grant GM07128. DNA sequencing was supported in part by NIH Cancer Center Grant CA013330. M.P. is a Wolfson-Royal Society research merit awardee.
We thank Young Nam Lee and Paul Bieniasz for the gifts of the CHKCG, pCR3.1/Rec and pCRVI/G-PR-Pol plasmids and Ralf Tonjes for plasmid pCGU1. We thank Larry Herbst for help with Western blots.
Footnotes
Published ahead of print on 12 November 2008.
REFERENCES
- 1.Accola, M. A., S. Hoglund, and H. G. Gottlinger. 1998. A putative alpha-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type 1 Gag precursor is crucial for viral particle assembly. J. Virol. 722072-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Accola, M. A., B. Strack, and H. G. Gottlinger. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 745395-5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adamson, C. S., S. D. Ablan, I. Boeras, R. Goila-Gaur, F. Soheilian, K. Nagashima, F. Li, K. Salzwedel, M. Sakalian, C. T. Wild, and E. O. Freed. 2006. In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (Bevirimat). J. Virol. 8010957-10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamson, C. S., and E. O. Freed. 2007. Human immunodeficiency virus type 1 assembly, release, and maturation. Adv. Pharmacol. 55347-387. [DOI] [PubMed] [Google Scholar]
- 5.Armbruester, V., M. Sauter, E. Krautkraemer, E. Meese, A. Kleiman, B. Best, K. Roemer, and N. Mueller-Lantzsch. 2002. A novel gene from the human endogenous retrovirus K expressed in transformed cells. Clin. Cancer Res. 81800-1807. [PubMed] [Google Scholar]
- 6.Arnaud, F., M. Caporale, M. Varela, R. Biek, B. Chessa, A. Alberti, M. Golder, M. Mura, Y. P. Zhang, L. Yu, F. Pereira, J. C. Demartini, K. Leymaster, T. E. Spencer, and M. Palmarini. 2007. A paradigm for virus-host coevolution: sequential counter-adaptations between endogenous and exogenous retroviruses. PLoS Pathog. 3e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnaud, F., P. R. Murcia, and M. Palmarini. 2007. Mechanisms of late restriction induced by an endogenous retrovirus. J. Virol. 8111441-11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbulescu, M., G. Turner, M. I. Seaman, A. S. Deinard, K. K. Kidd, and J. Lenz. 1999. Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans. Curr. Biol. 9861-868. [DOI] [PubMed] [Google Scholar]
- 9.Beimforde, N., K. Hanke, I. Ammar, R. Kurth, and N. Bannert. 2008. Molecular cloning and functional characterization of the human endogenous retrovirus K113. Virology 371216-225. [DOI] [PubMed] [Google Scholar]
- 10.Belshaw, R., V. Pereira, A. Katzourakis, G. Talbot, J. Paces, A. Burt, and M. Tristem. 2004. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 1014894-4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Best, S., P. Le Tissier, G. Towers, and J. P. Stoye. 1996. Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 382826-829. [DOI] [PubMed] [Google Scholar]
- 12.Bogerd, H. P., H. L. Wiegand, J. Yang, and B. R. Cullen. 2000. Mutational definition of functional domains within the Rev homolog encoded by human endogenous retrovirus K. J. Virol. 749353-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boller, K., O. Janssen, H. Schuldes, R. R. Tonjes, and R. Kurth. 1997. Characterization of the antibody response specific for the human endogenous retrovirus HTDV/HERV-K. J. Virol. 714581-4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boller, K., H. Konig, M. Sauter, N. Mueller-Lantzsch, R. Lower, J. Lower, and R. Kurth. 1993. Evidence that HERV-K is the endogenous retrovirus sequence that codes for the human teratocarcinoma-derived retrovirus HTDV. Virology 196349-353. [DOI] [PubMed] [Google Scholar]
- 15.Boller, K., K. Schonfeld, S. Lischer, N. Fischer, A. Hoffmann, R. Kurth, and R. R. Tonjes. 2008. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J. Gen. Virol. 89567-572. [DOI] [PubMed] [Google Scholar]
- 16.Bowzard, J. B., J. W. Wills, and R. C. Craven. 2001. Second-site suppressors of Rous sarcoma virus Ca mutations: evidence for interdomain interactions. J. Virol. 756850-6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bronson, D. L., E. E. Fraley, J. Fogh, and S. S. Kalter. 1979. Induction of retrovirus particles in human testicular tumor (Tera-1) cell cultures: an electron microscopic study. J. Natl. Cancer Inst. 63337-339. [PubMed] [Google Scholar]
- 18.Chen, F. C., and W. H. Li. 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum Genet. 68444-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dewannieux, M., S. Blaise, and T. Heidmann. 2005. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses. J. Virol. 7915573-15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dewannieux, M., F. Harper, A. Richaud, C. Letzelter, D. Ribet, G. Pierron, and T. Heidmann. 2006. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 161548-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dirksen, E. R., and J. A. Levy. 1977. Virus-like particles in placentas from normal individuals and patients with systemic lupus erythematosus. J. Natl. Cancer Inst. 591187-1192. [DOI] [PubMed] [Google Scholar]
- 22.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 321792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flockerzi, A., A. Ruggieri, O. Frank, M. Sauter, E. Maldener, B. Kopper, B. Wullich, W. Seifarth, N. Muller-Lantzsch, C. Leib-Mosch, E. Meese, and J. Mayer. 2008. Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genomics 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gay, B., J. Tournier, N. Chazal, C. Carriere, and P. Boulanger. 1998. Morphopoietic determinants of HIV-1 Gag particles assembled in baculovirus-infected cells. Virology 247160-169. [DOI] [PubMed] [Google Scholar]
- 25.Guo, X., J. Hu, J. B. Whitney, R. S. Russell, and C. Liang. 2004. Important role for the CA-NC spacer region in the assembly of bovine immunodeficiency virus Gag protein. J. Virol. 78551-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henderson, L. E., R. Sowder, G. Smythers, R. E. Benveniste, and S. Oroszlan. 1985. Purification and N-terminal amino acid sequence comparisons of structural proteins from retrovirus-D/Washington and Mason-Pfizer monkey virus. J. Virol. 55778-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes, J. F., and J. M. Coffin. 2004. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 1011668-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jern, P., and J. Coffin. 2008. Effects of retroviruses on host genome function. Annu. Rev. Genet. 42709-732. [DOI] [PubMed] [Google Scholar]
- 29.Kalter, S. S., R. J. Helmke, R. L. Heberling, M. Panigel, A. K. Fowler, J. E. Strickland, and A. Hellman. 1973. Brief communication: C-type particles in normal human placentas. J. Natl. Cancer Inst. 501081-1084. [DOI] [PubMed] [Google Scholar]
- 30.Kanamoto, T., Y. Kashiwada, K. Kanbara, K. Gotoh, M. Yoshimori, T. Goto, K. Sano, and H. Nakashima. 2001. Anti-human immunodeficiency virus activity of YK-FH312 (a betulinic acid derivative), a novel compound blocking viral maturation. Antimicrob. Agents Chemother. 451225-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller, P. W., M. C. Johnson, and V. M. Vogt. 2008. Mutations in the spacer peptide and adjoining sequences in Rous sarcoma virus Gag lead to tubular budding. J. Virol. 826788-6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kingston, R. L., and V. M. Vogt. 2005. Domain swapping and retroviral assembly. Mol. Cell 17166-167. [DOI] [PubMed] [Google Scholar]
- 33.Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J. P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, N. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J. C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R. H. Waterston, R. K. Wilson, L. W. Hillier, J. D. McPherson, M. A. Marra, E. R. Mardis, L. A. Fulton, A. T. Chinwalla, K. H. Pepin, W. R. Gish, S. L. Chissoe, M. C. Wendl, K. D. Delehaunty, T. L. Miner, A. Delehaunty, J. B. Kramer, L. L. Cook, R. S. Fulton, D. L. Johnson, P. J. Minx, S. W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J. F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409860-921. [DOI] [PubMed] [Google Scholar]
- 34.Lee, Y. N., and P. D. Bieniasz. 2007. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 3e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang, C., J. Hu, R. S. Russell, A. Roldan, L. Kleiman, and M. A. Wainberg. 2002. Characterization of a putative alpha-helix across the capsid-SP1 boundary that is critical for the multimerization of human immunodeficiency virus type 1 Gag. J. Virol. 7611729-11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lilly, F. 1967. Susceptibility to two strains of Friend leukemia virus in mice. Science 155461-462. [DOI] [PubMed] [Google Scholar]
- 37.Lower, R., K. Boller, B. Hasenmaier, C. Korbmacher, N. Muller-Lantzsch, J. Lower, and R. Kurth. 1993. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proc. Natl. Acad. Sci. USA 904480-4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lower, R., J. Lower, C. Tondera-Koch, and R. Kurth. 1993. A general method for the identification of transcribed retrovirus sequences (R-U5 PCR) reveals the expression of the human endogenous retrovirus loci HERV-H and HERV-K in teratocarcinoma cells. Virology 192501-511. [DOI] [PubMed] [Google Scholar]
- 39.Lower, R., R. R. Tonjes, C. Korbmacher, R. Kurth, and J. Lower. 1995. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J. Virol. 69141-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magin-Lachmann, C., S. Hahn, H. Strobel, U. Held, J. Lower, and R. Lower. 2001. Rec (formerly Corf) function requires interaction with a complex, folded RNA structure within its responsive element rather than binding to a discrete specific binding site. J. Virol. 7510359-10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magin, C., R. Lower, and J. Lower. 1999. cORF and RcRE, the Rev./Rex and RRE/RxRE homologues of the human endogenous retrovirus family HTDV/HERV-K. J. Virol. 739496-9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malet, I., M. Wirden, A. Derache, A. Simon, C. Katlama, V. Calvez, and A. G. Marcelin. 2007. Primary genotypic resistance of HIV-1 to the maturation inhibitor PA-457 in protease inhibitor-experienced patients. AIDS 21871-873. [DOI] [PubMed] [Google Scholar]
- 43.Medstrand, P., and J. Blomberg. 1993. Characterization of novel reverse transcriptase encoding human endogenous retroviral sequences similar to type A and type B retroviruses: differential transcription in normal human tissues. J. Virol. 676778-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medstrand, P., and D. L. Mager. 1998. Human-specific integrations of the HERV-K endogenous retrovirus family. J. Virol. 729782-9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melamed, D., M. Mark-Danieli, M. Kenan-Eichler, O. Kraus, A. Castiel, N. Laham, T. Pupko, F. Glaser, N. Ben-Tal, and E. Bacharach. 2004. The conserved carboxy terminus of the capsid domain of human immunodeficiency virus type 1 gag protein is important for virion assembly and release. J. Virol. 789675-9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mura, M., P. Murcia, M. Caporale, T. E. Spencer, K. Nagashima, A. Rein, and M. Palmarini. 2004. Late viral interference induced by transdominant Gag of an endogenous retrovirus. Proc. Natl. Acad. Sci. USA 10111117-11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murcia, P. R., F. Arnaud, and M. Palmarini. 2007. The transdominant endogenous retrovirus enJS56A1 associates with and blocks intracellular trafficking of Jaagsiekte sheep retrovirus Gag. J. Virol. 811762-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ono, M., T. Yasunaga, T. Miyata, and H. Ushikubo. 1986. Nucleotide sequence of human endogenous retrovirus genome related to the mouse mammary tumor virus genome. J. Virol. 60589-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmarini, M., J. M. Sharp, M. de las Heras, and H. Fan. 1999. Jaagsiekte sheep retrovirus is necessary and sufficient to induce a contagious lung cancer in sheep. J. Virol. 736964-6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pamilo, P., and M. Nei. 1988. Relationships between gene trees and species trees. Mol. Biol. Evol. 5568-583. [DOI] [PubMed] [Google Scholar]
- 51.Purdy, J. G., J. M. Flanagan, I. J. Ropson, K. E. Rennoll-Bankert, and R. C. Craven. 2008. Critical role of conserved hydrophobic residues within the major homology region in mature retroviral capsid assembly. J. Virol. 825951-5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raaum, R. L., K. N. Sterner, C. M. Noviello, C. B. Stewart, and T. R. Disotell. 2005. Catarrhine primate divergence dates estimated from complete mitochondrial genomes: concordance with fossil and nuclear DNA evidence. J. Hum. Evol. 48237-257. [DOI] [PubMed] [Google Scholar]
- 53.Ruda, V. M., S. B. Akopov, D. O. Trubetskoy, N. L. Manuylov, A. S. Vetchinova, L. L. Zavalova, L. G. Nikolaev, and E. D. Sverdlov. 2004. Tissue specificity of enhancer and promoter activities of a HERV-K(HML-2) LTR. Virus Res. 10411-16. [DOI] [PubMed] [Google Scholar]
- 54.Ruprecht, K., H. Ferreira, A. Flockerzi, S. Wahl, M. Sauter, J. Mayer, and N. Mueller-Lantzsch. 2008. Human endogenous retrovirus family HERV-K(HML-2) RNA transcripts are selectively packaged into retroviral particles produced by the human germ cell tumor line Tera-1 and originate mainly from a provirus on chromosome 22q11.21. J. Virol. 8210008-10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satta, Y., J. Klein, and N. Takahata. 2000. DNA archives and our nearest relative: the trichotomy problem revisited. Mol. Phylogenet. Evol. 14259-275. [DOI] [PubMed] [Google Scholar]
- 56.Scarlata, S., and C. Carter. 2003. Role of HIV-1 Gag domains in viral assembly. Biochim. Biophys. Acta 161462-72. [DOI] [PubMed] [Google Scholar]
- 57.Sfakianos, J. N., R. A. LaCasse, and E. Hunter. 2003. The M-PMV cytoplasmic targeting-retention signal directs nascent Gag polypeptides to a pericentriolar region of the cell. Traffic 4660-670. [DOI] [PubMed] [Google Scholar]
- 58.Simpson, G. R., C. Patience, R. Lower, R. R. Tonjes, H. D. Moore, R. A. Weiss, and M. T. Boyd. 1996. Endogenous D-type (HERV-K) related sequences are packaged into retroviral particles in the placenta and possess open reading frames for reverse transcriptase. Virology 222451-456. [DOI] [PubMed] [Google Scholar]
- 59.Sperber, G. O., T. Airola, P. Jern, and J. Blomberg. 2007. Automated recognition of retroviral sequences in genomic data—RetroTector. Nucleic Acids Res. 354964-4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sundquist, W. I., and C. P. Hill. 2007. How to assemble a capsid. Cell 13117-19. [DOI] [PubMed] [Google Scholar]
- 61.Swofford, D. L. 2003. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4B10. Sinauer Associates, Sunderland, MA.
- 62.Takahata, N. 1993. Allelic genealogy and human evolution. Mol. Biol. Evol. 102-22. [DOI] [PubMed] [Google Scholar]
- 63.Turner, G., M. Barbulescu, M. Su, M. I. Jensen-Seaman, K. K. Kidd, and J. Lenz. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. 111531-1535. [DOI] [PubMed] [Google Scholar]
- 64.Wright, E. R., J. B. Schooler, H. J. Ding, C. Kieffer, C. Fillmore, W. I. Sundquist, and G. J. Jensen. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 262218-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang, J., H. Bogerd, S. Y. Le, and B. R. Cullen. 2000. The human endogenous retrovirus K Rev response element coincides with a predicted RNA folding region. RNA 61551-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang, J., H. P. Bogerd, S. Peng, H. Wiegand, R. Truant, and B. R. Cullen. 1999. An ancient family of human endogenous retroviruses encodes a functional homolog of the HIV-1 Rev protein. Proc. Natl. Acad. Sci. USA 9613404-13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu, D., C. T. Wild, D. E. Martin, S. L. Morris-Natschke, C. H. Chen, G. P. Allaway, and K. H. Lee. 2005. The discovery of a class of novel HIV-1 maturation inhibitors and their potential in the therapy of HIV. Expert Opin. Investig. Drugs 14681-693. [DOI] [PubMed] [Google Scholar]
- 68.Zhou, J., C. H. Chen, and C. Aiken. 2006. Human immunodeficiency virus type 1 resistance to the small molecule maturation inhibitor 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acid substitutions at the CA-SP1 cleavage site in Gag. J. Virol. 8012095-12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou, J., C. H. Chen, and C. Aiken. 2004. The sequence of the CA-SP1 junction accounts for the differential sensitivity of HIV-1 and SIV to the small molecule maturation inhibitor 3-O-{3′,3′-dimethylsuccinyl}-betulinic acid. Retrovirology 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou, J., X. Yuan, D. Dismuke, B. M. Forshey, C. Lundquist, K. H. Lee, C. Aiken, and C. H. Chen. 2004. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J. Virol. 78922-929. [DOI] [PMC free article] [PubMed] [Google Scholar]