

Abstract
Recognition of virus infections by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I), and melanoma differentiation associated gene 5 (MDA5), activates signaling pathways, leading to the induction of inflammatory cytokines that limit viral replication. To determine the effects of PRR-mediated innate immune response on hepatitis B virus (HBV) replication, a 1.3mer HBV genome was cotransfected into HepG2 or Huh7 cells with plasmid expressing TLR adaptors, myeloid differentiation primary response gene 88 (MyD88), and TIR-domain-containing adaptor-inducing beta interferon (TRIF), or RIG-I/MDA5 adaptor, interferon promoter stimulator 1 (IPS-1). The results showed that expressing each of the three adaptors dramatically reduced the levels of HBV mRNA and DNA in both HepG2 and Huh7 cells. However, HBV replication was not significantly affected by treatment of HBV genome-transfected cells with culture media harvested from cells transfected with each of the three adaptors, indicating that the adaptor-induced antiviral response was predominantly mediated by intracellular factors rather than by secreted cytokines. Analyses of involved signaling pathways revealed that activation of NF-κB is required for all three adaptors to elicit antiviral response in both HepG2 and Huh7 cells. However, activation of interferon regulatory factor 3 is only essential for induction of antiviral response by IPS-1 in Huh7 cells, but not in HepG2 cells. Furthermore, our results suggest that besides NF-κB, additional signaling pathway(s) are required for TRIF to induce a maximum antiviral response against HBV. Knowing the molecular mechanisms by which PRR-mediated innate defense responses control HBV infections could potentially lead to the development of novel therapeutics that evoke the host cellular innate antiviral response to control HBV infections.
Hepatitis B virus (HBV) is a noncytopathic, hepatotropic virus belonging to the Hepadnaviridae family. Infection of HBV can be either transient or chronic in nature (13, 30). Transient infection frequently leads to acute hepatitis and, in rare cases, results in fatal, fulminant hepatitis (30). Chronic infection represents a major public health burden affecting an estimated 400 million individuals worldwide and carries a high risk for the development of chronic active hepatitis, cirrhosis, and primary hepatocellular carcinoma (43). As for many other viruses, the outcomes of HBV infections and pathogenesis of the associated diseases are determined by virus-host interactions, largely mediated by innate and adaptive immune responses (8, 73).
Virus infection elicits a rapid and potent innate immune response in mammalian cells to produce proinflammatory cytokines and chemokines that limit virus replication and coordinate adaptive antiviral immunity (50). Central to this cellular response is the virus-induced production of alpha/beta interferons (IFN-α/β), which play an indispensable role in host defense against virus infections (22, 45, 61). In the last decade, tremendous progresses have been made in uncovering the cellular pattern recognition receptors (PRRs) that sense diverse pathogen-associated molecular patterns and deciphering the molecular pathways that couple pathogen recognition to the induction of IFNs and other cytokines (50, 66, 71). Notably, mammalian cells sense virus infections predominantly via either endosomal Toll-like receptor 3 (TLR3), TLR7/8, and TLR9 or cytoplasmic caspase activation and recruitment domain (CARD)-containing DEx(D/H) box RNA helicases, including retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA5) (1, 79). While the four TLRs are activated by selective binding of viral double stranded RNA (TLR3), single-stranded poly-U RNA (TLR7/8), or unmethylated CpG DNA motifs present in viral genomes (TLR9) in the endosomes (2, 24, 39), RIG-I and MDA5 recognize cytoplasmic viral RNAs bearing distinguishable structural features from cellular RNA, such as the presence of triphosphates at the 5′ terminus (31, 51). As illustrated in Fig. 1, recognition of such virus-associated molecular patterns by each of the cellular PRRs recruits their distinct adaptor proteins, which activate signaling cascades to induce cytokine production in virus-infected cells. For examples, binding of viral RNA to RIG-I/MDA5 induces conformation changes that lead to the exposure of their CARD domains (65). Through homotypic CARD interaction with the IFN promoter stimulator 1 (IPS-1; also known as CARDIF, MAVS, and VISA) adaptor protein, the RIG-I/MDA5 is recruited onto the outer membrane of the mitochondria to form a macromolecular signaling complex that serves to activate downstream interferon regulatory factor 3 (IRF3), nuclear factor-κB (NF-κB), c-Jun/ATF2, and other transcription factors that stimulate the transcription of IFNs and certain IFN-stimulated genes (ISGs) (38, 44, 59, 77). Similarly, engagement of the TLRs with viral nucleic acids recruits distinct adaptor proteins TIR-domain-containing adaptor-inducing beta interferon (TRIF; for TLR3) or MyD88 (for TLR7/8 and TLR9), which initiates signaling pathways to activate critical transcription factors, such as NF-κB, IRF3, IRF7, or IRF5 among others, to induce expression of proinflammatory cytokines and other cellular genes (37).
FIG. 1.

Schematic representation of the major viral pattern recognition receptor-mediated signaling pathways. The three PRR adaptors and their relationship with the two downstream transcription factors (NF-κB and IRF3) and the activated target genes examined in the present study are illustrated. See the text for more detailed discussion.
Microarray analyses of host cellular gene expression profiles in response to infections of the variety of viruses in cultured cells and tissue samples obtained from infected people and animals in the last decade reveal that induction of ISG expression, an indicator of the activation of PRR-mediated host cellular antiviral responses, is observed in almost all of the virus infections examined, with rare exceptions (34, 72). One of such exceptions is HBV infection of chimpanzees (73). While it had been shown that induction of ISGs in the livers of chimpanzees and humans was a hallmark of hepatitis C virus (HCV) infections (5, 6, 27, 40, 60, 62), HBV infection of chimpanzees did not induce the expression of ISGs as the virus spreads throughout the liver (73). This surprising and unprecedented observation implies that HBV either fails to activate or has evolved ability to inhibit the activation of the PRR-mediated innate immune responses.
To resolve these issues and determine whether activation of PRR-mediated innate immunity in human hepatoma cells could inhibit HBV replication, we investigated the induction and antiviral effects of TLR- and RIG-I/MDA5-mediated host cellular innate immune responses against HBV in HepG2 and Huh7 cells via overexpression of the three PRR adaptors IPS-1, TRIF, and MyD88. Our results demonstrated that expressing each of the three adaptors potently inhibited HBV replication, predominantly through intracellular antiviral pathway(s) rather than secreted cytokines. Most strikingly, we found that, unlike IFN-α/β inhibition of HBV replication in mouse hepatocytes, where the cytokines prevented pregenomic RNA (pgRNA)-containing nucleocapsid formation (74), the innate immune response-elicited by expression of adaptor proteins in human hepatoma cells limited HBV replication by reducing the steady-state levels of viral mRNAs. Analysis of involved signaling pathways revealed that activation of NF-κB is essential for all three adaptors to elicit antiviral response in both HepG2 and Huh7 cells, but activation of IRF3 is only essential for induction of antiviral response by IPS-1 in Huh7 cells and not HepG2 cells. Our results thus suggest that although HBV fails to activate host cellular innate defense pathways, it is extremely sensitive to the antiviral effects elicited by PRR-mediated host cellular antiviral response in human hepatocyte-derived cells. Further analysis of the intracellular antiviral pathway could potentially lead to the identification of molecular targets for the development of antivirals that evoke the intricate cellular antiviral pathway(s) and eliminate HBV infections.
MATERIALS AND METHODS
Cell culture and kinase inhibitors.
HepG2 cells were obtained from ATCC and maintained in Dulbecco modified Eagle medium and F-12 medium (DMEM/F12) (Invitrogen) supplemented with 10% fetal bovine serum, 100 U of penicillin/ml, and 100 μg of streptomycin/ml. Huh7 cells were maintained as previously described (20). Mitogen-activated protein kinase (MAPK) inhibitors SB202190, U0126, and SP600125 were purchased from Calbiochem.
Plasmid construction.
Plasmids pHBV1.3 and pCMV-HBV were described previously (19). Plasmid pS that expresses HBV small (S) envelope proteins (HBsAg) was described previously and was kindly provided by Volker Bruss (University of Gottingen, Gottingen, Germany) (55). Plasmid pcHBs-V5, which expressed C-terminally V5-tagged HBsAg, was generated by insertion of a 0.8-kb PCR fragment spanning the HBsAg coding region into pcDNA3.1/V5-His-TOPO vector. Plasmids expressing C-terminally hemagglutinin (HA)-tagged MyD88, TRIF with deletion of carboxyl-terminal 68 amino acids (saTRIF), and constitutively active (or superactive) IRF3 (saIRF3) proteins were purchased from Invivogen (San Diego, CA). Plasmids expressing wild-type IκB-alpha (IκBα) and dominant-negative IκB-alpha (DN-IκBα) were obtained from Christoph Seeger (Fox Chase Cancer Center, Philadelphia, PA). Plasmids expressing other adaptors or cellular signaling proteins used in the present study were constructed via similar approaches described previously (35). Briefly, to obtain cDNA clones of human RIG-I, MDA5, CARD domains of RIG-1 and MDA5, IPS-1, and IRF3, first-strand cDNA was made from Huh7 cell-derived total RNA with oligo(dT)12-18 primer and Superscript III DNA polymerase (Invitrogen) by following the manufacturer's directions. Full-length cDNA for each of the forementioned genes with N-terminal FLAG tag was amplified by PCR (primer sequences are available upon request). The purified PCR fragments were digested with the restriction enzymes AflII and NotI and cloned into pcDNA5/FRT/ΔCAT vector that was derived from pCDNA5/FRT/CAT (Invitrogen) by removing the CAT-coding sequence with ApaI and XhoI digestion and self-ligation (35). The plasmid expressing the DN-IRF3 was generated by amplification of the coding region for the amino acid 55 to 452 of IRF3 (42) and cloned into pcDNA5/FRT/ΔCAT as described above. The identity of each cDNA clone was verified by nucleotide sequence analysis.
Cell transfection.
HepG2 and Huh7 cells were seeded into 35-mm-diameter collagen-coated dishes at a density of 1.2 × 106 cells per dish in antibiotic-free complete DMEM/F12. After 6 h, each well was transfected with a total of 4 μg of plasmid with Lipofectamine 2000 (Invitrogen) by following the manufacturer's directions. Transfected cells were harvested at the indicated times. The levels of HBV mRNAs and core-associated DNA were determined by Northern and Southern blot hybridization, respectively.
Nucleic acid analysis.
Intracellular viral core DNA was extracted as described previously (18). Briefly, cells from one 35-mm-diameter dish were lysed with 0.5 ml of lysis buffer containing 10 mM Tris-HCl (pH 8.0), 10 mM EDTA, 1% NP-40 (0.1% for Huh7 cells), and 2% sucrose at 37°C for 10 min. Cell debris and nuclei were removed by centrifugation, and the supernatant was mixed with 130 μl of 35% PEG-8000 containing 1.5 M NaCl. After 1 h of incubation in ice, viral nucleocapsids were pelleted by centrifugation at 12,000 × g for 10 min at 4°C, followed by 1 h digestion at 37°C in 400 μl of digestion buffer containing 0.5 mg of pronase (Calbiochem)/ml, 0.5% sodium dodecyl sulfate (SDS), 150 mM NaCl, 25 mM Tris-HCl (pH 8.0), and 10 mM EDTA. The digestion mixture was extracted twice with phenol, and DNA was precipitated with ethanol and dissolved in TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). One-third of the DNA sample from each plate was resolved by electrophoresis into a 1.5% agarose gel. Gel was then subjected to denaturalization in a solution containing 0.5 M NaOH and 1.5 M NaCl, followed by neutralization in a buffer containing 1 M Tris-HCl (pH 7.4) and 1.5 M NaCl. DNA was then blotted onto Hybond-XL membrane (GE Healthcare) in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer.
Total cellular RNA was extracted with TRIzol reagents (Invitrogen), by following the instructions of the manufacturer. A portion (10 μg) of total RNA was resolved in 1.5% agarose gel containing 2.2 M formaldehyde and transferred onto Hybond-XL membrane in 20× SSC buffer.
For the detection of HBV DNA and RNA, membranes were probed with either an [α-32P]UTP (800 Ci/mmol; Perkin-Elmer)-labeled minus- or plus-strand-specific full-length HBV riboprobe. Hybridization was carried out in 5 ml of EKONO hybridization buffer (Genotech) with 1 h of prehybridization at 65°C and overnight hybridization at 65°C, followed by a 1-h wash with 0.1× SSC and 0.1% SDS at 65°C. The membrane was exposed to a phosphorimager screen, and hybridization signals were quantified with QuantityOne software (Bio-Rad).
Western blot analysis.
Transfected cells in a 35-mm-diameter dish were washed once with phosphate-buffered saline (PBS) buffer and lysed in 200 μl of 1× Lamini buffer. A total of 20 μl of the cell lysate was resolved on an SDS-10% polyacrylamide gel and transferred onto polyvinylidene difluoride membrane (Bio-Rad). The membrane was blocked with PBS containing 0.1% Tween 20 and 5% nonfat dry milk and probed with antibodies against Flag tag (Sigma), HA tag (Clontech), TRIF (Invivogen), and β-actin (Chemicon International). Bound antibodies were revealed by horseradish peroxidase-labeled secondary antibodies and visualized with an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech) according to the protocol of the manufacturer.
Reverse transcription-PCR.
For semiquantitative detection of IFN-β, IFN-λ1, ISG56, MxA, and β-actin mRNA, total cellular RNA was extracted with TRIzol, and cDNA was synthesized with oligo(dT)12-18 primer and Superscript III DNA polymerase (Invitrogen) by following the manufacturer's directions. The PCRs were carried out in a 25-μl reaction mixture with the Advantage cDNA PCR kit (Clontech). The PCR annealing temperatures selected varied depending on the primers selected for amplification. The primer sequences are available upon request.
NF-κB DNA-binding activity assay.
Cell nuclear extracts were obtained by using a nuclear extract kit (Active Motif), and the protein concentration was determined by using a BCA assay (Pierce). A 2.5-μg portion of nuclear extract was subjected to NF-κB p65 DNA-binding enzyme-linked immunosorbent assay (ELISA) (Active Motif) according to the manufacturer's protocol.
RESULTS
Activation of the host cellular innate immune response inhibits HBV replication in HepG2 and Huh7 cells.
Study of HBV and host cell interactions is hampered by the lack of cell culture systems for efficient viral infection. As an alternative, HBV genome replication can be initiated by transfection of HBV DNA that is able to transcribe pregenomic RNA into a few human hepatocyte-derived tumor cell lines, such as HepG2 and Huh7 cells. However, as for many tumor-derived cell lines, the two cell lines are not completely competent in innate immune response. For example, neither HepG2 nor Huh7 cells express TLR3 receptors and thus do not respond to the treatment with double-stranded RNA (dsRNA) (41). Moreover, activation of RIG-I and/or MDA5-mediated cellular innate immunity in cultured cells is usually achieved by transfection of their cognate ligands, such as 5′-triphosphate RNA and dsRNA, respectively. However, cytoplasm-delivered dsRNA could potentially interact with other cellular dsRNA binding proteins, such as dsRNA-dependent protein kinase (PKR) and adenosine deaminase act on RNA 1 (ADAR1), and might directly affect viral replication (14, 54). To overcome these limitations and avoid the confounding effects of RNA transfection, the viral PRR-mediated host cellular innate immune responses in hepatocyte-derived cells were activated by overexpression of the three major adaptor proteins for TLRs (TRIF and MyD88) and RIG-I/MDA5 (IPS-1) (Fig. 1), which efficiently activate cellular responses similar to that initiated by binding of nucleic acid ligands to their cognate PRRs (12, 59). Because overexpression of full-length TRIF induces apoptosis, we utilized in the present study a mutant or superactivated (saTRIF) that lacks the C-terminal 68 amino acids but activates promoters containing NF-κB and ISRE motifs at levels comparable to wild-type TRIF, without induction of apoptosis (36). In addition, to independently evaluate the antiviral effects of the RIG-I and MDA5 pathways, the innate immune responses mediated by the two PRRs were activated by overexpression of either the full-length RIG-I and MDA5 protein or the CARD domains derived from the two receptors (78).
Accordingly, HepG2 cells were cotransfected with a 1.3mer HBV genome and control vector or plasmids expressing full-length RIG-I or MDA5, the CARD domains of RIG-I and MDA5 (designated as RN230 and MN300, respectively), and the adaptor proteins MyD88, saTRIF, and IPS-1. As a control, one set of HBV genome transfected cells were treated with 500 IU of IFN-α/ml that displays almost a maximum antiviral effect of the cytokine against HBV (data not shown). Cells were harvested 4 days after transfection. Intracellular HBV mRNA and core-associated HBV DNA replicative intermediates were analyzed by Northern and Southern blot hybridization, respectively. Expression of desired proteins in transfected cells was confirmed by Western blot analyses (Fig. 2A, lower panel). The results demonstrated that except for cells cotransfected with plasmid expressing full-length RIG-I, the steady-state levels of HBV mRNAs were reduced in all other cells that were treated with IFN-α or that overexpressed full-length MDA5, RN230, or MN300 or each of the three adaptors (Fig. 2A, upper panel, see also the left panel of Fig. 2C for quantitative comparison). Interestingly, the levels of core-associated HBV DNA were decreased in proportion to HBV mRNA reductions in the cells under the corresponding treatment conditions (Fig. 2A, middle panel, and compare the left two panels of Fig. 2C). These results suggested that the primary effect of the antiviral responses either induced by IFN-α treatment or elicited by overexpression of PRR adaptors and CARDs of RIG-I and MDA5 was to downregulate viral RNAs. However, the possibility that the innate immune responses also inhibited additional steps in HBV nucleocapsid assembly and DNA replication could not be ruled out. Similarly, a strong inhibition on HBV replication, as demonstrated by the reduced levels of both HBV mRNA and DNA, were observed in Huh7 cells cotransfected with plasmids expressing each of the three PRR adaptors (Fig. 2B and right two panels of Fig. 2C). Furthermore, no evidence of cytotoxicity was observed for any experimental set, as determined by microscopic observation and quantitatively measured by cotransfection of cells with plasmids expressing green fluorescent protein (GFP) and individual adaptors or control vector. The results from the later experiment demonstrated that the percentage of GFP-positive cells in cultures cotransfected with plasmids expressing GFP and each of the three adaptor proteins did not differ from that in cells cotransfected with control vector and GFP-expressing plasmid (data not shown).
FIG. 2.

Overexpression of the CARD domain of RIG-I or MDA5 and three adaptor proteins—IPS-1, TRIF, and MyD88—potently inhibits HBV replication in human hepatocyte-derived tumor cell lines. (A) A plasmid that encodes pgRNA of wild-type HBV (pHBV1.3) was cotransfected with control plasmid (lane 1) or plasmids that express RIG-I (lane 3), CARD of RIG-I (RN230, lane 4), MDA5 (lane 5), CARD of MDA5 (MN300, lane 6), IPS-1 (lane 7), TRIF (lane 8), or MyD88 (lane 9) into HepG2 cells. In these transfection experiments, 2 μg of pHBV1.3 was cotransfected with 2 μg of control plasmid or 1 μg of control plasmid plus 1 μg of plasmid expressing proteins under investigation into a 35-mm dish of cells. Lane 1 was loaded with sample derived from pHBV1.3 and control plasmid cotransfected cells, and lane 2 is loaded with samples derived from pHBV1.3 and control plasmid cotransfected cells treated with 500 IU of IFN-α/ml, which is added 12 h after transfection. Cells were harvested at 4 day after transfection, and the levels of viral RNAs and core-associated DNA were determined by Northern (upper panel) and Southern (middle panel) blot hybridization analyses, respectively. For RNA analysis, each lane was loaded with 10 μg of total RNA. rRNAs (28S and 18S) were presented as loading controls. The positions of the HBV 3.5-, 2.4-, and 2.1-kb RNAs are indicated. For DNA analysis, HBV core DNA was probed with a genome-length, minus-strand-specific HBV riboprobe. The positions of relaxed circular (RC), single-stranded (SS) DNAs are indicated. Proper expression of intended proteins by the transfected plasmids was confirmed by Western blot analysis with the antibodies described in Materials and Methods, and the levels of β-actin serve as loading controls (lower panel). (B) Huh7 cells were cotransfected with 2 μg of pHBV1.3 and 2 μg of control plasmid (lane 1 and 2) or 1 μg of control plasmid plus 1 μg of plasmid expressing IPS-1 (lane 3), TRIF (lane 4) and MyD88 (lane 5). One set of the control plasmid transfected cells (lane 2) was treated with 500 IU of IFN-α/ml at 12 h after transfection. Cells were harvested at 4 day after transfection, and the levels of viral RNAs and core-associated DNA were determined as described above. (C) Levels of HBV mRNAs and DNA in panels A and B were determined by using a phosphorimager with QuantityOne software (Bio-Rad) and plotted as the percentage of the RNA or DNA levels in control cells that were cotransfected with pHBV1.3 and vector plasmids (lane 1).
Altogether, the results described above clearly demonstrated that overexpression of saTRIF and IPS-1 induced a strong antiviral response that noncytopathically inhibited HBV replication in both HepG2 and Huh7 cells. In agreement with a previous report by Xiong et al., the overexpression of MyD88 also inhibited HBV replication (76), albeit to a lesser extent, compared to the other two adaptors (Fig. 2C). Moreover, activation of either RIG-I or MDA5 pathway by overexpression of their respective CARDs in HepG2 cells induced a robust antiviral response against HBV (Fig. 2A, lanes 4 and 6). In corroboration with their ability to activate innate immune response upon overexpression (53), only full-length MDA5, but not RIG-I, was able to inhibit HBV replication in HepG2 cells (Fig. 2, lanes 3 and 5). This is most possibly due to the distinct structural features of the two PRRs. RIG-I contains a C-terminal repressor domain which intramolecularly interacts with the CARDs to prevent its autoactivation. Binding of its agonists induces RIG-I multimerization and conformation change that relieves autorepression and exposes the CARDs. On the contrary, MDA5 lacks such an internal repressor domain and, consequently, induces innate immune response upon overexpression (53, 78).
PRR-mediated innate immune response reduces the steady-state levels of viral RNAs via posttranscriptional mechanisms.
To determine whether the observed reduction of HBV RNAs is due to transcriptional inhibition or posttranscriptionally accelerated decay of viral RNA, we intended to distinguish whether the adaptor-induced reduction of HBV mRNAs was an HBV promoter-dependent or an HBV RNA sequence-dependent phenomenon. To this end, plasmids expressing each of the three PRR adaptors or control vector were cotransfected into HepG2 cells with either pCMVHBV in which the HBV pgRNA transcription is under the control of a cytomegalovirus immediate-early (CMV-IE) promoter or plasmid pS in which the 2.1-kb HBV subgenomic mRNA is transcribed under the direction of a simian virus 40 (SV40) early promoter (illustrated in Fig. 3A). Cells were harvested 2 days posttransfection, and the levels of HBV mRNA were determined by Northern blot assay. As shown in Fig. 3B and C, the levels of both HBV pgRNA in pCMVHBV transfected cells and 2.1-kb mRNA in pS transfected cells were dramatically reduced upon the overexpression of saTRIF and IPS-1, but to a lesser extent in MyD88-overexpressing cells. These results essentially corroborated with the observations obtained in pHBV1.3 transfected cells as shown in Fig. 2A and thus suggested that the innate immunity elicited by the PRR adaptors either inhibited the transcriptional activity of the CMV and SV40 promoters or posttranscriptionally reduced the levels of HBV RNAs in an HBV RNA sequence-dependent manner.
FIG. 3.

Reduction of HBV RNA levels induced by the innate immune responses does not depend on the nature of the promoters. (A) Schematic representation of the RNA species and the corresponding promoter that directs the RNA transcription in the plasmids used in the present study. The numbers indicate the HBV DNA sequence with 1 at the unique EcoRI site of HBV genome. (B) HepG2 cells were cotransfected with 2 μg of pCMV-HBV and 2 μg of control plasmid (lanes 1 to 3) or 1 μg of control plasmid plus 1 μg of plasmid expressing IPS-1 (lane 4), TRIF (lane 5), and MyD88 (lane 6). The control plasmid cotransfected cells were left untreated (lane 1) or treated with 500 IU of IFN-α/ml (lane 2) and 400 ng of IFN-λ1/ml (lane 3), respectively. Cells were harvested at 4 day after transfection, and the levels of viral RNAs were determined by Northern blot hybridization. rRNAs (28S and 18S) were presented as loading controls. The positions of the HBV 3.5-, 2.4-, and 2.1-kb RNAs are indicated. (C) HepG2 cells were cotransfected with 2 μg of pS and 2 μg of control plasmid (lane 1) or 1 μg of control plasmid plus 1 μg of plasmid expressing IPS-1 (lane 2), TRIF (lane 3), and MyD88 (lane 4), respectively. Cells were harvested at 2 day after transfection, and the levels of HBV 2.1-kb mRNA were determined by Northern blot hybridization. rRNAs (28S and 18S) were presented as loading controls. (D) HepG2 cells were cotransfected with 2 μg of pcHBs-V5 and 2 μg of control plasmid (lane 1) or 1 μg of control plasmid plus 1 μg of plasmid expressing IPS-1 (lane 2), TRIF (lane 3), and MyD88 (lane 4), respectively. Cells were harvested at 2 day after transfection, and the levels of 1-kb HBV mRNA and 0.8-kb neomycin phosphotransferase II (NTP II) mRNA were determined by Northern blot hybridization. rRNAs (28S and 18S) were used as loading controls. The relative RNA level in each sample is expressed as the percentage of RNA level in control cells cotransfected with vector plasmid (lane 1 of Fig. 3B, C, and D) and is given underneath each of the blots.
To investigate these two possibilities, plasmid pcHBs-V5 that transcribed a 1.0-kb truncated HBV mRNA spanning only the coding region of HBsAg under the control of a CMV-IE promoter and 0.8-kb neomycin phosphotransferase II (NTP-II) mRNA under the direction of a SV40 early promoter (illustrated in Fig. 3A) were cotransfected into HepG2 cells, with the control vector or plasmids expressing each of the three adaptors. As shown in Fig. 3D, the levels of both HBsAg and NTP-II mRNAs in HepG2 cells were not significantly affected by any of the three adaptors. These results thus suggest that the reduction of HBV mRNAs by the three adaptors in HepG2 cells is most likely due to posttranscriptional decay of viral RNAs. Interestingly, because HBV pgRNA and 2.1-kb mRNA (Fig. 3B and C), but not the 1.0-kb truncated HBsAg mRNA (Fig. 3D), are efficiently reduced in HepG2 cells that overexpress the PRR adaptors, it is reasonable to conclude that RNA sequence element(s) located in the 3′ overlapping region of the three HBV mRNA species may be critical for the selective reduction of viral RNAs.
Analysis of cellular pathways activated by overexpression of the adaptors.
As illustrated in Fig. 1, recognition of pathogen-associated molecular patterns by TLRs stimulates the recruitment of a set of intracellular TIR-domain-containing adaptors, including MyD88 and TRIF. MyD88 is a universal adaptor for all TLRs with the exception of TLR3 and activates MAPKs (ERK, JNK, and p38) and transcription factor NF-κB to control the expression of inflammatory cytokine genes in many types of cells. TRIF is recruited by TLR3 and TLR4 to activate MAPKs, NF-κB, and transcription factor IRF3, all of which are required for activation of IFN-β and IFN-λ1 genes (47). Similarly, IPS-1 mediates RIG-I and MDA5 signaling to activate multiple intracellular signaling pathways, including MAPKs, NF-κB, and IRF3, which induce IFNs and other cellular genes (59). To determine whether the proper cellular responses are activated by the expression of the adaptors in the hepatoma cell lines under the condition of HBV replication, the DNA-binding activity of NF-κB and induction of IFN-β, IFN-λ1, ISG56, and MxA mRNAs were determined in the HBV genome-transfected HepG2 and Huh7 cells. As shown in Fig. 4, the results reveal the following observations. First, as expected, all three adaptors activate NF-κB, as measured by p65 DNA-binding activity with a commercial ELISA kit (Active Motif). Moreover, the activation can be efficiently inhibited by a DN-IκBα, which bears an N-terminal 39-amino-acid deletion and thus cannot be phosphorylated by IKK-α/β (Fig. 4A) (67). Second, based on the induction of ISG56 (Fig. 4B), which depends on activation of IRF3, but not NF-κB (11, 15), it can be deduced that only IPS-1 and saTRIF, but not MyD88, efficiently activated IRF3. Third, mRNAs for both IFN-β and IFN-λ1 were induced by IPS-1 and saTRIF, but not MyD88. Consistently, MxA gene expression, which relies on the JAK-STAT signaling pathway, was only induced in cells in which IFNs were induced. Finally, as expected, the constitutively active form of IRF3 (saIRF3), generated by the substitution of the Ser-Thr residues in the carboxyl terminus with the phosphomimetic Asp (42), potently induced ISG56 and IFNs and, as a consequence, the MxA gene is also induced (28). In summary, these results suggest that overexpression of the three PRR adaptors elicited the authentic innate immune responses in human hepatocyte-derived cells, essentially as observed in other cell types (78).
FIG. 4.
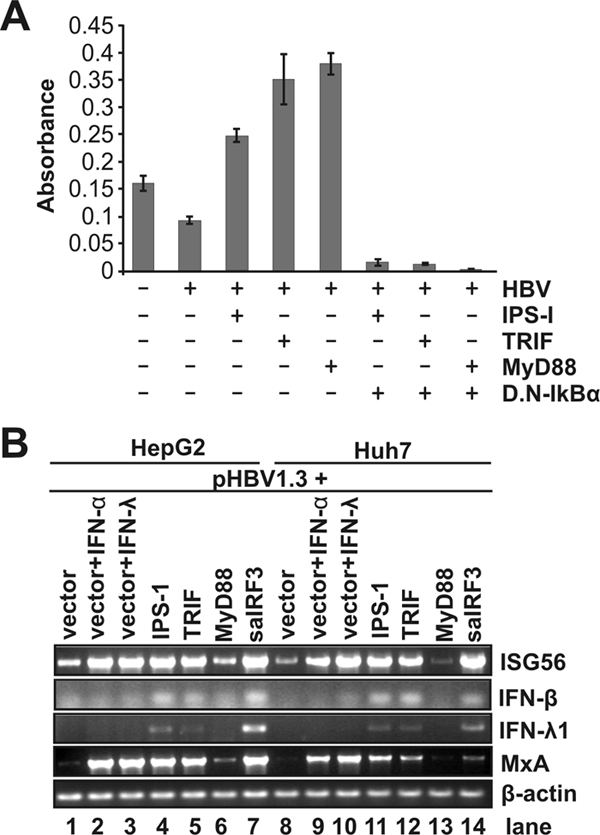
Expression of the three adaptor proteins activates desired intracellular signaling pathways. (A) NF-κB DNA-binding assay. Huh7 cells were untransfected or transfected with the indicated plasmids. After 2 days, 2.5 μg of the nuclear extract was subjected to the p65 DNA-binding ELISA according to the manufacturer's direction. (B) HepG2 (lanes 1 to 7) or Huh7 (lanes 8 to 14) cells were cotransfected with 2 μg of pHBV1.3 and 2 μg of control plasmid (lanes 1 to 3 and lanes 8 to 10) or 1 μg of control plasmid plus 1 μg of plasmid expressing IPS-1 (lanes 4 and 11), TRIF (lanes 5 and 12), MyD88 (lanes 6 and 13), and constitutively active IRF3 (saIRF3, lanes 7 and 14), respectively. Cells were harvested 2 days after transfection, and the levels of ISG56, IFN-β, IFN-λ1, MxA, and β-actin mRNAs were determined by reverse transcription-PCR assay.
Inhibition of HBV replication by PRR-mediated innate immune response is an intracellular event.
It has been shown that, in addition to the secretion of IFNs, chemokines, and other proinflammatory cytokines, the PRR-activated cellular innate immune response could also induce the expression of several ISGs and other cellular genes that might directly inhibit viral infections (57, 58). Moreover, although both IFN-α and IFN-λ inhibited HBV replication in Huh7 and HepG2 cells, the antiviral potency of the cytokines was not as great as that of the PRR adaptor-elicited innate immunity (Fig. 2A). It would therefore be interesting to know whether the observed antiviral effects elicited by the adaptors depend on the secreted cytokines or intracellular proteins.
To investigate these two possibilities, cultured media were harvested between 24 and 96 h posttransfection from HepG2 or Huh7 cells transfected with plasmids expressing each of the three adaptors or control plasmid and applied onto pHBV1.3 transfected HepG2 and Huh7 cells, respectively. As controls, the pHBV1.3 transfected cells were left untreated or treated with 500 IU of IFN-α/ml or 400 ng of IFN-λ1/ml. As shown in Fig. 5, in agreement with the results presented above, IFN-α and IFN-λ1 modestly reduced the levels of HBV RNAs in both HepG2 and Huh7 cells (Fig. 5, lanes 2 and 3). However, it is surprising that the viral RNA levels were not affected or reduced fewer than 20% in HepG2 and Huh7 cells treated with conditioned medium harvested from the cells transfected with plasmid expressing TRIF, MyD88, or saIRF3 (Fig. 5, lanes 6 to 8). In contrast, treatment of cells with the conditioned medium harvested from the IPS-1-overexpressed cells resulted in a reduction of HBV mRNA levels in Huh7 cells by ca. 33%, which is similar with what was observed in Huh7 cells treated with either IFN-α or IFN-λ (Fig. 5, lane 5), but apparently at a lesser extent compared to that observed in cells transfected with IPS-1-expressing plasmid, which reduced the level of viral RNA by 80% (Fig. 2B, lane 3, and Fig. 2C).
FIG. 5.
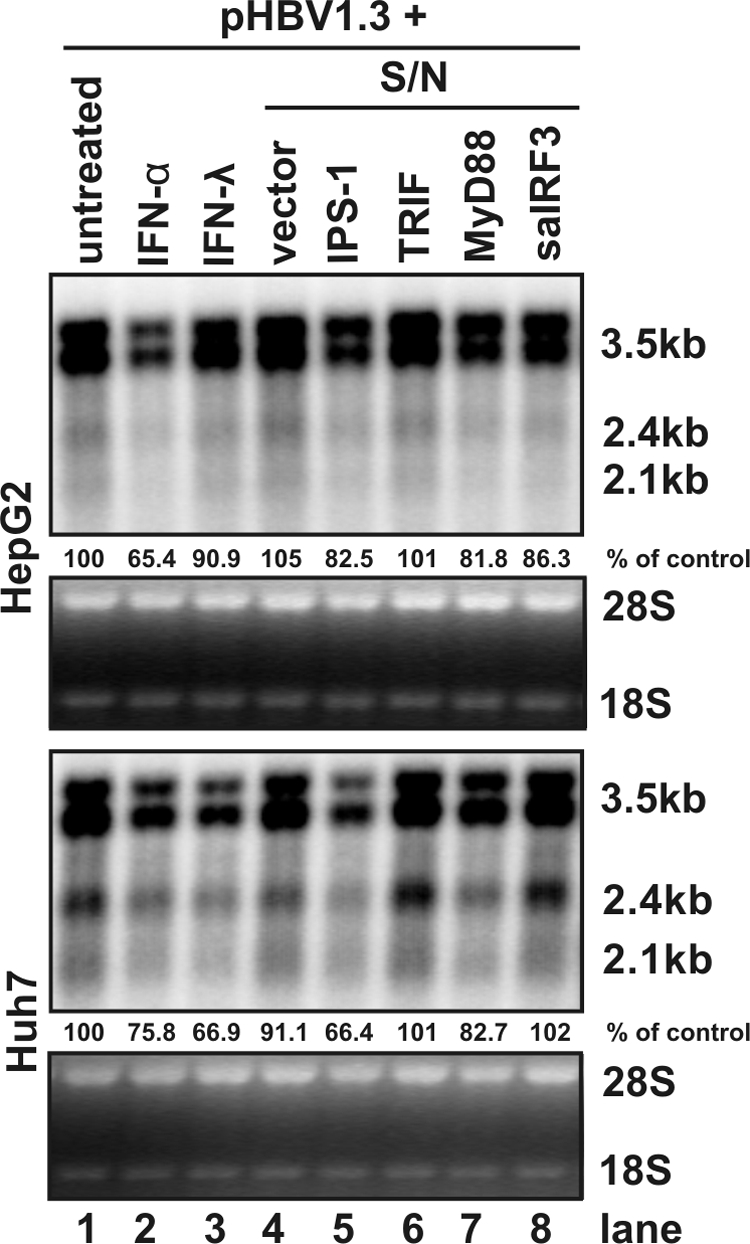
Antiviral effects induced by the innate immune response are intracellular events but not mediated by secreted cytokines. HepG2 (upper panel) and Huh7 (lower panel) cells were transfected with 1.6 μg of pHBV1.3 per well in 12-well plates. At 12 h after the transfection, culture media were replaced with fresh DMEM/F12 (lane 1), fresh DMEM/F12 containing 500 IU of IFN-α/ml (lane 2) or 400 ng of IFN-λ1/ml (lane 3), or conditioned media prepared by combination of equal volumes of fresh DMEM/F12 with medium harvested from HepG2 and Huh7 cells transfected with control plasmid (lane 4) or plasmid expressing IPS-1 (lane 5), TRIF (lane 6), MyD88 (lane 7), or saIRF3 (lane 8). The transfected cells were cultured for an additional 48 h with the same medium change at 24 h under the condition described above and then harvested for total RNA extraction. The levels of HBV RNA were determined by Northern blot hybridization. rRNAs (28S and 18S) were presented as loading controls. Relative RNA level in each sample is expressed as the percentage of RNA level in control cells cotransfected with pHBV1.3 and vector plasmid, but left untreated (lane 1), and is presented underneath each of the blots.
Hence, these results suggest that the observed strong inhibition of HBV replication by the three PRR adaptors is predominantly mediated by inducible intracellular factor(s) rather than secreted cytokines. It will be interesting to determine the intracellular signaling pathways that mediated the antiviral responses elicited by the PRR adaptors, which will ultimately lead to the identification of cellular factors that decrease the levels of HBV RNAs.
Role of MAPK-mediated signaling in the inhibition of HBV replication.
As mentioned above, MAPKs, including ERK, JNK, and p38, are activated essentially in all of the PRR-mediated innate immune responses and play pivotal roles in the induction of proinflammatory cytokines (7, 80). To examine whether any of the three major MAPK-mediated signal transduction pathways was required for the inhibition of HBV replication by the three adaptors, HepG2 cells were cotransfected with pHBV1.3 and plasmids expressing each of the three adaptors and control plasmid. At 12 h after the transfection, cells were left untreated or treated with 25 μM SB202190, 10 μM U0126, or 25 μM SP600125, selective inhibitors of p38 MAPK, ERK, or JNK, respectively. The concentrations of the kinase inhibitors used in the present study were adopted from previous reports demonstrating that the desired kinases were inhibited in HepG2 cells under these conditions (23, 75, 81). The cells were harvested at day 4 after transfection, and the levels of HBV RNAs were measured by Northern blot hybridization. Consistent with results presented in Fig. 2, cotransfection of pHBV1.3 with plasmid expressing IPS-1, saTRIF, or MyD88 reduced the levels of HBV RNA (Fig. 6A, lanes 1 to 4). Interestingly, treatment of HBV genome-transfected cells with p38, ERK, or JNK inhibitors in the absence of PRR adaptor overexpression reduced the level of HBV pgRNA by ca. 30, 70, and 60%, respectively (Fig. 6, compare lanes 1 to lanes 5, 9, and 13, respectively). This result implied that the basal level activities of the three MAPKs might be required for the maximum transcription of HBV mRNAs in HepG2 cells. However, as shown in Fig. 6B, overexpression of the three adaptors resulted in similar extents of HBV RNA reductions in the absence or presence of the MAPK inhibitors and thus suggested that none of the inhibitors is able to attenuate the inhibitory effects of the three adaptors on HBV replication.
FIG. 6.

The three major MAPKs are not involved in the inhibition of HBV replication by the innate immune response. (A) HepG2 cells were cotransfected with 2 μg of pHBV1.3 and 2 μg of control plasmid (lanes 1, 5, 9, and 13) or 1 μg of control plasmid plus 1 μg of plasmid expressing IPS-1 (lanes 2, 6, 10, and 14), TRIF (lanes 3, 7, 11, and 15), and MyD88 (lanes 4, 8, 12, and 16). Twelve hours after transfection, cells were left untreated (lanes 1 to 4) or treated with 25 μM MAPK p38 inhibitor SB202190 (lanes 5 to 8), 10 μM ERK inhibitor U0126 (lanes 9 to 12), or 25 μM JNK inhibitor SP600125 (lanes 13 to 16). The dimethyl sulfoxide concentration in all experiment groups was normalized at 0.1%. Culture media and the inhibitor were replaced every other day, and cells were harvested at day 4 after transfection. Total RNA was extracted and analyzed by Northern blot hybridization. rRNAs served as loading control. The RNA level in each sample is expressed as the percentage of RNA level in control cells cotransfected with pHBV1.3 and vector plasmid, but left untreated (lane 1). (B) RNA levels in the adaptor-expressing plasmids transfected cells were plotted as the percentage of that in cells cotransfected with pHBV1.3 and vector plasmid and left untreated (lane 1) or treated with the indicated kinase inhibitors in the absence of adaptor protein overexpression (lanes 5, 9, or 13), respectively.
Role of NF-κB and IRF3-mediated signaling in the inhibition of HBV replication.
In addition to the MAPKs, NF-κB and IRF3 are two key transcription factors for PRR-mediated induction of proinflammatory cytokines, IFNs, and many other cellular genes (29). To determine their roles in the antiviral responses elicited by the three PRR adaptors, the activation of NF-κB and the function of activated IRF3 can be selectively inhibited by the well-characterized DN-IκBα and DN-IRF3, respectively (28). To provide a basis for further pathway analysis, we first examined the effects of disrupting the NF-κB and IRF3-mediated signaling pathways on HBV replication without overexpression of PRR adaptors. As shown in Fig. 7, the results demonstrated that expression of either wild-type or dominant-negative IκB and IRF3 did not affect the levels of HBV RNAs in the two cell lines. However, it is interesting that overexpression of saIRF3 activated a similar innate immune response (Fig. 4B, compare lanes 7 and 14) and induced ISG56 expression (Fig. 7, lane 3) in both HepG2 and Huh7 cells but only dramatically reduced levels of HBV RNA and DNA in Huh7 cells (Fig. 7, upper and lower panels, lane 3, and data not shown). The reason for the cell-line-specific antiviral response induced by saIRF3 is currently not known.
FIG. 7.
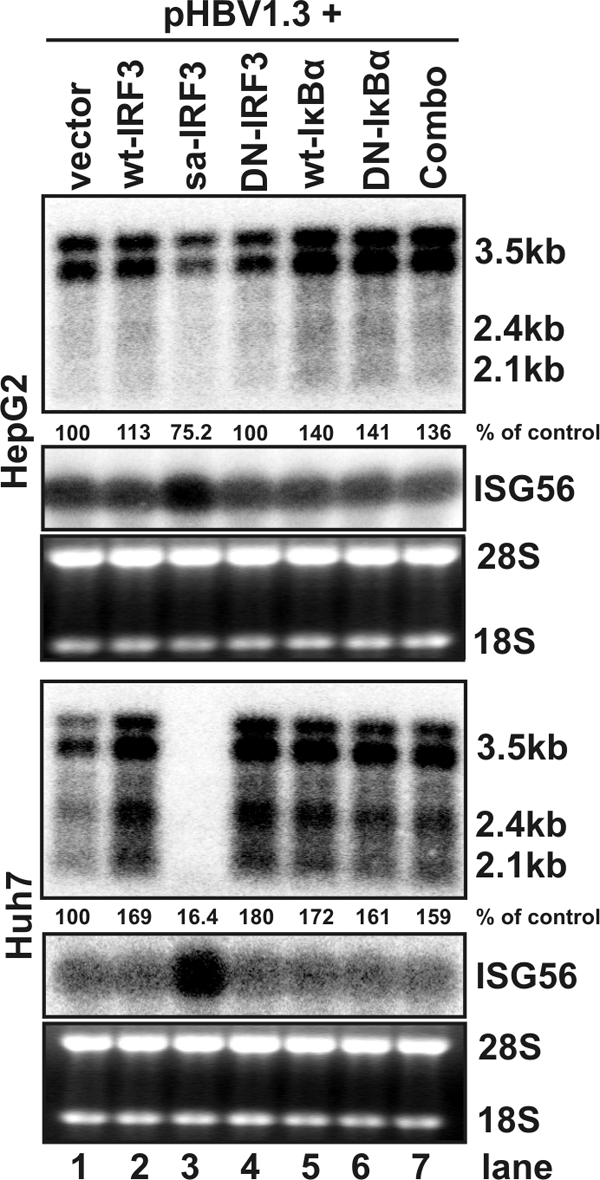
Antiviral effects of IRF3 and NF-κB-mediated signaling pathways in HepG2 and Huh7 cells. HepG2 (upper panel) or Huh7 (lower panel) cells were cotransfected with 2 μg of pHBV1.3 and 2 μg of control plasmid (lane 1) or plasmid expressing wild-type IRF3 (lane 2), saIRF3 (lane 3), DN-IRF3 (lane 4), wild-type IκBα (lane 5), DN-IκBα (lane 6), or DN-IRF3 and DN-IκB-α in combination (lane 7). Cells were harvested at 4 day after transfection, and the levels of viral RNAs were determined by Northern blot hybridization analysis. Induction of ISG56 mRNA serves as an indicator of IRF3 activation in transfected cells. rRNAs (28S and 18S) were presented as loading controls. The positions of the HBV 3.5-, 2.4-, and 2.1-kb RNAs are indicated. The relative RNA level in each sample is expressed as the percentage of RNA level in control cells cotransfected with pHBV1.3 and vector plasmid (lane 1) and is given underneath each of the blots.
We next investigated the role of the two transcription factor-mediated signaling pathways in the PRR adaptor-elicited innate immunity against HBV. HepG2 and Huh7 cells were either left untreated or transfected with control plasmid or cotransfected with pHBV1.3 and plasmids expressing each of the three adaptors and plasmids expressing the DN-IRF3 and/or DN-IκBα. Cells were harvested at 4 days after transfection, and the levels of viral RNAs were determined by Northern blot hybridization analysis. Induction of ISG56 mRNA (Fig. 8A and B, middle panel) served as an indicator for IRF3 activation in the transfected cells. The results from these experiments (Fig. 8) revealed the following. First, as shown in Fig. 2, TRIF and IPS-1 induced a strong reduction of viral mRNAs (Fig. 8A and B, top panel) in both HepG2 and Huh7 cells, but the antiviral effects of MyD88 are less potent (Fig. 8A and B, compare lane 3 to lanes 4 to 6). Second, it was DN-IκBα, but not DN-IRF3, that restored the levels of HBV RNA in the two cell lines transfected with MyD88 (Fig. 8A and B, compare lanes 6, 9, and 12). Third, although ISG56 mRNA was induced by IPS-1 and TRIF in both HepG2 and Huh7 cells and, as expected, the induction was inhibited by expression of DN-IRF3, the levels of HBV mRNAs were only restored by DN-IRF3 in Huh7, but not HepG2 cells that overexpressed IPS-1(Fig. 8A and B, compare lanes 4 and 7). In contrast, cotransfection of plasmid expressing DN-IκBα efficiently attenuated the antiviral effects of IPS-1 in both HepG2 and Huh7 cells (Fig. 8A and B, compare lanes 4 and 10). Finally, TRIF induced antiviral response can only be partially attenuated by expression of DN-IκBα in the two cell lines, but expression of DN-IRF3 had no effect (Fig. 8A and B, compare lanes 5, 8, and 11), indicating that additional unknown signaling pathway(s) are required to mediate TRIF-induced antiviral response against HBV in human hepatoma cells.
FIG. 8.

Role of IRF3 and NF-κB in mediating the antiviral response elicited by host cellular innate immunity. HepG2 (A) or Huh7 (B) cells in a 35-mm-diameter dish were either left untreated (lane 1), transfected with 4 μg of control plasmid (lane 2), or cotransfected with 4 μg of plasmids containing 2 μg of pHBV1.3, 1 μg of plasmid expressing each of the three adaptors, and 1 μg of plasmid expressing DN-IRF3 and/or DN-IκBα, plus 1 μg of control plasmid (omitted when DN-IRF3 and DN-IκBα were in combination). Cells were harvested 4 days after transfection, and the levels of viral RNAs were determined by Northern blot hybridization. Induction of ISG56 mRNA serves as an indicator of IRF3 activation in transfected cells. rRNAs (28S and 18S) were presented as loading controls. The positions of the HBV 3.5-, 2.4-, and 2.1-kb RNAs are indicated. The RNA level in each sample is expressed as the percentage of that in control cells cotransfected with pHBV1.3 and vector plasmid (lane 3) and is given underneath the blots.
Taken together, the results presented here suggest that NF-κB is required for all three adaptors to elicit antiviral response in both HepG2 and Huh7 cells. However, although it was efficiently activated by TRIF or IPS-1 in both HepG2 and Huh7 cell, as indicated by ISG56 mRNA induction and inhibition by DN-IRF3, the IRF3 is only essential for induction of an antiviral response by IPS-1 in Huh7 cells.
DISCUSSION
Our work presented here shows that either IFN treatment or overexpression of the adaptors that mediate the signaling of the major viral PRRs noncytopathically inhibited HBV replication in human hepatocyte-derived cell lines (Fig. 2). Moreover, three lines of evidence support the notion that the PRR adaptor-elicited antiviral response was mediated by intracellular factors, rather than secreted cytokines. First, the adaptor-induced antiviral response was far more potent than that induced by high concentrations of IFN-α or IFN-λ (Fig. 2 and Fig. 5). Second, treatment of HBV genome-transfected HepG2 and Huh7 cells with culture medium harvested from the adaptor-expressing plasmid-transfected HepG2 and Huh7, respectively, did not significantly inhibit HBV replication (Fig. 5). Finally, analyses of the involved signal transduction pathways in the adaptor-induced antiviral response suggested that activation of IRF3 is only essential for induction of antiviral response by IPS-1 in Huh7 cells but is not essential for all three adaptors to induce antiviral response in HepG2 cells (Fig. 8). It is well known that IRF3 is essential for the induction of type I and type III IFNs by both IPS-I and TRIF. Hence, IFNs should not be the mediators of adaptor-induced antiviral response in HepG2 cells (Fig. 8). In addition, it is worth noting that the primary effect of either IFNs or host cellular innate immunity elicited by the PRR adaptors on HBV replication in HepG2 and Huh7 cells is to reduce the steady-state levels of viral RNAs but not to inhibit viral DNA replication (Fig. 2 and 3).
Previous studies with HBV transgenic mice or duck HBV-infected primary duck hepatocytes indicated that there are several distinct intracellular mechanisms that can be activated by innate and/or adaptive immune responses to limit hepadnavirus replication. First, it was convincingly demonstrated that IFN-α/β treatment of HBV replicating mouse hepatocytes or duck HBV replicating primary duck hepatocytes and chicken hepatoma cells does not affect the level of viral RNA but selectively inhibits the formation of pgRNA-containing nucleocapsids. As a consequence, viral DNA replication is inhibited in the cells treated with the cytokines (21, 56, 74). Second, it was shown that adoptive transfer of HBV surface antigen (HBsAg)-specific cytotoxic T lymphocytes (CTLs) or injection of interleukin-2 (IL-2) resulted in a posttranscriptional downregulation of viral mRNA in IFN-γ and tumor necrosis factor alpha-dependent mechanisms (17, 68). Third, acute infection of lymphocytic choriomeningitis virus or repeated injection of IFN-α/β inducer poly(I-C) into HBV transgenic mice inhibited HBV RNA transcription (70). Finally, a single intravenous injection of ligands specific for TLR3, TLR4, TLR5, TLR7, and TLR9 into HBV transgenic mice noncytolytically inhibited HBV replication within 24 h in an IFN-α/β-dependent manner (32). Thus far, despite several reports suggesting that IFN treatment of human-hepatocyte-derived cells inhibited HBV RNA transcription or posttranscriptionally reduced viral mRNA levels, the inhibition of pgRNA-containing capsid formation by IFNs or other cytokines had not yet been convincingly demonstrated in any of human cell lines (52, 69). Hence, it is possible that the molecular mechanism by which the innate immunity controls HBV replication is species specific.
Analysis of signaling pathways required for each of adaptors to elicit antiviral responses against HBV revealed that activation of NF-κB is essential for all three adaptors to elicit antiviral response in both HepG2 and Huh7 cells. However, activation of IRF3 is only essential for the induction of antiviral response by IPS-1 in Huh7 cells and not HepG2 cells. It is paradoxical that despite the fact that expression of ISG56, IFN-β and IFN-λ1was as efficiently induced in HepG2 cells as in Huh7 cells (Fig. 4B, compare lanes 7 and 14), saIRF3 only elicited robust antiviral response in Huh7 cells (Fig. 7, upper and lower panels, lane 3). Consistent with this observation, the IRF3 is only essential for induction of an antiviral response by IPS-1 in Huh7 cells and not in HepG2 cells. These findings suggest that IPS-1 might downregulate the levels of HBV mRNA via distinct mechanisms in HepG2 and Huh7 cells (Fig. 7 and 8). Moreover, our results also suggest that additional signaling pathway(s) other than MAPKs, NF-κB, and IRF3 are required for TRIF to induce a maximum antiviral effect against HBV.
In our effort to determine whether the observed reduction of HBV RNAs upon overexpression of the three adaptors is via the transcriptional and/or posttranscriptional mechanisms, we found that both HBV pgRNA and 2.1-kb subgenomic RNA can be efficiently reduced by the three adaptors in an HBV promoter-independent manner. However, the level of a 1.0-kb truncated HBV mRNA that spans only the coding region of HBV small envelope protein (HBsAg) was not affected by the three adaptors in HepG2 cells (Fig. 3). These results suggest that the reduction of HBV mRNAs by all three adaptors in HepG2 cells is most likely due to posttranscriptional decay of viral RNAs and RNA sequence element(s) located in the 3′ overlapping region of HBV mRNAs is critical for the selective reduction of viral RNA. Although it is not yet known whether the same or distinct effector molecules are induced by the different PRR adaptors in HepG2 cells to downregulate HBV RNA, microRNAs (miRNAs) and/or lupus-associated antigen (La) are two cellular factors that might possibly accelerate the posttranscriptional decay of viral RNAs and thus deserve a serious consideration in future studies.
MicroRNAs are small noncoding RNAs of approximately 22 nucleotides in length. They are derived from cellular transcripts and sequence specifically bind to their targeting mRNAs to result in either mRNA cleavage or translational repression (3, 4, 33). miRNAs have been demonstrated to play an important role in defending the hosts against virus infection in plants and invertebrates (10), but it has been argued that in mammals this sequence-specific innate antiviral immunity has been replaced by a sequence-nonspecific, dsRNA-triggered, IFN-mediated innate host defense mechanism (9, 49). However, recent studies suggest that cellular miRNAs can be induced by TLR signaling and IFN treatment to regulate innate immune signal transduction or even directly inhibit the replication of HCV (48, 46, 64, 63). It is, therefore, entirely possible that certain cellular miRNAs could be induced by the innate immune response and directly catalyze viral RNA decay or indirectly modulate cellular RNA decay enzyme expression.
Alternatively, as mentioned above, selective decay of HBV mRNA has been previously observed in the livers of HBV transgenic mice injected with HBsAg-specific CTLs or cytokines such as IL-2. It was shown that the process is mediated by tumor necrosis factor alpha and IFN-γ (17, 68). In an attempt to identify host factors involved in degradation of the viral RNA, the mouse La protein (mLa) was identified as an HBV RNA-specific binding protein, which presumably binds and stabilizes HBV RNA in the nuclei of hepatocytes (25). It was found that the CTLs or IL-2 treatment resulted in the processing of full-length mLa, which render viral RNA vulnerable for degradation by cellular nucleases. Moreover, the mLa binding site was mapped to a predicted stem-loop structure within an element 91 nucleotides long located at the 5′ end of the posttranscriptional regulatory element of HBV (16, 26). Interestingly, our results presented in Fig. 3 demonstrated that the nucleotide sequence in the nontranslational region of HBV 2.1-kb mRNA, where the mLa binding site is located, is critical for HBV RNA decay induced by the three PRR adaptors. Hence, it will be interesting to determine whether the adaptors induce the processing of human La antigen in HepG2 and Huh7 cells and examine the role of human La in HBV RNA decay in the innate immune response elicited by each of the three adaptors.
In summary, although it is not yet known how the PRR adaptor-elicited cellular innate immunity downregulates the levels of HBV RNAs, the results presented here clearly indicate the existence of an intricate antiviral mechanism in human hepatocytes that could be activated under certain conditions and potently inhibit HBV replication in a noncytopathic fashion. Noncytopathic inhibition of viral gene expression is an attractive therapeutic means of controlling infections, especially in the case of HBV, where the 3.5-kb RNA serves as the template for reverse transcription of the encapsidated viral DNA genome. Further understanding the nature of these antiviral mechanisms should provide valuable insights into novel strategies for the development of antivirals that evoke the antiviral response to eliminate HBV infection.
Acknowledgments
We thank Pamela Norton and Anand Mehta for critical reading of the manuscript.
This study was supported by a grant from the National Institutes of Health (AI061441) and by the Hepatitis B Foundation through an appropriation of the Commonwealth of Pennsylvania. H.G. and J.-T.G. are Bruce Witte fellow and scholar of the Hepatitis B Foundation, respectively.
Footnotes
Published ahead of print on 29 October 2008.
REFERENCES
- 1.Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124783-801. [DOI] [PubMed] [Google Scholar]
- 2.Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413732-738. [DOI] [PubMed] [Google Scholar]
- 3.Ambros, V. 2004. The functions of animal microRNAs. Nature 431350-355. [DOI] [PubMed] [Google Scholar]
- 4.Bartel, D. P. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116281-297. [DOI] [PubMed] [Google Scholar]
- 5.Bigger, C. B., K. M. Brasky, and R. E. Lanford. 2001. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J. Virol. 757059-7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bigger, C. B., B. Guerra, K. M. Brasky, G. Hubbard, M. R. Beard, B. A. Luxon, S. M. Lemon, and R. E. Lanford. 2004. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 7813779-13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chi, H., S. P. Barry, R. J. Roth, J. J. Wu, E. A. Jones, A. M. Bennett, and R. A. Flavell. 2006. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. USA 1032274-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chisari, F. V., and C. Ferrari. 1995. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 1329-60. [DOI] [PubMed] [Google Scholar]
- 9.Cullen, B. R. 2006. Is RNA interference involved in intrinsic antiviral immunity in mammals? Nat. Immunol. 7563-567. [DOI] [PubMed] [Google Scholar]
- 10.Ding, S. W., and O. Voinnet. 2007. Antiviral immunity directed by small RNAs. Cell 130413-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elco, C. P., J. M. Guenther, B. R. Williams, and G. C. Sen. 2005. Analysis of genes induced by Sendai virus infection of mutant cell lines reveals essential roles of interferon regulatory factor 3, NF-κB, and interferon but not Toll-like receptor 3. J. Virol. 793920-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Funami, K., M. Sasai, H. Oshiumi, T. Seya, and M. Matsumoto. 2008. Homo-oligomerization is essential for Toll/IL-1 receptor domain-containing adaptor molecule-1-mediated NF-κB and IRF-3 activation. J. Biol. Chem. 28318283-18291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganem, D., and A. M. Prince. 2004. Hepatitis B infection: natural history and clinical consequences. N. Engl. J. Med. 3501118-1129. [DOI] [PubMed] [Google Scholar]
- 14.Garcia, M. A., J. Gil, I. Ventoso, S. Guerra, E. Domingo, C. Rivas, and M. Esteban. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 701032-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grandvaux, N., M. J. Servant, B. tenOever, G. C. Sen, S. Balachandran, G. N. Barber, R. Lin, and J. Hiscott. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 765532-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guang, S., and J. E. Mertz. 2005. Pre-mRNA processing enhancer (PPE) elements from intronless genes play additional roles in mRNA biogenesis than do ones from intron-containing genes. Nucleic Acids Res. 332215-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guilhot, S., L. G. Guidotti, and F. V. Chisari. 1993. Interleukin 2 downregulates hepatitis B virus gene expression in transgenic mice by a posttranslational mechanism. J. Virol. 677444-7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo, H., W. S. Mason, C. E. Aldrich, J. R. Saputelli, D. S. Miller, A. R. Jilbert, and J. E. Newbold. 2005. Identification and characterization of avihepadnaviruses isolated from exotic anseriformes maintained in captivity. J. Virol. 792729-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo, H., T. Zhou, D. Jiang, A. Cuconati, G. H. Xiao, T. M. Block, and J. T. Guo. 2007. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-akt signal transduction pathway. J. Virol. 8110072-10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo, J. T., V. V. Bichko, and C. Seeger. 2001. Effect of alpha interferon on the hepatitis C virus replicon. J. Virol. 758516-8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo, J. T., M. Pryce, X. Wang, M. I. Barrasa, J. Hu, and C. Seeger. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J. Virol. 771885-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haller, O., G. Kochs, and F. Weber. 2006. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344119-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hassan, M., H. Ghozlan, and O. Abdel-Kader. 2005. Activation of c-Jun NH2-terminal kinase (JNK) signaling pathway is essential for the stimulation of hepatitis C virus (HCV) nonstructural protein 3 (NS3)-mediated cell growth. Virology 333324-336. [DOI] [PubMed] [Google Scholar]
- 24.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 3031526-1529. [DOI] [PubMed] [Google Scholar]
- 25.Heise, T., L. G. Guidotti, V. J. Cavanaugh, and F. V. Chisari. 1999. Hepatitis B virus RNA-binding proteins associated with cytokine-induced clearance of viral RNA from the liver of transgenic mice. J. Virol. 73474-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heise, T., L. G. Guidotti, and F. V. Chisari. 1999. La autoantigen specifically recognizes a predicted stem-loop in hepatitis B virus RNA. J. Virol. 735767-5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helbig, K. J., D. T. Lau, L. Semendric, H. A. Harley, and M. R. Beard. 2005. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology 42702-710. [DOI] [PubMed] [Google Scholar]
- 28.Heylbroeck, C., S. Balachandran, M. J. Servant, C. DeLuca, G. N. Barber, R. Lin, and J. Hiscott. 2000. The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J. Virol. 743781-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honda, K., and T. Taniguchi. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6644-658. [DOI] [PubMed] [Google Scholar]
- 30.Hoofnagle, J. H., and A. M. di Bisceglie. 1997. The treatment of chronic viral hepatitis. N. Engl. J. Med. 336347-356. [DOI] [PubMed] [Google Scholar]
- 31.Hornung, V., J. Ellegast, S. Kim, K. Brzozka, A. Jung, H. Kato, H. Poeck, S. Akira, K. K. Conzelmann, M. Schlee, S. Endres, and G. Hartmann. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314994-997. [DOI] [PubMed] [Google Scholar]
- 32.Isogawa, M., M. D. Robek, Y. Furuichi, and F. V. Chisari. 2005. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 797269-7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson, R. J., and N. Standart. 2007. How do microRNAs regulate gene expression? Sci. STKE 2007re1. [DOI] [PubMed] [Google Scholar]
- 34.Jenner, R. G., and R. A. Young. 2005. Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 3281-294. [DOI] [PubMed] [Google Scholar]
- 35.Jiang, D., H. Guo, C. Xu, J. Chang, B. Gu, L. Wang, T. M. Block, and J. T. Guo. 2008. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 821665-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser, W. J., and M. K. Offermann. 2005. Apoptosis induced by the Toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 1744942-4952. [DOI] [PubMed] [Google Scholar]
- 37.Kawai, T., and S. Akira. 2006. TLR signaling. Cell Death Differ. 13816-825. [DOI] [PubMed] [Google Scholar]
- 38.Kawai, T., K. Takahashi, S. Sato, C. Coban, H. Kumar, H. Kato, K. J. Ishii, O. Takeuchi, and S. Akira. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6981-988. [DOI] [PubMed] [Google Scholar]
- 39.Krug, A., A. R. French, W. Barchet, J. A. Fischer, A. Dzionek, J. T. Pingel, M. M. Orihuela, S. Akira, W. M. Yokoyama, and M. Colonna. 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21107-119. [DOI] [PubMed] [Google Scholar]
- 40.Lanford, R. E., B. Guerra, H. Lee, D. Chavez, K. M. Brasky, and C. B. Bigger. 2006. Genomic response to interferon-alpha in chimpanzees: implications of rapid downregulation for hepatitis C kinetics. Hepatology 43961-972. [DOI] [PubMed] [Google Scholar]
- 41.Li, K., Z. Chen, N. Kato, M. Gale, Jr., and S. M. Lemon. 2005. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 28016739-16747. [DOI] [PubMed] [Google Scholar]
- 42.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 182986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMahon, B. J. 2005. Epidemiology and natural history of hepatitis B. Semin. Liver Dis. 25(Suppl. 1)3-8. [DOI] [PubMed] [Google Scholar]
- 44.Meylan, E., J. Curran, K. Hofmann, D. Moradpour, M. Binder, R. Bartenschlager, and J. Tschopp. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 4371167-1172. [DOI] [PubMed] [Google Scholar]
- 45.Muller, U., U. Steinhoff, L. F. Reis, S. Hemmi, J. Pavlovic, R. M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science 2641918-1921. [DOI] [PubMed] [Google Scholar]
- 46.O'Connell, R. M., K. D. Taganov, M. P. Boldin, G. Cheng, and D. Baltimore. 2007. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 1041604-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Onoguchi, K., M. Yoneyama, A. Takemura, S. Akira, T. Taniguchi, H. Namiki, and T. Fujita. 2007. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 2827576-7581. [DOI] [PubMed] [Google Scholar]
- 48.Pedersen, I. M., G. Cheng, S. Wieland, S. Volinia, C. M. Croce, F. V. Chisari, and M. David. 2007. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449919-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pfeffer, S., and O. Voinnet. 2006. Viruses, microRNAs, and cancer. Oncogene 256211-6219. [DOI] [PubMed] [Google Scholar]
- 50.Pichlmair, A., and C. Reis e Sousa. 2007. Innate recognition of viruses. Immunity 27370-383. [DOI] [PubMed] [Google Scholar]
- 51.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314997-1001. [DOI] [PubMed] [Google Scholar]
- 52.Rang, A., S. Gunther, and H. Will. 1999. Effect of interferon alpha on hepatitis B virus replication and gene expression in transiently transfected human hepatoma cells. J. Hepatol. 31791-799. [DOI] [PubMed] [Google Scholar]
- 53.Saito, T., R. Hirai, Y. M. Loo, D. Owen, C. L. Johnson, S. C. Sinha, S. Akira, T. Fujita, and M. Gale, Jr. 2007. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 104582-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schormann, W., A. Kraft, D. Ponsel, and V. Bruss. 2006. Hepatitis B virus particle formation in the absence of pregenomic RNA and reverse transcriptase. J. Virol. 804187-4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schultz, U., J. Summers, P. Staeheli, and F. V. Chisari. 1999. Elimination of duck hepatitis B virus RNA-containing capsids in duck interferon-alpha-treated hepatocytes. J. Virol. 735459-5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sen, G. C., and G. A. Peters. 2007. Viral stress-inducible genes. Adv. Virus Res. 70233-263. [DOI] [PubMed] [Google Scholar]
- 58.Sen, G. C., and S. N. Sarkar. 2007. The interferon-stimulated genes: targets of direct signaling by interferons, double-stranded RNA, and viruses. Curr. Top. Microbiol. Immunol. 316233-250. [DOI] [PubMed] [Google Scholar]
- 59.Seth, R. B., L. Sun, C. K. Ea, and Z. J. Chen. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122669-682. [DOI] [PubMed] [Google Scholar]
- 60.Smith, M. W., Z. N. Yue, M. J. Korth, H. A. Do, L. Boix, N. Fausto, J. Bruix, R. L. Carithers, Jr., and M. G. Katze. 2003. Hepatitis C virus and liver disease: global transcriptional profiling and identification of potential markers. Hepatology 381458-1467. [DOI] [PubMed] [Google Scholar]
- 61.Stetson, D. B., and R. Medzhitov. 2006. Type I interferons in host defense. Immunity 25373-381. [DOI] [PubMed] [Google Scholar]
- 62.Su, A. I., J. P. Pezacki, L. Wodicka, A. D. Brideau, L. Supekova, R. Thimme, S. Wieland, J. Bukh, R. H. Purcell, P. G. Schultz, and F. V. Chisari. 2002. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 9915669-15674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taganov, K. D., M. P. Boldin, and D. Baltimore. 2007. MicroRNAs and immunity: tiny players in a big field. Immunity 26133-137. [DOI] [PubMed] [Google Scholar]
- 64.Taganov, K. D., M. P. Boldin, K. J. Chang, and D. Baltimore. 2006. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 10312481-12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takahasi, K., M. Yoneyama, T. Nishihori, R. Hirai, H. Kumeta, R. Narita, M. Gale, Jr., F. Inagaki, and T. Fujita. 2008. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 29428-440. [DOI] [PubMed] [Google Scholar]
- 66.Takeuchi, O., and S. Akira. 2008. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2017-22. [DOI] [PubMed] [Google Scholar]
- 67.Traenckner, E. B., H. L. Pahl, T. Henkel, K. N. Schmidt, S. Wilk, and P. A. Baeuerle. 1995. Phosphorylation of human I κB-alpha on serines 32 and 36 controls I κB-alpha proteolysis and NF-κB activation in response to diverse stimuli. EMBO J. 142876-2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsui, L. V., L. G. Guidotti, T. Ishikawa, and F. V. Chisari. 1995. Posttranscriptional clearance of hepatitis B virus RNA by cytotoxic T lymphocyte-activated hepatocytes. Proc. Natl. Acad. Sci. USA 9212398-12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tur-Kaspa, R., L. Teicher, O. Laub, A. Itin, D. Dagan, B. R. Bloom, and D. A. Shafritz. 1990. Alpha interferon suppresses hepatitis B virus enhancer activity and reduces viral gene transcription. J. Virol. 641821-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uprichard, S. L., S. F. Wieland, A. Althage, and F. V. Chisari. 2003. Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc. Natl. Acad. Sci. USA 1001310-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vilcek, J. 2006. Fifty years of interferon research: aiming at a moving target. Immunity 25343-348. [DOI] [PubMed] [Google Scholar]
- 72.Wieland, S., R. Thimme, R. H. Purcell, and F. V. Chisari. 2004. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 1016669-6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wieland, S. F., and F. V. Chisari. 2005. Stealth and cunning: hepatitis B and hepatitis C viruses. J. Virol. 799369-9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wieland, S. F., A. Eustaquio, C. Whitten-Bauer, B. Boyd, and F. V. Chisari. 2005. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc. Natl. Acad. Sci. USA 1029913-9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu, J. H., L. C. Hong, Y. Y. Tsai, H. W. Chen, W. X. Chen, and T. S. Wu. 2006. Mitogen-activated protein kinase (MAPK) signalling pathways in HepG2 cells infected with a virulent strain of Klebsiella pneumoniae. Cell Microbiol. 81467-1474. [DOI] [PubMed] [Google Scholar]
- 76.Xiong, W., X. Wang, X. Liu, L. Xiang, L. Zheng, and Z. Yuan. 2004. Interferon-inducible MyD88 protein inhibits hepatitis B virus replication. Virology 319306-314. [DOI] [PubMed] [Google Scholar]
- 77.Xu, L. G., Y. Y. Wang, K. J. Han, L. Y. Li, Z. Zhai, and H. B. Shu. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19727-740. [DOI] [PubMed] [Google Scholar]
- 78.Yoneyama, M., M. Kikuchi, K. Matsumoto, T. Imaizumi, M. Miyagishi, K. Taira, E. Foy, Y. M. Loo, M. Gale, Jr., S. Akira, S. Yonehara, A. Kato, and T. Fujita. 2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 1752851-2858. [DOI] [PubMed] [Google Scholar]
- 79.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5730-737. [DOI] [PubMed] [Google Scholar]
- 80.Zaru, R., N. Ronkina, M. Gaestel, J. S. Arthur, and C. Watts. 2007. The MAPK-activated kinase Rsk controls an acute Toll-like receptor signaling response in dendritic cells and is activated through two distinct pathways. Nat. Immunol. 81227-1235. [DOI] [PubMed] [Google Scholar]
- 81.Zheng, Y., J. Li, D. L. Johnson, and J. H. Ou. 2003. Regulation of hepatitis B virus replication by the ras-mitogen-activated protein kinase signaling pathway. J. Virol. 777707-7712. [DOI] [PMC free article] [PubMed] [Google Scholar]