

Abstract
Positive transcription elongation factor b (P-TEFb), composed of cyclin-dependent kinase 9 (CDK9) and cyclin T, is a global transcription factor for eukaryotic gene expression, as well as a key factor for human immunodeficiency virus (HIV) transcription elongation. P-TEFb phosphorylates the carboxyl-terminal domain (CTD) of the large subunit of RNA polymerase II (RNAP II), facilitating the transition from nonprocessive to processive transcription elongation. Recently, the bromodomain protein Brd4 has been shown to interact with the low-molecular-weight, active P-TEFb complex and recruit P-TEFb to the HIV type 1 long terminal repeat (LTR) promoter. However, the subsequent events through which Brd4 regulates CDK9 kinase activity and RNAP II-dependent transcription are not clearly understood. Here we provide evidence that Brd4 regulates P-TEFb kinase activity by inducing a negative pathway. Moreover, by analyzing stepwise initiation and elongation complexes, we demonstrate that P-TEFb activity is regulated in the transcription complex. Brd4 induces phosphorylation of CDK9 at threonine 29 (T29) in the HIV transcription initiation complex, inhibiting CDK9 kinase activity. P-TEFb inhibition is transient, as Brd4 is released from the transcription complex between positions +14 and +36. Removal of the phosphate group at T29 by an incoming phosphatase released P-TEFb activity, resulting in increased RNAP II CTD phosphorylation and transcription. Finally, we present chromatin immunoprecipitation studies showing that CDK9 with phosphorylated T29 is associated with the HIV promoter region in the integrated and transcriptionally silent HIV genome.
The largest subunit of eukaryotic RNA polymerase II (RNAP II) has a carboxyl-terminal domain (CTD) consisting of tandem repeats of the consensus heptad peptide Tyr-Ser-Pro-Thr-Ser-Pro-Ser, which is conserved among most eukaryotes (3, 17, 30). The CTD is phosphorylated at Ser 2 (Ser 2P) and Ser 5 (Ser 5P) as RNAP II progresses through the transcription initiation and elongation. At least three cyclin-dependent kinases are capable of phosphorylating the RNAP II CTD in the regulation of different stages of mRNA synthesis. The first kinase, cyclin-dependent kinase 7 (CDK7), along with MAT1 and cyclin H, forms a complex known as CDK-activating kinase which is part of the general transcription factor TFIIH (9, 23, 35, 45). As analyzed both in vitro and in vivo, TFIIH facilitates promoter clearance by preferentially phosphorylating Ser 5 of the RNAP II CTD within the promoter region (18, 36, 40). The second kinase, CDK9, is a cdc2-like serine/threonine kinase that was initially isolated by screening a human cDNA library by using oligonucleotide probes to identify CDK-related proteins (12). Consistent with the process of selection, the 43-kDa CDK9 protein shares a high degree of homology with cdc2, cdk2, cdk3, and cdk5. Understanding the function of CDK9 was facilitated by the discovery that CDK9 was the kinase subunit of positive transcription elongation factor b (P-TEFb), which supports transcription elongation (32). The primary cyclin partner of CDK9 is cyclin T1, but complexes may also include cyclin T2a, T2b, or cyclin K (10, 31). P-TEFb preferentially phosphorylates Ser 2 of the RNAP II CTD (11, 47). CDK8, the third kinase, forms a complex with cyclin C; it has been reported to interact with the RNAP II holoenzyme and MED/SRB-containing complexes, such as TRAP/SMCC and NAT, which negatively regulate activated but not basal transcription (20, 21, 34, 39). CDK8 appears to phosphorylate the CTD primarily on Ser 5 within the heptapeptide repeat, although Ser 2 phosphorylation has also been reported (13, 33, 39).
Brd4 is a mammalian bromodomain protein that binds to acetylated chromatin (4). Brd4 is a ubiquitously expressed nuclear protein of 200 kDa that plays a role in the regulation of cell growth (5, 6, 24). Brd4 or a Brd4-like protein has been reported to be contained in a human transcriptional mediator complex, suggesting that Brd4 may also participate in transcription (16). It has been reported that proteomic analysis revealed that Brd4 interacts with the active, low-molecular-weight form of P-TEFb (14). Although an early study suggested that Brd4 interacts directly with the cyclin T subunit of P-TEFb through its bromodomain (14), a recent report further suggests that a conserved region in the CTD of Brd4 mediates its specific interaction with P-TEFb (2). About half of cellular P-TEFb associates with Brd4, and the other half exists in a high-molecular-weight complex with the 7SK snRNA and the HEXIM1 protein (25, 28, 44, 46). In the low-molecular-weight complex, Brd4 has been shown to recruit P-TEFb to eukaryotic promoters, including the human immunodeficiency virus type 1 (HIV-1) long terminal repeat (LTR) promoter (43). Consistent with these findings, Ni et al. demonstrated that P-TEFb is critical for the transition of RNAP II into mature transcription elongation complex (TEC) (29). Within three minutes of P-TEFb inhibition, most RNAP II was restricted to within 150 bp of the transcription initiation site of the active Drosophila melanogaster Hsp70 gene. More recently, it has been shown that Brd4 specifically recruits P-TEFb to G1 growth-associated genes (26, 42). The subsequent events through which Brd4 affects the kinase activity of P-TEFb and regulates RNAP II-dependent transcription are not clearly understood.
The results presented in this study offer a unique new perspective on Brd4 and its interaction with the P-TEFb complex. Our results demonstrate that Brd4 inhibits CDK9 kinase activity by inducing the phosphorylation of threonine 29, an inhibitory phosphorylation site in CDK9. We also investigated the dynamic association of Brd4 with HIV-1 transcription complexes. Brd4 is released from transcription complexes between positions +14 and +36. Importantly, relief of the Brd4 inhibition of the CDK9 kinase activity increases transcription from the HIV-1 LTR. Chromatin immunoprecipitation (ChIP) analysis demonstrated that CDK9 with phospho-T29 (T29P CDK9) is associated with the HIV promoter region in the integrated and transcriptionally silent HIV genome. These results suggest that Brd4-induced T29P may play an important part in HIV latency.
MATERIALS AND METHODS
Purification of P-TEFb and Brd4.
CDK9 T29 mutants were produced and P-TEFb (CDK9/cyclinT1) protein complexes were purified as described previously (48). Brd4 was expressed and purified as described previously (14).
Production of T29P CDK9 antibody.
Antisera were raised and affinity purified against a CDK9 T29 phosphopeptide. Enzyme-linked immunosorbent assays using peptides containing the phosphorylated or nonphosphosphorylated form of CDK9 demonstrated the specificity of the antibody.
CTD kinase assay.
The CTD kinase assays were performed by mixing 50 ng glutathione S-transferase (GST)-CTD, 50 ng P-TEFb, 100 μM ATP, and 10 μCi [γ-32P]ATP and incubating for 60 min at 30°C. The total reaction mixture volume was 20 μl, and the final conditions were 50 mM Tris-HCl (pH 7.5), 5 mM dithiothreitol, 5 mM MnCl2, and 4 mM MgCl2. The phosphorylated GST-CTD was precipitated with glutathione-Sepharose beads and fractionated by electrophoresis on 8% sodium sulfate (SDS)-polyacrylamide gels, followed by autoradiography.
CDK9 autophosphorylation assay.
Autophosphorylation assays were performed by mixing 50 ng P-TEFb and 10 μCi [γ-32P]ATP in buffer (50 mM Tris-HCl [pH 7.5], 5 mM dithiothreitol, 5 mM MnCl2, and 4 mM MgCl2) and incubating for 60 min at 30°C. 32P-labeled CDK9 was immunoprecipitated with specific anti-CDK9 antibody and analyzed by electrophoresis on 4-to-20% SDS-polyacrylamide gels, followed by autoradiography.
Western blotting for T29P.
In vitro kinase assays were performed by incubating 50 ng P-TEFb with 100 μM ATP in the presence of different concentration of Brd4 for 60 min at 30°C. The phosphorylated CDK9 was fractionated by electrophoresis on a 4-to-20% SDS-protein gel and then transferred to a polyvinylidene difluoride membrane. The membrane was blocked by incubation in phosphate-buffered saline-Tween (PBS-T) buffer containing 0.2% Tween 20 and 10% milk for 2 to 3 h. Primary antibody (anti-T29P at 1:4,000) incubation was done in PBS-T buffer containing 0.2% Tween 20 and 10% milk overnight at 4°C. The membrane was rinsed two times in 20 ml PBS-T buffer containing 0.2% Tween 20 and then washed three times, for 10 min for the first time and 5 min the second and third times, with 50 ml PBS-T buffer containing 0.2% Tween 20. Secondary antibody (antirabbit antibody at 1:5,000) incubation was performed in PBS-T buffer containing 0.1% Tween 20 for 60 min at room temperature. The membrane was rinsed two times in 20 ml PBS-T buffer containing 0.1% Tween 20 and then washed five times, for 10 min the first time and 5 min the second through fifth times, with 50 ml PBS-T buffer containing 0.1% Tween 20.
IP of T29P.
In vitro kinase assays were performed by incubating 50 ng P-TEFb with 100 μM ATP in the presence or absence of Brd4 for 60 min at 30°C. Radio IP assay buffer was added to stop the reaction, and the volume was adjusted to 0.5 ml. Five micrograms T29P CDK9 antibody (rabbit) was added, and the reaction mixture gently rocked on a rocking platform overnight at 4°C. Protein A-Sepharose beads were added to the antigen/antibody mixture and incubated for 1 h at 4°C on a rocking platform. The protein A-antigen-antibody complex was collected by centrifugation at 12,000 × g for 20 s. The supernatant was removed, and the pellet was resuspended in 1 ml radio IP assay buffer by gentle vortexing, followed by incubation for 20 min at 4°C on a rocking platform. The wash step was repeated a total of four times. The immunoprecipitated T29P CDK9 was fractionated by electrophoresis on a 4-to-20% SDS-polyacrylamide gel, followed by Western blot analysis with anti-CDK9 antibody (mouse).
Purification of preinitiation complexes (PICs) and analysis of protein components of PICs.
Purification of PICs was carried out by using biotinylated templates as described previously (47). Briefly, PICs were assembled by incubating 500 ng biotinylated DNA templates with HeLa nuclear extract and then purified with streptavidin-coated magnetic beads. The beads were washed, and the protein components of PICs were analyzed by Western blotting with specific antibodies. The purified PICs were walked to position +U14 by incubation with 50 μM CTP, GTP, and UTP for 5 min at 30°C and then washed extensively with 1× in vitro transcription buffer (IVT buffer). The TECs stalled at U14 were walked stepwise along the DNA by repeated incubation with different sets of three nucleoside triphosphates (NTPs) and then washed extensively with 1× IVT buffer to remove the unincorporated NTPs. The protein compositions of TECs stalled at different stages were analyzed by fractionation by electrophoresis on 4-to-20% SDS-polyacrylamide gels, followed by Western blot analyses with specific antibodies.
In vitro transcription assay.
In vitro transcription reactions were performed as previously described (47). Briefly, reactions were set up by mixing templates, HeLa nuclear extracts, 50 μM ATP, 50 μM CTP, 50 μM GTP, 1.25 μM UTP, 20 μCi[α-32P]UTP, and 10 units of RNasin (Promega) in 1× IVT buffer. In vitro transcription reactions of purified TECs were performed by suspending the complexes in 1× IVT buffer, 50 μM ATP, 50 μM CTP, 50 μM GTP, 1.25 μM UTP, 20 μCi[α-32P]UTP, and 10 units of RNasin (Promega). The radiolabeled transcripts were fractionated by electrophoresis on 6% denaturing polyacrylamide gels and detected by autoradiography.
Luciferase assay.
HeLa cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. The plasmids containing wild-type (WT) or mutant CDK9 were cotransfected into 50 to 80% confluent HeLa cells with plasmids containing HIV LTR-driven luciferase. The cells were cultured for a further 48 h, and luciferase activity was assayed. The number of viable cells was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method.
IP of T29P for ChIP assay.
Following the in vitro kinase reactions, reaction mixtures were diluted to 400 μl with IP buffer (50 mM HEPES, pH 7.3, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS). Five micrograms of T29P antibody was added, and the IP mix incubated overnight on a rotator. The magnetic beads were blocked by being washed 2× with sonication buffer and resuspended in the same volume. The beads were blocked with single-stranded DNA by adding 1/30 volume of sonicated single-stranded DNA (10 mg/ml) and incubated overnight with rotation at 4°C. Approximately 30 to 40 μl of magnetic beads were added per IP mixture, and then the mixtures were rotated for about 1 to 1.5 h at 4°C. The beads were washed as follows: once with IP buffer, twice with IP buffer with 500 mM NaCl, twice with LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0), and twice with Tris-EDTA buffer. The immunoprecipitates were denatured with SDS and separated on a 12.5% SDS-polyacrylamide gel electrophoresis gel.
ChIP assay.
ChIP assays were carried out by using 5 μg of anti-CDK9, anti-T29P, normal immunoglobulin G (IgG), or anti-RNAP II antibody, following the methods described previously (48). After proteins were cross-linked to DNA in TZM-bl cells using 1.0% formaldehyde, chromatin was sonicated five times for 20 s each, generating DNA fragments with 250 to 1,000 bp. The supernatants were diluted with ChIP dilution buffer to a total volume of 400 μl, and the different antibodies indicated above were added. After overnight rotation at 4°C, the immune complexes were collected by the addition of protein A-coated magnetic beads (Dynal; Invitrogen). After extensive washes, the immune complexes were eluted with 1% SDS-NaHCO3 solution for 1 to 2 h at room temperature. The eluted complexes were treated with NaCl-RNase A solution overnight to reverse cross-linked protein-DNA complexes. After the cross-linking was reversed, the DNA was purified by using a PCR purification kit (BiONEER) and amplified by PCR using PCR primers specific for the HIV-1 LTR.
RESULTS
Brd4 inhibits CDK9 kinase activity by inducing autophosphorylation of threonine 29 in CDK9.
Given the recent report that Brd4 is associated with at least a fraction of the low-molecular-weight complex of P-TEFb (14, 43), we were interested in determining if Brd4 regulated P-TEFb kinase activity. To examine the effect of Brd4 on P-TEFb kinase activity, purified Brd4 was incubated with affinity-purified P-TEFb and the CTD of the large subunit of RNAP II as substrate. Interestingly, the results presented in Fig. lA demonstrate that Brd4 inhibits WT CDK9 kinase activity on the RNAP II CTD in a dose-dependent manner (lanes 1 to 4). At the highest concentration of Brd4 tested, a 60% reduction in kinase activity was observed. In view of our recent results showing that Tax inhibits CDK9 through autophosphorylation at threonine 29 (48), two CDK9 amino acid substitution mutants, T29A, a threonine-to-alanine substitution at amino acid 29, and T29E, a threonine-to-glutamic acid substitution, were included in the analysis. The T29A mutant blocks phosphorylation at T29, while T29E mimics the effect of phosphorylation at T29. In contrast to the effect of Brd4 on WT P-TEFb, Brd4 did not inhibit the CDK9 kinase activity of the T29A mutant (Fig. 1A, lanes 5 to 8). We also point out that the kinase activity of P-TEFb was essentially equivalent for the WT and the T29A substitution mutant (Fig. 1A, lanes 1 and 5). Two observations were made with the T29E amino acid substitution mutant. First, the kinase activity of T29E was significantly less than that of the WT or T29A (Fig. 1A, compare lanes 1, 5, and 9), consistent with our interpretation that T29 phosphorylation inhibits CDK9 kinase activity. Second, Brd4 did not inhibit the residual kinase activity of T29E (Fig. 1A, lanes 9 to 12), suggesting that the amino acid substitution mimicked the inhibitory effect induced by Brd4.
FIG. 1.
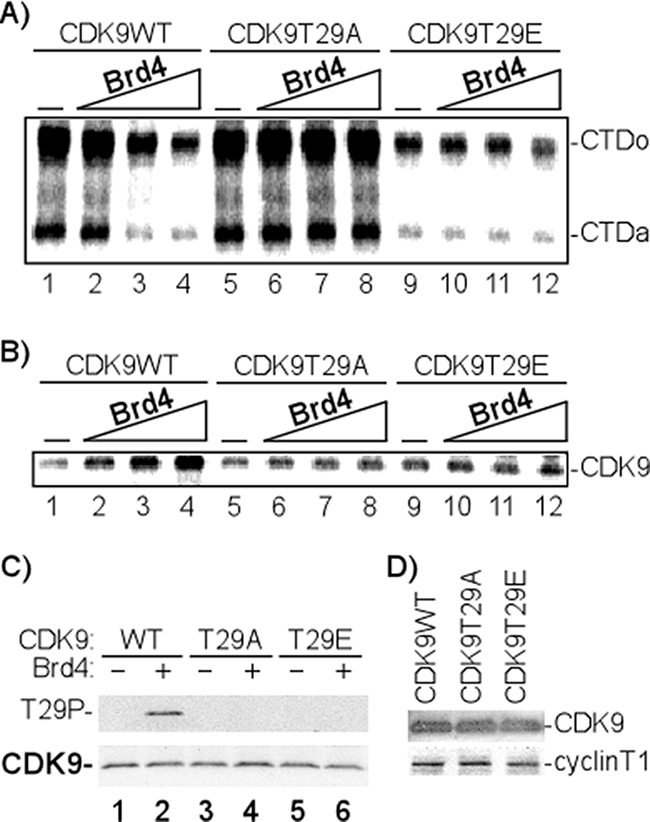
Brd4 regulates CDK9 kinase activity through phosphorylation of Thr 29. (A) Effect of Brd4 on CTD phosphorylation by P-TEFb (CDK9). In vitro kinase assays were performed by incubating 50 ng GST-CTD; 50 ng P-TEFb, composed of WT CDK9 or mutants (T29A or T29E); and cyclin T1, with [γ-32P]ATP in the absence (−) or presence of increasing concentrations of Brd4 (30, 60, and 120 ng). The 32P-labeled GST-CTD was precipitated with glutathione-Sepharose beads and fractionated by electrophoresis on an 8% SDS-polyacrylamide gel, followed by autoradiography. The hypophosphorylated (CTDa) and hyperphosphorylated (CTDo) forms of CTD are indicated. The results shown are representative of three independent experiments performed under similar conditions. (B) Effect of Brd4 on CDK9 autophosphorylation. In vitro kinase assays were performed by incubating 50 ng P-TEFb with [γ-32P]ATP in the absence (−) or presence of increasing concentrations of Brd4 (30, 60, and 120 ng). 32P-labeled CDK9 was immunoprecipitated with anti-CDK9 antibody and analyzed by electrophoresis on a 4-to-20% SDS-polyacrylamide gel, followed by autoradiography. The results shown are representative of three independent experiments performed under similar conditions. (C) CDK9 T29 phosphorylation in the presence of Brd4. In vitro kinase assays were performed by incubating 50 ng P-TEFb, composed of WT CDK9 or mutants (T29A or T29E) and cyclin T1, with ATP in the absence (−) or presence (+) of 120 ng Brd4. CDK9 T29 phosphorylation was analyzed by electrophoresis on a 4-to-20% SDS-polyacrylamide gel, followed by Western blotting with anti-CDK9 or anti-T29P antibody. (D) Proteins used in the kinase assays whose results are shown in panels A and B were analyzed by Western blotting with anti-CDK9 or anti-cyclin T1, respectively.
In a parallel assay, using [γ-32P]ATP as a radioactive tracer, we analyzed the phosphorylation of CDK9 in response to incubation with Brd4. The results presented in Fig. 1B demonstrate that Brd4 induces the phosphorylation of WT CDK9 (lanes 1 to 4) but not the T29A or T29E mutant (lanes 5 to 12). These results are consistent with the hypothesis that Brd4 induces phosphorylation of CDK9 at T29. To confirm this hypothesis, antisera were raised against a CDK9 T29 phosphopeptide. Following preliminary screening of antisera from immunized animals by Western blotting, hyperimmune antisera were affinity purified. The specificity of the antibody was demonstrated by two assays, Western blotting (Fig. 1C) and enzyme-linked immunosorbent assay using peptides containing the phosphorylated or nonphosphorylated form of CDK9 (data not shown). To demonstrate that Brd4 induces the phosphorylation of CDK9 at T29, in vitro kinase assays were performed by incubating 50 ng P-TEFb, composed of WT or mutant (T29A or T29E) CDK9 and cyclin T1, with ATP in the absence or presence of 120 ng Brd4. Western blot analysis of CDK9 was then performed with anti-CDK9 or anti-T29P. The results presented in Fig. 1C demonstrate that while equivalent amounts of CDK9 were present in each reaction mixture (Fig. 1C, lower panel), the T29P-specific antibody reacted only with WT CDK9 incubated with Brd4 (lane 2). These results suggest that T29 was phosphorylated when CDK9 was incubated with Brd4. Western blot analyses demonstrated that equal amounts of the input WT and mutant CDK9 complexes were used in the above-described kinase assays (Fig. 1D).
Phosphorylation of CDK9 at T29 inhibits transcription.
We speculated that phosphorylation of CDK9 at T29, which inhibits phosphorylation of the RNAP II CTD, would inhibit transcription activity. To test this point experimentally, nuclear extracts were cleared with CDK9 antisera and then reconstituted with P-TEFb containing WT, T29A, or T29E CDK9. The HIV basal promoter template was added to the reconstituted extracts in the presence of NTPs and [32P]UTP. Following incubation, transcription was analyzed by quantitation of the nucleotide-168 transcript. The results presented in Fig. 2A demonstrate several important points. First, reconstitution of the extract with WT CDK9 stimulated the basal transcription of HIV (Fig. 2A, lanes 3 and 4). Similarly, the addition of CDK9 containing a threonine-to-alanine amino acid substitution at T29 stimulated the basal transcription of HIV (lanes 5 and 6). In contrast, the addition of P-TEFb containing CDK9 with the amino acid substitution T29E, which mimics phosphorylation at T29, failed to stimulate the basal transcription of HIV (lanes 7 and 8). These results provide direct evidence that phosphorylation of CDK9 at T29 inhibits transcription activity. Western blot analyses with anti-CDK9 and anti-cyclin T1 antibodies demonstrated that equivalent amounts of the P-TEFb complexes (WT, T29A, or T29E CDK9/cyclinT1) were added to the reconstituted in vitro transcription reaction mixtures (Fig. 2A, lower panel).
FIG. 2.
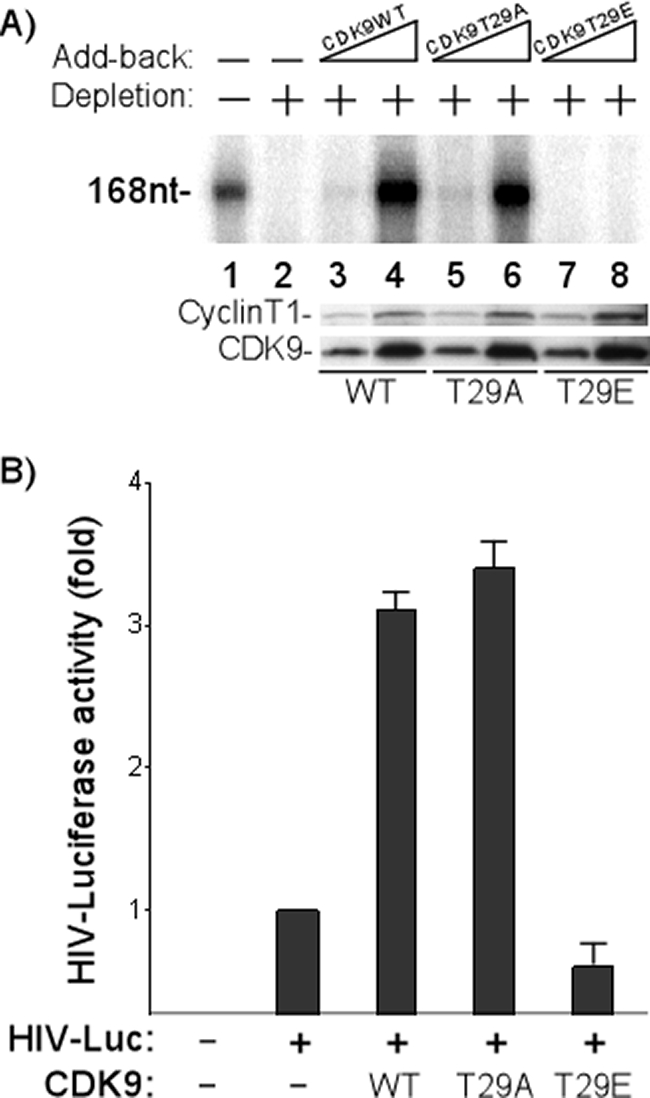
CDK9 T29 phosphorylation inhibits transcription activity of P-TEFb in vitro and in vivo. (A) CDK9 T29 phosphorylation inhibits in vitro transcription. P-TEFb was depleted (+; −, not depleted) from HeLa nuclear extracts with anti-CDK9 antibody. Increasing concentrations of baculovirus-purified P-TEFb (50 ng and 150 ng) containing WT, T29A, or T29E CDK9 were then added back to the depleted extracts (−, no add-back). In vitro transcription reactions were set up by incubating the HIV template, reconstituted nuclear extracts, 50 μM ATP, 50 μM CTP, 50 μM GTP, 1.25 μM UTP, 20 μCi[α-32P]UTP, and 10 units of RNasin (Promega) in 1× IVT buffer. The radiolabeled transcripts (168 nucleotides [168nt]) (47) were fractionated by electrophoresis on a 6% denaturing polyacrylamide gel, followed by autoradiography. Western blot analyses of WT and mutant P-TEFb complexes added back to the depleted extracts are shown in the lower panels. (B) CDK9 T29 phosphorylation regulates P-TEFb transcription activity in vivo. The plasmids containing CDK9 WT or mutants were cotransfected into HeLa cells with a plasmid containing the HIV LTR-driven luciferase (HIV-Luc). The cells were cultured for 48 h and luciferase activity was assayed. Each result shown is the average of the results of four experiments with the standard error indicated. +, present; −, absent.
The biological significance of CDK9 regulation in HIV transcription was also analyzed in vivo. The plasmid containing WT CDK9 or the T29A or T29E mutant was cotransfected into HeLa cells with the HIV LTR-driven luciferase plasmid. Previous experiments have shown that the transfected CDK9 is incorporated with cyclin T into P-TEFb complexes (48). The transfected cells were then cultured for 48 h, and the luciferase activity was assayed. The results shown in Fig. 2B demonstrate that the overexpression of WT or T29A CDK9 increased basal HIV promoter activity up to threefold. In contrast, the overexpression of T29E CDK9 failed to increase the basal transcription of HIV (Fig. 2B), consistent with the transcriptionally inactive nature of this mutant. In fact, a slight reduction in HIV promoter activity was observed in the presence of the T29E mutant.
Association of Brd4 and P-TEFb with the HIV initiation and elongation complexes.
Given the effect of Brd4 on P-TEFb kinase activity and the inactivity of the T29E P-TEFb in vitro and in vivo, it was of interest to analyze the interaction of Brd4, P-TEFb, and T29P in the PIC and TEC. We utilized the HIV basal promoter in these in vitro binding and transcription assays since it has been shown that this template requires Brd4 and P-TEFb (43). The results presented in Fig. 3A demonstrate that both Brd4 and CDK9 (P-TEFb) are incorporated into the HIV PIC formed following the incubation of template DNA with HeLa nuclear extract (lane 2). The addition of triphosphates to allow the initiation complex to elongate to +14 nucleotides did not result in any significant changes in the levels of Brd4 or CDK9 associated with TEC (lane 3). When the complex was elongated to +36 nucleotides, we found that Brd4 was released while CDK9 remained associated with the complex (lane 4). These results suggest that there is a significant transition in the protein composition of the transcription complex between positions +14 and +36.
FIG. 3.
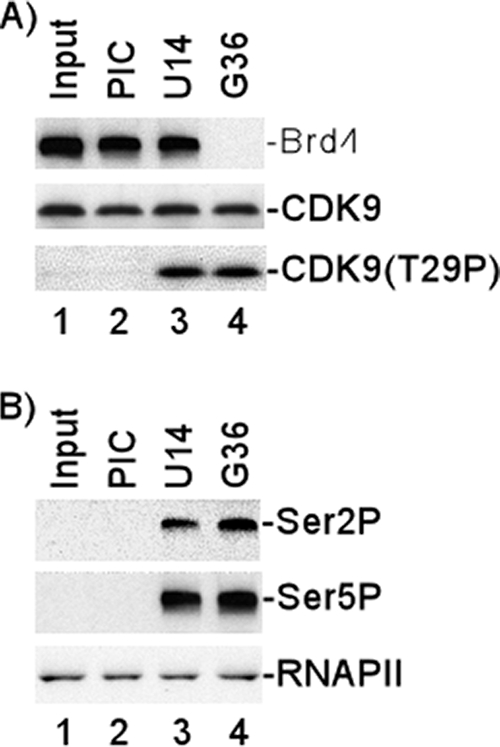
Association of Brd4 with HIV transcription complexes and CDK9 T29 phosphorylation during transcription. HIV-1 PICs were assembled by incubating biotinylated HIV-1 templates with HeLa nuclear extract and then purified with streptavidin-coated magnetic beads. The purified PICs were incubated with 50 μM ATP for 10 min and then washed extensively with 1× IVT buffer. The PICs were walked to position +U14 by incubation with 50 μM CTP, GTP, and UTP for 5 min at 30°C and then washed extensively with 1× IVT buffer. The TECs stalled at U14 were walked stepwise along the DNA by repeated incubation with different sets of three NTPs and then washed extensively with 1× IVT buffer to remove the unincorporated NTPs. (A) Protein compositions of PICs and TECs stalled at different stages were analyzed by fractionation by electrophoresis on 4-to-20% SDS-polyacrylamide gels, followed by Western blot analyses with antibody against Brd4, CDK9, or T29P CDK9. (B) Protein compositions of PICs and TECs stalled at different stages were analyzed by fractionation by electrophoresis on 4% SDS-polyacrylamide gels, followed by Western blot analyses with antibody against Ser 2P RNAP II CTD or Ser 5P RNAP II CTD. Western blot analysis with specific antibody (N-20) against the amino terminus of the largest subunit of RNAP II shown in bottom panel demonstrated equal amounts of RNAP II in the PICs and TECs.
In parallel Western blots, we analyzed the phosphorylation of CDK9 T29 (Fig. 3A, lower panel). Interestingly, CDK9 was not phosphorylated at T29 in the PIC but was phosphorylated at T29 in TECs isolated at positions +14 and +36.
We also analyzed the phosphorylation of the RNAP II CTD with antisera specific for Ser 2P and Ser 5P (Fig. 3B). The results demonstrated that phosphorylation of the RNAP II CTD at Ser 2 was observed in the elongation complexes stalled at positions +14 and +36 (Fig. 3B, top panel). CTD phosphorylation at Ser 5 is also significantly enhanced as the PIC converts to an elongation complex between positions +1 and +14 (Fig. 3B, middle panel). To compare RNAP II of PICs and TECs, the protein compositions of PICs and TECs stalled at different stages were analyzed by fractionation by electrophoresis on 4-to-20% SDS-polyacrylamide gels, followed by Western blot analysis with specific antibody (N-20) against the amino terminus of the largest subunit of RNAP II (Fig. 3B, bottom panel). The results demonstrate that RNAP II stays constant as the PIC converts to an elongation complex between positions +1 and +14.
A phosphatase is recruited to the TEC.
The results presented in Fig. 3A suggest that even though Brd4 is released, phosphorylation of CDK9 at T29 remains. These results prompted us to examine whether a phosphatase might be recruited into the complex after Brd4 is released, to dephosphorylate T29 and activate transcription. To examine this possibility, the HIV template DNA was incubated with HeLa nuclear extract and the PICs purified by centrifugation and mild washing. Subsequently, the complexes were elongated to position +36 or +51. As shown in Fig. 4A, reincubation of the elongation complexes with RNAP II-depleted nuclear extract made a distinct difference in the phosphorylation of CDK9 at T29. In the elongation complexes which were not reincubated with extract, CDK9 T29 was phosphorylated in complexes stalled at positions +36 and +51 (Fig. 4A, lanes 3 and 5). In contrast, when complexes stalled at position +36 were reincubated with RNAP II-depleted nuclear extract and then elongated to position +51, a significant decrease in T29 phosphorylation in TECs stalled at position +51 was observed (Fig. 4A, lanes 4 and 6).
FIG. 4.

Analysis of T29P in HIV transcription initiation complex and TECs. (A) State of CDK9 T29 phosphorylation. The purified PICs were walked to position +U14 by incubation with 50 μM CTP, GTP, and UTP for 5 min at 30°C and then washed extensively with 1× IVT buffer. The TECs stalled at U14 were walked stepwise along the DNA by repeated incubation with different sets of three NTPs and then washed extensively with 1× IVT buffer to remove the unincorporated NTPs. TECs stalled at G36 were reincubated without (G36) or with (G36*) RNAP II-depleted extract and then assayed directly or elongated stepwise to position +51 (A51, A51*). The protein compositions of PICs and TECs that were stalled at different stages were analyzed by Western blotting with anti-T29P, anti-CDK9, anti-cyclin T1, or anti-Brd4. (B) PP2A was recruited into TECs. The purified PICs were walked to position +U14 by incubation with 50 μM CTP, GTP, and UTP for 5 min at 30°C and then washed extensively with 1× IVT buffer. The TECs stalled at U14 were walked stepwise along the DNA by repeated incubation with different sets of three NTPs and then washed extensively with 1× IVT buffer to remove the unincorporated NTPs. TECs stalled at U14 or G36 were reincubated with RNAP II-depleted extracts (U14* or G36*, respectively) and then washed extensively with 1× IVT buffer. The protein compositions of PICs and TECs that were stalled at different stages were analyzed by Western blotting with anti-PP2A or anti-PP1. (C) Analysis of RNAP II CTD phosphorylation. The protein compositions of PICs and TECs that were stalled at different stages were analyzed by Western blotting with antibody against Ser 2P RNAP II CTD or Ser 5P RNAP II CTD. (D) Recruitment of phosphatase correlates with HIV transcription in vitro. Runoff transcripts (168nt) from TECs that were stalled at different stages were fractionated by electrophoresis on a 6% denaturing polyacrylamide gel, followed by autoradiography. (E and F) Purified PP2A dephosphorylates T29P during transcription elongation. The purified PICs were walked to position +U14 by incubation with 50 μM CTP, GTP, and UTP for 5 min at 30°C and then washed extensively with 1× IVT buffer. The TECs stalled at U14 were walked stepwise along the DNA by repeated incubation with different sets of three NTPs and then washed extensively with 1× IVT buffer to remove the unincorporated NTPs. TECs stalled at G36 were reincubated without (G36) or with (G36*) purified PP2A and then elongated stepwise to position +51 (A51, A51*). The protein compositions of PICs and TECs that were stalled at different stages were analyzed by Western blotting with anti-T29P (E) or anti-Ser 2P RNAP II CTD or anti-Ser 5P RNAP II CTD (F). (G) PP2A was required for the basal transcription of HIV. PP2A was added back in increasing concentrations (25 ng and 50 ng) to PP2A-depleted extract (+; −, not depleted), and then in vitro transcription assays were performed by incubating HIV templates with the reconstituted extracts. The radiolabeled transcripts (168nt) were fractionated by electrophoresis on a 6% denaturing polyacrylamide gel and detected by autoradiography. Western blot analyses of the PP2A-depleted extract are shown in the lower panels.
In view of the results presented in Fig. 4A, which suggest that a phosphatase might be recruited to the elongation complex, we assayed for the presence of phosphatase PP2A or PP1 by Western blotting. The results presented in Fig. 4B demonstrate that if the stalled +36 elongation complex was reincubated with nuclear extract, specific binding of phosphatase PP2A was observed (lane 6). Interestingly, PP2A binding was not observed in the stalled +14 complex, U14*, which was reincubated with nuclear extract (lane 5), suggesting that there is a transition in the complex between positions +14 and +36 which allows the association of the phosphatase with the elongation complex. Interestingly, even though the PP2A is incorporated into the complex at position +36, T29 phosphorylation is not lost until position +51. While it was present in the nuclear extract (Fig. 4B, lane 1), phosphatase PP1 was not detected in association with the elongation complexes (Fig. 4B, lanes 5 and 6).
We next analyzed RNAP II CTD Ser 2P levels in the various transcription complexes. Consistent with the loss of T29P in the elongation complex stalled at position +51, which originated from the TEC stalled at position +36 which had been reincubated with nuclear extract (Fig. 4A, lane 6), we observed a distinct increase in the level of CTD Ser 2P in this complex (Fig. 4C, lane 6). In contrast, RNAP II CTD phosphorylation at Ser 5 by CDK7 (TFIIH) stayed constant (Fig. 4C, bottom panel). We also noted a significant increase in the transcription activity of the complex which had been reincubated with extract (Fig. 4D, lane 4).
The results presented above suggest that PP2A might be involved in the dephosphorylation of T29P CDK9. To test this possibility directly, transcription complexes stalled at position +36 were formed and then incubated with purified PP2A. The results presented in Fig. 4E demonstrate that PP2A is, in fact, capable of dephosphorylating T29P CDK9. Similar to the results with whole-cell extracts (Fig. 4A), phosphorylation at T29 was reduced to background levels when the +36 complex was incubated with purified PP2A and elongated to position +51 (Fig. 4E, lane 6). Of interest, but consistent with the results presented in Fig. 4A, phosphatase PP2A was not able to dephosphorylate T29P CDK9 in the +36 complex (Fig. 4E, lane 4). Finally, similar to the results obtained with the whole-cell extracts (Fig. 4C), incubation of the +36 TEC with PP2A followed by elongation to position +51 resulted in an increase in the CDK9 kinase activity assayed by RNAP II CTD phosphorylation at Ser 2 (Fig. 4F, top panel, lanes 5 and 6). In contrast, RNAP II CTD phosphorylation at Ser 5 by CDK7 (TFIIH) stayed constant in the absence and presence of PP2A (Fig. 4F, bottom panel, lanes 5 and 6).
To determine the functional significance of PP2A, PP2A was depleted from nuclear extract with anti-PP2A antibody (Fig. 4G, lower panel). The specificity of the depletion was demonstrated by the fact that phosphatase PP1 was not depleted (Fig. 4G, lower panel). In vitro transcription assays were then performed by incubating HIV templates with PP2A-depleted extracts (Fig. 4G, upper panel). The results demonstrated that the basal transcription of HIV decreased when PP2A was depleted from extracts (lanes 1 and 2). Importantly, when PP2A was added back to the extract, the basal transcription of HIV was restored (lanes 1 to 4). These experiments demonstrate that PP2A plays an important role in the basal transcription of HIV.
P-TEFb CDK9 associated with latent HIV promoter is phosphorylated at T29.
Our studies have thus far demonstrated that Brd4 induces an autophosphorylation of CDK9 at T29, that T29 phosphorylation inhibits transcription, and that CDK9 is transiently phosphorylated at T29 in in vitro transcription complexes. To determine if the T29P CDK9 component of P-TEFb is found associated with transcription complexes in vivo, ChIP assays were carried out with TZM-bl cells, which contain separate integrated copies of the luciferase and β-galactosidase genes under the control of the HIV-1 basal promoter. The constitutive level of integrated HIV promoter activity in the TZM-bl cell line is extremely low, similar to the low expression level observed in cells latently infected with HIV. For these ChIP assays, we utilized the anti-T29 phosphopeptide sera characterized by Western blotting, as shown in Fig. 1C. To demonstrate the specificity of the T29P-specific CDK9 antisera for IP, control IPs were performed. P-TEFb was incubated with increasing concentrations of Brd4 protein. The reaction mixtures were then immunoprecipitated with anti-T29P CDK9 antisera and analyzed by Western blotting with anti-CDK9 antibody. The results presented in Fig. 5A demonstrate that the antibody specifically immunoprecipitates T29P CDK9. While the antibody failed to immunoprecipitate unphosphorylated T29 CDK9 which had not been incubated with Brd4 (lane 1), the T29P antibody precipitated CDK9 which had been incubated with Brd4 (lanes 2 to 5). In general, the amount of T29P CDK9 increased with increasing concentrations of Brd4 protein.
FIG. 5.
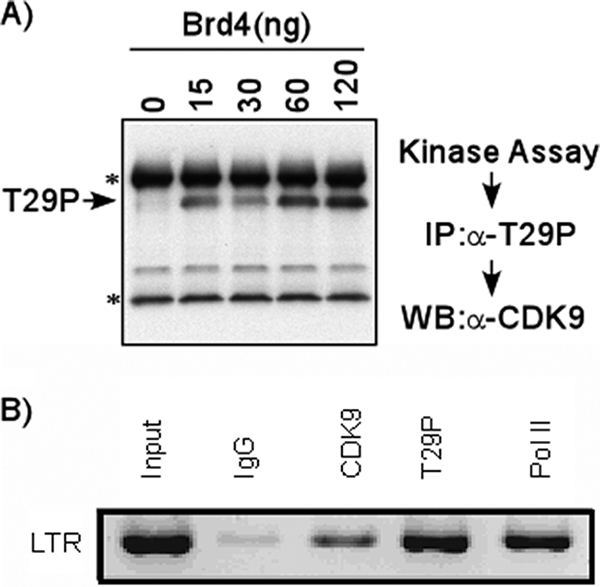
T29P CDK9 associated with latent HIV promoter in vivo. (A) Specificity of T29P CDK9 antibody for IP. In vitro kinase assays were performed by incubating P-TEFb with ATP in the presence of different concentrations of Brd4. The phosphorylated CDK9 was then immunoprecipitated with T29P CDK9 antibody (rabbit) and fractionated by electrophoresis on a 4-to-20% SDS-polyacrylamide gel, followed by Western blot analysis with anti-CDK9 antibody (mouse). Bands indicated by asterisks represent heavy and light chains of the antibody used for IP. α, anti; WB, Western blot. (B) T29P CDK9 is associated with latent HIV promoter in vivo. ChIP assays were carried out with TZM-bl cells, which contain separate integrated copies of the luciferase and β-galactosidase genes under the control of the HIV-1 promoter. Cross-linked extracts were immunoprecipitated with 5 μg of control IgG, anti-CDK9, anti-T29P CDK9, or anti-RNAP II (Pol II) antibody. PCR analysis with primers specific for the HIV LTR region was performed on the precipitated DNA.
We next performed ChIP assays on the TZM-bl cells. Using primers specific for the HIV LTR region, PCR amplification was performed to analyze the precipitated DNAs. The results shown in Fig. 5B demonstrate that in contrast to the result for control IgG (second lane), a positive signal was observed in lanes immunoprecipitated with CDK9 and RNAP II antibodies (third and fifth lanes). Of significant interest, we also observed a strong signal in the lane precipitated with the T29P CDK9 antiserum (fourth lane). The relatively high level of T29P CDK9 present on the HIV promoter is consistent with the low level of viral transcription observed in these cells.
DISCUSSION
P-TEFb, composed of CDK9 and its regulatory partner cyclin T1 (32), is dynamically controlled by both positive and negative regulators to maintain a functional equilibrium appropriate for the growth or differentiation state of the cell. Two distinct P-TEFb complexes have been identified. A significant portion of P-TEFb is found in a high-molecular-weight complex in which CDK9 kinase activity is inactivated by the coordinated actions of 7SK snRNA and HEXIM1 protein (25, 28, 44, 46). The second major P-TEFb complex is the low-molecular-weight complex in which P-TEFb is found associated with positive regulators, such as Brd4 (2, 14, 43). Several mechanisms exist to regulate P-TEFb activity; for example, the ability to mobilize P-TEFb from the inactive high-molecular-weight complex, which involves the dissociation of HEXIM1, is apparently essential for stress-induced transcription (25, 28, 44, 46). CDK9 kinase activity is also regulated by cyclin T1 levels (22). Liou et al. showed that while CDK9 levels remained constant, cyclin T1 protein expression in freshly isolated monocytes was very low, increased early during macrophage differentiation, and decreased to low levels after about 1 week in culture. The kinase and transcription activities of TAK/P-TEFb paralleled the changes in cyclin T1 protein expression. The results presented in this study demonstrate that P-TEFb kinase activity may also be regulated by Brd4 through a novel autophosphorylation pathway.
Depending upon cell growth conditions, as much as one-half of cellular P-TEFb associates with Brd4, and the other half exists in a complex with the 7SK snRNA and the HEXIM1 protein (25, 28, 44, 46). Interestingly, it appears that multiple domains of Brd4 may interact with the cyclin T subunit of P-TEFb (2, 14). In the low-molecular-weight complex, Brd4 is generally regarded as a positive transcription regulator which recruits P-TEFb to eukaryotic promoters, including the HIV-1 LTR promoter (43). Our results suggest, however, that once bound to the promoter, Brd4 transiently inhibits P-TEFb kinase activity through autophosphorylation of T29, most likely to facilitate the organization of an efficient elongation complex. Once Brd4 is released from the elongation complex between positions +1 and +14, P-TEFb activation requires the incorporation of a phosphatase into the complex to dephosphorylate T29P. One candidate phosphatase is PP2A. In this study, we show that PP2A is recruited to the TEC and is able to remove phosphate from CDK9 at T29, leading to a transition from a transcriptionally silent state to a basal transcription state. PP2A accounts for a majority of the serine-threonine phosphatase activity in cells and has been implicated in the regulation of many signaling pathways and phenotypes (15, 19). Recently, evidence for the involvement of PP2A in the regulation of HIV transcription and replication has been reported (7, 8, 37). The catalytic subunit of PP2A (PP2Ac) is able to increase the basal activity of the HIV-1 promoter and markedly enhances the promoter's response to phorbol myristate acetate (8). Interestingly, PP2A has also been shown to dephosphorylate CDK9 in Tat-activated HIV-1 transcription, inhibiting Tat transactivation (1). It is not clear at this point whether PP2A is the sole phosphatase to act on T29P or whether others may functionally substitute.
Thus, our results suggest that P-TEFb plays a mediating role in the basal transcription of HIV, regulating transcription in the initial stages of transcription in a negative manner through a Brd4-induced autophosphorylation of CDK9 at T29 (Fig. 6A). We envision this step as a regulatory step which allows functional initiation complexes to form in preparation for transcription elongation. Once Brd4 is released from the TEC, the removal of T29P would allow the positive function of P-TEFb in transcription elongation to be realized. It is possible that the Brd4-induced T29 phosphorylation may play a role in HIV latency (Fig. 6B). Indeed, we found by ChIP assay analysis that the CDK9 associated with inactive and latent HIV promoters is phosphorylated at T29. It is possible that in the latent state, the release of Brd4 and subsequent dephosphorylation of CDK9 do not occur, leading to a block in the basal transcription of HIV.
FIG. 6.

Model for Brd4 regulation of P-TEFb activity. (A and B) Schematic diagrams of basal transcription of HIV and HIV latency are shown. Brd4 recruits P-TEFb to the HIV promoter. However, as the complex proceeds from initiation to elongation, Brd4 induces autophosphorylation of P-TEFb at threonine 29 (T29P), inhibiting P-TEFb CTD phosphorylation. In the case of basal transcription, Brd4 exits the complex, allowing the phosphatase PP2A to enter the complex, dephosphorylate P-TEFb, and relieve the inhibition of its kinase activity. This relieves the block to elongation and allows basal gene expression (A). In the case of HIV latency, P-TEFb remains phosphorylated at T29 and elongation cannot proceed (B).
It will be of interest to determine how phosphorylation at T29 inhibits CDK9 kinase activity. One possibility is that T29P induces a conformational change in CDK9. Certainly, this has been shown for the phosphorylation of other sites in CDK9. Phosphorylation at T186, located at the tip of a flexible T-loop in CDK9, induces a major conformational change of the T-loop which allows the entry of the substrate and ATP into the kinase catalytic pocket (38). Alternatively, phosphorylation at T29 may facilitate the binding of an inhibitory protein to CDK9. The phosphorylation-induced binding would be akin to the increase in HEXIM1 and 7SK RNA binding to CDK9 following phosphorylation at T186. We consider this possibility less likely, however, since T29 phosphorylation inhibits the kinase activity of purified P-TEFb.
HIV latency is associated with a lack of proviral gene expression. Quite likely, the maintenance of proviral latency is the end product of several different pathways that function to downregulate HIV transcription. For example, it has been reported that the association of NF-κB p50-histone deacetylase 1 complexes with the HIV LTR is an important step for viral latency (41). In this model, NF-κB p50-histone deacetylase 1 complexes constitutively bind the latent HIV LTR and induce histone deacetylation and repressive changes in the chromatin structure of the HIV LTR, changes that impair the recruitment of RNAP II and transcriptional initiation. In this study, we show that Brd4-induced T29 phosphorylation in CDK9 may also contribute to HIV latency. We have presented direct evidence that T29P decreases P-TEFb CTD kinase activity and inhibits the transcriptional activity of P-TEFb. Moreover, using ChIP assays, we demonstrated that high levels of T29P P-TEFb are associated with the transcriptionally latent viral genome in TZM-bl cells. If the phosphate group is removed from T29 by an incoming phosphatase, such as PP2A, CDK9 is thus released from repression and phosphorylates Ser 2 of the RNAP II CTD. The processivity of the bound polymerase II is thus increased, resulting in basal transcription (Fig. 6A). If the phosphate group is not removed from T29, CDK9 kinase activity is inhibited. Due to the absence of cyclin T1-CDK9-mediated Ser 2 phosphorylation of the bound RNAP II CTD, HIV genes stay transcriptionally silent, leading to HIV latency (Fig. 6B). Given that the Brd4/P-TEFb complex has been implicated in the transcriptional regulation of a variety of cellular genes, it would be of interest to determine if CDK9 phosphorylation at T29 is a universal mechanism of transcription inhibition. Along these lines, it is of interest that Montanuy et al. recently reported a common mechanism of elongation control at the c-myc and and HIV-1 genes with an essential role for the TATA box and specific modulatory contributions of upstream regulatory sequences (27).
Footnotes
Published ahead of print on 29 October 2008.
REFERENCES
- 1.Ammosova, T., K. Washington, Z. Debebe, J. Brady, and S. Nekhai. 2005. Dephosphorylation of CDK9 by protein phosphatase 2A and protein phosphatase-1 in Tat-activated HIV-1 transcription. Retrovirology 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bisgrove, D. A., T. Mahmoudi, P. Henklein, and E. Verdin. 2007. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA. 10413690-13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buratowski, S. 2003. The CTD code. Nat. Struct. Biol. 10679-680. [DOI] [PubMed] [Google Scholar]
- 4.Dey, A., F. Chitsaz, A. Abbasi, T. Misteli, and K. Ozato. 2003. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA. 1008758-8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dey, A., J. Ellenberg, A. Farina, A. E. Coleman, T. Maruyama, S. Sciortino, J. Lippincott-Schwartz, and K. Ozato. 2000. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G2-to-M transition. Mol. Cell. Biol. 206537-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farina, A., M. Hattori, J. Qin, Y. Nakatani, N. Minato, and K. Ozato. 2004. Bromodomain protein Brd4 binds to GTPase-activating SPA-1, modulating its activity and subcellular localization. Mol. Cell. Biol. 249059-9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faulkner, N. E., J. M. Hilfinger, and D. M. Markovitz. 2001. Protein phosphatase 2A activates the HIV-2 promoter through enhancer elements that include the pets site. J. Biol. Chem. 27625804-25812. [DOI] [PubMed] [Google Scholar]
- 8.Faulkner, N. E., B. R. Lane, P. J. Bock, and D. M. Markovitz. 2003. Protein phosphatase 2A enhances activation of human immunodeficiency virus type 1 by phorbol myristate acetate. J. Virol. 772276-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feaver, W. J., O. Gileadi, Y. Li, and R. D. Kornberg. 1991. CTD kinase associated with yeast RNA polymerase II initiation factor b. Cell 671223-1230. [DOI] [PubMed] [Google Scholar]
- 10.Fu, T. J., J. Peng, G. Lee, D. H. Price, and O. Flores. 1999. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J. Biol. Chem. 27434527-34530. [DOI] [PubMed] [Google Scholar]
- 11.Garber, M. E., T. P. Mayall, E. M. Suess, J. Meisenhelder, N. E. Thompson, and K. A. Jones. 2000. CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 Tat-P-TEFb complex to TAR RNA. Mol. Cell. Biol. 206958-6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grana, X., A. De Luca, N. Sang, Y. Fu, P. P. Claudio, J. Rosenblatt, D. O. Morgan, and A. Giordano. 1994. PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retinoblastoma protein in vitro. Proc. Natl. Acad. Sci. USA 913834-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hengartner, C. J., V. E. Myer, S. M. Liao, C. J. Wilson, S. S. Koh, and R. A. Young. 1998. Temporal regulation of RNA polymerase II by Srb10 and Kin28 cyclin-dependent kinases. Mol. Cell 243-53. [DOI] [PubMed] [Google Scholar]
- 14.Jang, M. K., K. Mochizuki, M. Zhou, H. S. Jeong, J. N. Brady, and K. Ozato. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19523-534. [DOI] [PubMed] [Google Scholar]
- 15.Janssens, V., and J. Goris. 2001. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353417-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang, Y. W., P. Veschambre, H. Erdjument-Bromage, P. Tempst, J. W. Conaway, R. C. Conaway, and R. D. Kornberg. 1998. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. USA 958538-8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobor, M. S., and J. Greenblatt. 2002. Regulation of transcription elongation by phosphorylation. Biochim. Biophys. Acta 1577261-275. [DOI] [PubMed] [Google Scholar]
- 18.Komarnitsky, P., E. J. Cho, and S. Buratowski. 2000. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 142452-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lechward, K., O. S. Awotunde, W. Swiatek, and G. Muszynska. 2001. Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim. Pol. 48921-933. [PubMed] [Google Scholar]
- 20.Leclerc, V., J. P. Tassan, P. H. O'Farrell, E. A. Nigg, and P. Leopold. 1996. Drosophila Cdk8, a kinase partner of cyclin C that interacts with the large subunit of RNA polymerase II. Mol. Biol. Cell 7505-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao, S. M., J. Zhang, D. A. Jeffery, A. J. Koleske, C. M. Thompson, D. M. Chao, M. Viljoen, H. J. van Vuuren, and R. A. Young. 1995. A kinase-cyclin pair in the RNA polymerase II holoenzyme. Nature 374193-196. [DOI] [PubMed] [Google Scholar]
- 22.Liou, L. Y., C. H. Herrmann, and A. P. Rice. 2002. Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J. Virol. 7610579-10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu, H., L. Zawel, L. Fisher, J. M. Egly, and D. Reinberg. 1992. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature 358641-645. [DOI] [PubMed] [Google Scholar]
- 24.Maruyama, T., A. Farina, A. Dey, J. Cheong, V. P. Bermudez, T. Tamura, S. Sciortino, J. Shuman, J. Hurwitz, and K. Ozato. 2002. A mammalian bromodomain protein, Brd4, interacts with replication factor C and inhibits progression to S phase. Mol. Cell. Biol. 226509-6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michels, A. A., A. Fraldi, Q. Li, T. E. Adamson, F. Bonnet, V. T. Nguyen, S. C. Sedore, J. P. Price, D. H. Price, L. Lania, and O. Bensaude. 2004. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 232608-2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mochizuki, K., A. Nishiyama, M. K. Jang, A. Dey, A. Ghosh, T. Tamura, H. Natsume, H. Yao, and K. Ozato. 2008. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 2839040-9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montanuy, I., R. Torremocha, C. Hernandez-Munain, and C. Sune. 2008. Promoter influences transcription elongation: TATA-box element mediates the assembly of processive transcription complexes responsive to cyclin-dependent kinase 9. J. Biol. Chem. 2837368-7378. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen, V. T., T. Kiss, A. A. Michels, and O. Bensaude. 2001. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414322-325. [DOI] [PubMed] [Google Scholar]
- 29.Ni, Z., A. Saunders, N. J. Fuda, J. Yao, J. R. Suarez, W. W. Webb, and J. T. Lis. 2008. P-TEFb is critical for the maturation of RNA polymerase II into productive elongation in vivo. Mol. Cell. Biol. 281161-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palancade, B., and O. Bensaude. 2003. Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur. J. Biochem. 2703859-3870. [DOI] [PubMed] [Google Scholar]
- 31.Peng, J., Y. Zhu, J. T. Milton, and D. H. Price. 1998. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 12755-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price, D. H. 2000. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 202629-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rickert, P., J. L. Corden, and E. Lees. 1999. Cyclin C/CDK8 and cyclin H/CDK7/p36 are biochemically distinct CTD kinases. Oncogene 181093-1102. [DOI] [PubMed] [Google Scholar]
- 34.Rickert, P., W. Seghezzi, F. Shanahan, H. Cho, and E. Lees. 1996. Cyclin C/CDK8 is a novel CTD kinase associated with RNA polymerase II. Oncogene 122631-2640. [PubMed] [Google Scholar]
- 35.Rossignol, M., I. Kolb-Cheynel, and J. M. Egly. 1997. Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J. 161628-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy, R., J. P. Adamczewski, T. Seroz, W. Vermeulen, J. P. Tassan, L. Schaeffer, E. A. Nigg, J. H. Hoeijmakers, and J. M. Egly. 1994. The MO15 cell cycle kinase is associated with the TFIIH transcription-DNA repair factor. Cell 791093-1101. [DOI] [PubMed] [Google Scholar]
- 37.Ruediger, R., N. Brewis, K. Ohst, and G. Walter. 1997. Increasing the ratio of PP2A core enzyme to holoenzyme inhibits Tat-stimulated HIV-1 transcription and virus production. Virology 238432-443. [DOI] [PubMed] [Google Scholar]
- 38.Russo, A. A., P. D. Jeffrey, and N. P. Pavletich. 1996. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 3696-700. [DOI] [PubMed] [Google Scholar]
- 39.Sun, X., Y. Zhang, H. Cho, P. Rickert, E. Lees, W. Lane, and D. Reinberg. 1998. NAT, a human complex containing Srb polypeptides that functions as a negative regulator of activated transcription. Mol. Cell 2213-222. [DOI] [PubMed] [Google Scholar]
- 40.Trigon, S., H. Serizawa, J. W. Conaway, R. C. Conaway, S. P. Jackson, and M. Morange. 1998. Characterization of the residues phosphorylated in vitro by different C-terminal domain kinases. J. Biol. Chem. 2736769-6775. [DOI] [PubMed] [Google Scholar]
- 41.Williams, S. A., L. F. Chen, H. Kwon, C. M. Ruiz-Jarabo, E. Verdin, and W. C. Greene. 2006. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 25139-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang, Z., N. He, and Q. Zhou. 2008. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 28967-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang, Z., J. H. Yik, R. Chen, N. He, M. K. Jang, K. Ozato, and Q. Zhou. 2005. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19535-545. [DOI] [PubMed] [Google Scholar]
- 44.Yang, Z., Q. Zhu, K. Luo, and Q. Zhou. 2001. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414317-322. [DOI] [PubMed] [Google Scholar]
- 45.Yankulov, K. Y., and D. L. Bentley. 1997. Regulation of CDK7 substrate specificity by MAT1 and TFIIH. EMBO J. 161638-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yik, J. H., R. Chen, R. Nishimura, J. L. Jennings, A. J. Link, and Q. Zhou. 2003. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 12971-982. [DOI] [PubMed] [Google Scholar]
- 47.Zhou, M., M. A. Halanski, M. F. Radonovich, F. Kashanchi, J. Peng, D. H. Price, and J. N. Brady. 2000. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol. Cell. Biol. 205077-5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou, M., H. Lu, H. Park, J. Wilson-Chiru, R. Linton, and J. N. Brady. 2006. Tax interacts with P-TEFb in a novel manner to stimulate human T-lymphotropic virus type 1 transcription. J. Virol. 804781-4791. [DOI] [PMC free article] [PubMed] [Google Scholar]