

Abstract
Marek's disease virus (MDV), the etiologic agent of Marek's disease, is a potent oncogenic herpesvirus. MDV is highly contagious and elicits a rapid onset of malignant T-cell lymphomas in chickens within several weeks after infection. MDV genome codes an oncoprotein, Meq, which shares resemblance with the Jun/Fos family of bZIP transcription factors. Similar to Jun, the leucine zipper region of Meq allows the formation of homo- and heterodimers. Meq homo- and heterodimers have different DNA binding affinities and transcriptional activity; therefore, they may differentially regulate transcription of viral and cellular genes. In this study we investigated the role of Meq homodimers in the pathogenicity of MDV by generating a chimeric meq gene, which contains the leucine zipper region of the yeast transcription factor GCN4 (meqGCN). A recombinant virus (rMd5-MeqGCN) containing the chimeric meqGCN gene in place of parental meq was generated with overlapping cosmid clones of Md5, a very virulent MDV strain. The rMd5-MeqGCN virus replicated in vitro and in vivo but was unable to transform T cells in infected chickens. These data provide the first in vivo evidence that Meq homodimers are not sufficient for MDV-induced transformation.
Marek's disease virus (MDV), the etiologic agent of Marek's disease, is a potent oncogenic herpesvirus that elicits a rapid onset of malignant T-cell lymphomas in chickens within several weeks of infection, resulting in mortality. MDV is classified as an alphaherpesvirus based on viral genome organization and sequence but shares biological characteristics with gammaherpesviruses, such as its tropism and its ability to transform lymphocytes (33). Of the previously described serotypes of MDV, now classified as Gallid herpesvirus 2 (GaHV-2) or MDV serotype 1 (MDV-1), GaHV-3 or MDV-2, and the Meleagrid herpesvirus 1 (MeHV-1) or turkey herpesvirus, only MDV-1 is oncogenic. In addition, a number of different pathotypes exist within MDV-1 ranging from mild to very virulent plus (3, 39).
The search for candidate viral oncogenes in the MDV genome led to the discovery of meq, which is abundantly expressed in MDV transformed cells and is encoded only by the genome of MDV-1. The meq gene is named after the EcoQ fragment where it is located (i.e., Marek's EcoQ), and two copies are found in the viral genome within the terminal repeat long (TRL) and internal repeat long (IRL) regions (28, 32, 33, 37). Meq, a 339-amino-acid nuclear phosphoprotein, is a bZIP (basic-region leucine zipper) protein, which shares significant homology, in the bZIP domain, with the proto-oncogene c-Jun, a transcription factor of the AP-1 (activating protein) complex (13, 22, 24). AP-1 transcription factors are a group of well-described proteins that are characterized by their ability to bind and regulate sequence-specific gene elements, AP-1 sites, which are found in many genes associated with cell proliferation (35). Transformation by deregulated expression of c-Jun or its viral counterpart v-Jun is well documented, and therefore the shared homology between Meq and Jun is intriguing (38). In vitro data support the oncogenic nature of Meq, which can promote anchorage-independent growth, cell cycle progression, and antiapoptosis (23, 24). Recently, in vitro expression of Meq was shown to upregulate genes similar to those upregulated by v-Jun, suggesting that Meq transforms via a v-Jun transforming pathway (20). In addition, knockdown of c-jun diminishes Meq's transforming ability in vitro, strongly suggesting that a Meq/Jun partnership plays a key role in Meq's oncogenic properties. However, the most convincing evidence for Meq's oncogenic property was the characterization of a Meq-null recombinant MDV virus (rMd5ΔMeq) which replicated in chickens and did not induce tumors (25). Significantly, the Meq-null virus also provided better protection than currently available vaccines in chickens upon challenge with the most virulent strains of MDV (18), pointing to a potential strategy for further vaccine improvement, wherein more subtle mutation(s) of Meq are engineered, to abolish its transforming ability, while retaining its ability of establishing infection in vivo and associated antigenicity. To this end, it is important to further dissect the transforming and replication functions of Meq, which at present remain largely unexplored.
Like Jun, the leucine zipper region of Meq allows the formation of homodimers and heterodimers (21). It has been shown that dimerization partners of bZIP proteins are important determinants of DNA binding specificity and therefore transcriptional regulation. For example, different Jun dimers have been shown to play distinct roles in transformation, i.e., anchorage- or serum-independent growth (38). Again, similarly to Jun, the DNA binding properties of Meq depend on its dimerization partner. Previous characterization of the in vitro DNA binding properties of Meq revealed that Meq-Jun heterodimers bind AP-1 sequences, whereas Meq homodimers in addition to AP-1 sequences also bind sequences found at the MDV origin of replication (Ori) (21, 29). Transcriptional analysis of Meq on the AP-1 containing meq promoter and the MDV pp14/38 bidirectional promoter which contains the MDV Ori revealed that Meq transactivates the meq promoter but represses the bidirectional pp14/38 promoter. Because Meq has been shown to bind the MDV Ori, it is possible that Meq homodimers repress the pp38/14 promoter by binding this sequence element. In addition to regulating pp38 expression, an MDV protein highly expressed during lytic infection, Meq was also shown to bind the ICP4 promoter by chromatin immunoprecipitation analyses. Therefore, Meq may also potentially regulate the expression of an additional lytic protein ICP4 (21, 29). This, together with luciferase reporter data, suggests that Meq heterodimers activate AP-1 containing promoters, therefore potentially activating genes associated with cell proliferation, whereas Meq homodimers may repress genes associated with lytic infection and consequently may be involved in regulating lytic or latent infection. Collectively, these data point to a role of Meq heterodimers in transformation and Meq homodimers in the regulation of viral replication. In order to delineate the functions of Meq and address the role of Meq homodimers in MDV pathogenesis, a recombinant Meq mutant virus (rMd5-MeqGCN) expressing a chimeric meq gene (MeqGCN) was constructed by substituting the parental Meq leucine zipper with that of the leucine zipper of the yeast protein GCN4. The leucine zipper region of GCN4 allows for the formation of only homodimers (16), thus conferring homodimerization of MeqGCN. The homodimerization and DNA binding and transactivation/repression properties of MeqGCN were tested in vitro. Recombinant virus expressing MeqGCN was studied in vitro and in vivo and compared to parental rMd5 and rMd5ΔMeq viruses. In vitro, rMd5-MeqGCN replicated similarly to parental rMd5; however, infection of chickens with rMd5-MeqGCN did not result in tumor formation. We therefore present the first in vivo evidence that Meq homodimers are not sufficient for MDV induced transformation. Our data also showed that subtle mutations of Meq are effective in generating a nonvirulent recombinant MDV, paving the way to the development of improved recombinant vaccine based on Meq mutations.
MATERIALS AND METHODS
Cells and viruses.
Primary duck embryonic fibroblasts (DEF) were used for virus propagation, virus reactivation assay, growth curves and DNA transfections. Recombinant viruses were generated from cosmids derived from a very virulent MDV strain, Md5 (30). The previously described rMd5ΔMeq was also used in the present study (25). Chicken embryonic fibroblasts (CEF) were inoculated with rMd5-MeqGCN virus to obtain viral DNA used for transfection to recover a revertant virus (rMd5-MeqGCNR). The DF-1 cell line was used for luciferase assays and coimmunoprecipitation experiments (15). DEF and CEF were maintained in Leibowitz-McCoy (LM) medium supplemented with 5% bovine calf serum and penicillin-streptomycin at 37°C. The DF-1 cell line was maintained in LM medium supplemented with 4% fetal bovine serum and penicillin-streptomycin at 37°C.
Cosmids.
Previously described cosmids SN5, P89, SN16, A6, and B40, encompassing the entire genome of the very virulent strain Md5 (30), were used to generate a recombinant Md5 (rMd5) virus and a recombinant Md5 with a chimeric Meq gene containing the leucine zipper from GCN4 (Fig. 1 and 2).
FIG. 1.
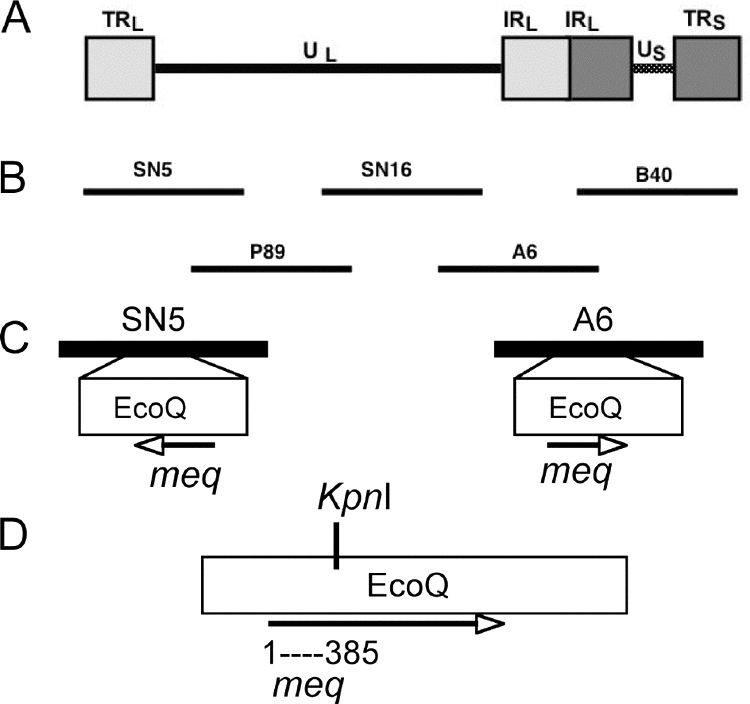
Cosmid clones used to recover recombinant viruses. (A) Organization of the serotype 1 MDV genome. (B) Schematic representation of the overlapping cosmid clones used to reconstitute recombinant viruses rMd5 and rMeqGCN, derived from a very virulent strain of MDV (Md5). (C) Location of EcoQ fragment and Meq gene in cosmids SN5 and A6. (D) Location of KpnI site described in Materials and Methods, located within the EcoQ fragment at nucleotide 385 of the meq gene.
FIG. 2.

Schematic representation of the meq gene. The DNA binding basic regions (BR1 and BR2) and transactivation domains are depicted. The leucine zipper (LZ) sequence of parental meq and GCN4 used to replace parental meq LZ in rMd5-MeqGCN are shown. Asterisks indicate the conserved leucine sites.
To generate mutations in the meq gene, a 741-bp DNA fragment referred herein as meq-KpnI, which contains the coding sequence for meq nucleotides 1 to 385, was mutated by a series of subcloning steps and overlapping PCR. The meq-KpnI fragment is located within the EcoQ fragment of the Md5 genome (Fig. 1). First, the EcoQ fragment was released from previously described cosmids A6 and SN5 by using the recA assisted restriction endonuclease (RARE) method (7). Briefly, recA and the primers SR1116 (5′-GAA TCG GAT TTG GAA TAA CCG AAT TCG GTG ATA TAA AGA C-3′) and SR1117 (5′-GAC ATT ACA AGA ATA GTT TGA ATT CTC GGG ATA ATC TCC C-3′) were used to protect the flanking EcoRI sites of the EcoQ fragment during the EcoRI methylation reaction. The unmethylated EcoRI sites were digested with EcoRI releasing the EcoQ fragment, which was subsequently cloned into pCR2.1 (Invitrogen, Carlsbad, CA). The digested A6 and SN5 cosmids were self-ligated, generating A6ΔEcoQ and SN5ΔEcoQ and HB101 competent bacteria transduced using Gigapack III (Stratagene, La Jolla, CA) according to the manufacturer's recommendations. Next, the meq-KpnI fragment was released from pCR2.1-EcoQ by digestion with KpnI, and the fragment was gel purified, using the QIAEX II gel extraction kit (Qiagen, Valencia, CA), and cloned into KpnI-digested pCR2.1 vector. Overlapping PCR, in a combination of three PCRs, was performed to replace the leucine zipper region of meq with the leucine zipper region of GCN4. Two primary PCRs were performed to generate the 5′ and 3′ ends of meq-KpnI fragment. The 5′ reaction was performed using the primers M13R (5′-CAG GAA ACA GCT ATG AC-3′) and SR1192-GCN4 leucine zipper reverse (5′-CTC ATT TTC CAA GTG ATA ATT TTT CGA AAG CAA TTC TTC AAC CTT GTC TTC AAG TTT GTC TAC ATA GTC CGT CTG CTT CCT-3′). 3′-end PCR was performed using the primers SR1190 (5′-GAC CGA GAT AGG GTT GAG TG-3′) and SR1193-GCN4 leucine zipper forward (5′-AAA AAT TAT CAC TTG GAA AAT GAG GTT GCC AGA TTA AAG AAA TTA GTT GGC GAA CGC CGT GTA CAG TTG GCT TGT CAT GAG CCA-3′). Both amplicons were gel purified, mixed together, and used as templates in a third PCR with the primers M13R and SR1190, generating a full meq-KpnI fragment containing the GCN4 leucine zipper in place of the meq leucine zipper (meq-GCN4-KpnI). The meq-GCN4-KpnI fragment was subsequently cloned into pCRBlunt (Invitrogen) transformed into Escherichia coli TOP 10 cells, and positive clones were identified by restriction digestion and fully sequenced. The meq-GCN4-KpnI fragment was then released by KpnI digestion and cloned into pcr2.1-EcoQΔKpnI, replacing the parental meq-KpnI fragment contained in this region. The newly generated EcoQ fragment (EcoQ-MeqGCN) containing the mutated meq leucine zipper (MeqGCN) was then transferred into cosmids A6ΔEcoQ and SN5ΔEcoQ, using RARE. The EcoRI site of cosmids A6ΔEcoQ and SN5ΔEcoQ was protected during the methylation reaction using recA and primer SR1130 (5′-GAA TCG GAT TTG GAA TAA CCG AAT TCT CGG GAT AAT CTC CCG ATG G-3′), subjected to EcoRI restriction digestion to linearize the cosmids, and subsequently ligated to the EcoQ-MeqGCN fragment, yielding cosmids SN5-MeqGCN and A6-MeqGCN. After ligation and transduction, clones containing the Eco-MeqGCN fragment in the correct orientation were identified by PCR, and selected positive colonies were sequenced across the junction regions. The integrity of the SN5-MeqGCN and A6-MeqGCN cosmids was confirmed by evaluating their restriction digestion pattern.
Transfections.
Parental and mutant cosmid DNA were digested with NotI to release the viral insert and purified by phenol-chloroform extraction and ethanol precipitation before transfection. To generate rMd5 and rMd5-MeqGCN, 500 ng of cosmids P89, SN16, B40, SN5, and A6 or P89, SN16, B40, SN5-MeqGCN, and A6-MeqGCN, respectively, were used to transfect 1.2 × 106 DEF in 60-mm dishes by the calcium phosphate procedure. At 5 days posttransfection, cells were trypsin treated, seeded onto a 100-mm dish, and monitored for cytopathic effects. Viral stocks of recovered viruses were subsequently made in DEF for further analysis.
Revertant virus.
To generate a revertant virus containing the parental meq gene, gel-purified parental Md5 EcoQ fragment, together with proteinase K-digested and phenol-chloroform extracted rMd5-MeqGCN genomic DNA, was used to cotransfect CEF by the calcium phosphate procedure. After viral plaques were evident, the cells were overlaid with 0.9% Bacto agar in growth media, and individual plaques were harvested by trypsinization. Cells from each plaque were divided into two aliquots: one was used to infect DEF, and the other was used for DNA extraction and PCR analysis. The presence of parental meq or GCN4 leucine zipper sequences in individual plaques was detected by PCR using the SR1118-meq start primer (5′-GAT CCC GGG GAG ATG TCT CAG GAG CCA GAG C-3′) and the leucine zipper specific reverse primer SR3073-meq leucine zipper (5′-GTC CTT AGA TCT CGA ATT TCC -3′) SR3074-GCN4 leucine zipper (5′-CTA ATT TCT TTA ATC TGG CAA C-3′) for parental and GCN4 sequences, respectively.
IFA and IHC.
Confluent DEF monolayers were infected with rMd5 or rMd5-MeqGCN and, when viral plaques were apparent, cells were fixed with ice-cold acetone-alcohol (6:4), and the expression of Meq was evaluated by indirect immunofluorescence assay (IFA) using rabbit polyclonal anti-Meq antibodies (1:300). Lymphocytes collected for reactivation assays were deposited on a microscope slide using a cytospin centrifuge, fixed with ice-cold acetone/alcohol (6:4), and the expression of pp38 was evaluated by IFA using the H19 pp38 specific monoclonal antibody (1:400). For immunohistochemistry (IHC), lymphoid organs (thymus, spleen, and bursa of Fabricius), and feather follicles from infected and uninfected chickens were embedded in optimal cutting temperature compound (Tissue-Tek OCT; Sakura Finetek, Torrance, CA), immediately frozen in liquid nitrogen, and stored at −80°C until use. Cryostat sections (6 to 8 μm thick) of tissue were prepared, fixed with cold acetone at −20°C for 5 min, and air dried. Immunostaining was carried out with H19 pp38 monoclonal antibody (1:3,000) and the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions.
MeqGCN homodimerization.
For in vitro homodimerization assays, parental meq and MeqGCN were cloned into the pcDNA3.1 (Invitrogen) eukaryotic expression vector. Both meq and MeqGCN genes were cloned in frame with hemagglutinin (HA) and Flag tags to aid in protein detection (12). The resulting plasmids were denoted as pHA-MeqGCN, pFlag-MeqGCN, pHA-Meq, and pFlag-Meq.
DF-1 cells were cotransfected with 2 μg of either pFlag-Meq or pFlag-MeqGCN and 2 μg of pHA-Meq-, pHA-MeqGCN, pCMV-cJun, or pRCAS-cFos using Fugene 6 (Roche, Mannheim, Germany). pCMV-cJun and pRCAS-cFos were generous gifts from Junnlin Liu, M. D. Anderson Cancer Center). Cells were harvested 48 h after transfection and lysed in radioimmunoprecipitation assay buffer (10 mM Tris-HCl [pH 7.4], 1% NP-40, 0.1% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride). Cell lysates (500 μl) were immunoprecipitated with 20 μl of Flag antibody-conjugated agarose beads (Sigma, St. Louis, MO). Meq/c-Jun or Meq/c-Fos heterodimers were immunoprecipitated with 3 μg of either anti-cFos antibody (Santa Cruz, Santa Cruz, CA) or anti-cJun antibody (Santa Cruz) for 2 h. The immune complex was then captured by the addition 20 μl of a protein A- and protein G-Sepharose bead mixture and rocked for an additional 2 h at 4°C. Beads were washed four times with radioimmunoprecipitation assay buffer, boiled for 5 min in 20 μl of 2× sodium dodecyl sulfate (SDS) sample buffer (125 mM Tris-HCl [pH 6.8], 4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.6% bromophenol blue), and subjected to SDS-polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride membranes. After blocking for 1 h at room temperature with 5% skim milk in TBST (20 mM Tris-HCl [pH 7.5], 137 mM NaCl, 0.05% Tween 20), the membranes were incubated with primary antibodies for 2 h at room temperature. The membranes were washed with TBST three times for 10 min each time at room temperature and then incubated with horseradish peroxidase-conjugated antibodies for 1 h at room temperature. Membranes were washed three times with TBST and visualized with enhanced chemiluminescence reagent (Amersham-Pharmacia, Piscataway, NJ). The final dilutions of the primary antibodies for immunoblotting were 1:500 for anti-HA tag antibody (Covance, Berkeley, CA), 1:4,000 for anti-Meq (20), 1 μg/ml for anti-cFos (Santa Cruz), and 1 μg/ml for anti-cJun (Santa Cruz).
Expression of recombinant Meq and MeqGCN proteins.
Recombinant parental Meq and MeqGCN proteins were produced using a baculovirus expression system (Invitrogen). Parental meq and meqGCN genes were cloned into a modified pFAST-BAC vector, which carries a Flag tag. One hundred million Sf9 cells were infected with recombinant baculoviruses, expressing either Flag-tagged Meq (Flag-Meq) or Flag-tagged MeqGCN (Flag-MeqGCN). Cells were harvested 48 h after infection and lysed in lysis buffer (50 mM Tris-HCl [pH 7.5], 500 mM NaCl, 2% glycerol) supplemented with protease inhibitor cocktail (Roche). Cleared lysates were incubated overnight with 100 μl of Flag antibody-conjugated agarose beads, followed by four washes with lysis buffer. Recombinant proteins were then eluted with 3× Flag peptides according to the manufacturer's protocol. The purity and concentration of protein was measured by SDS-polyacrylamide gel electrophoresis and Coomassie blue staining using bovine serum albumin as a standard.
EMSA.
Electrophoretic mobility shift assay (EMSA) analysis was performed with baculovirus-expressed recombinant purified Meq proteins. The DNA binding activity of Flag-Meq and Flag-MeqGCN was tested with two different probes, an AP-1 consensus oligonucleotide (5′-CGC TTG ATG AGT CAG CCG GAA-3′; Promega, Madison, WI) and a probe derived from the MDV Ori (5′-TGC TCA TTT GCA TAC ACA TCA CGT GAT AGT-3′). Probes were labeled at the 5′ ends with [32P]ATP and polynucleotide kinase according to the manufacturer's instructions (Promega). Purified recombinant Meq or MeqGCN proteins (160 ng) were incubated for 20 min at 30°C in gel shift buffer (Promega), followed by an additional 20 min incubation with approximately 10,000-cpm-labeled probe. For competition reactions, purified proteins were first incubated with unlabeled probes for 20 min at 30°C prior to incubation with labeled probes. Reaction products were resolved on nondenaturing 6% polyacrylamide-Tris-glycine gels and visualized using a Fuji Film Bas-1800 II phosphorimaging system.
Luciferase assays.
The transactivation and repression activities of the MeqGCN protein were evaluated on MDV promoters by luciferase assay. The meq promoter region (nucleotides −355 to −1) was cloned into the pGL3 vector (Promega) to form the pGL3-meq promoter. The pp38 and pp14 promoters were cloned into the pGL3 reporter vector and have been previously described (21). Three tandem copies of the MDV Ori sequence (TGC TCA TTT GCA TAC ACA TCA CGT GAT AGT) found within the pp38/pp14 bidirectional promoter were cloned into the pGL2-TATA luciferase vector (kindly provided by Stephen Safe, Texas A&M University) to generate pGL2-3XOri. The meq and MeqGCN genes were cloned into the pcDNA 3.1 Zeo vector (Invitrogen) to generate expression vectors, pcDNA-Meq, and pcDNA-MeqGCN. DF-1 cells (105) were seeded in 12-well plates at 16 h prior to transfection and incubated at 37°C under standard conditions. Transfections were performed by using siPORT XP-1 (Ambion, Austin, TX); 500 ng of pcDNA (empty vector control), pcDNA-Meq, or pcDNA-MeqGCN; and either 250 ng of pGL3-meq promoter vector, pGL2-Ori, or 500 ng of pp38 and pp14 promoter vectors, according to manufacturer's recommendations. Cell lysates were harvested at 48 h posttransfection with active lysis buffer (Promega), and luciferase activity was measured, using a Biotek Clarity luminometer (Biotek, Winooski, VT). The protein concentration in each transfected sample was measured by a Bradford assay (Bio-Rad, Hercules, CA), and the luciferase activity was normalized to protein concentration. Assays were performed in triplicate, and three independent experiments were performed for each reporter vector tested. The transactivation or transrepression activity was expressed as the fold difference relative to the empty pcDNA vector control. The results of all three experiments were analyzed by one-way analysis of variance, followed by a Tukey's HSD test using the SPSS version 14.0 software (SPSS, Inc., Chicago, IL). For all analysis, a P value of ≤0.05 was considered statistically significant.
Colony formation in soft agar.
DF-1 cells were transfected with pcDNA, pcDNA-Meq, or pcDNA-MeqGCN using siPORT XP-1 (Ambion). Approximately 48 h after transfection the transfected DF-1 cells were selected with 300 μg of zeocin (Invitrogen)/ml for 4 weeks. The expression of Meq was confirmed by IFA using Meq antibody as described above. Pools of resistant cells (5 × 103) were seeded in 0.33% agarose containing LM media with 150 μg of zeocin/ml and 10% FBS overlaid on a 0.5% agarose in a 35-mm plate. After 3 weeks of culture, the colonies were examined under a light microscope and photographed using ×12 magnification. Three different fields were randomly selected, and colonies greater than 100 μm were counted. Two independent experiments were performed, and each experiment was performed in triplicate.
Southern blot.
DNA from rMd5, rMd5-MeqGCN, and rMd5ΔMeq-infected DEF was isolated by proteinase K digestion, followed by phenol-chloroform extraction and ethanol precipitation. Portions (3 μg) of each DNA sample were digested with either EcoRI or PstI, separated on a 1% agarose gel, and transferred to nylon membranes. [32P]dCTP-labeled probes representing the complete MDV genome (cosmids SN5, P89, SN16, A6, and B40) or EcoQ fragment (2,456-bp fragment) were generated by random priming using a High Prime DNA labeling kit (Roche) and used to hybridize to viral DNA according to standard protocols (34).
In vitro growth kinetics.
To compare the growth characteristics of rMd5 and rMd5-MeqGCN, DEF seeded on 35-mm plates were inoculated with approximately 50 PFU of each virus. On days 2, 4, and 6 after inoculation, the infected cells were trypsinized, serial dilutions were inoculated onto DEF monolayers and seeded onto 35-mm plates, and plaques at the different dilutions were counted 7 days later.
Pathogenesis experiments. (i) Experiment 1.
To study the effect of Meq homodimers on viral replication, 4-week-old specific-pathogen-free (SPF) chickens (Hy-Vac, Adel, Iowa) were randomly sorted into experimental groups of nine chickens each. One group remained as a noninoculated control, whereas the other groups were inoculated subcutaneously with 3,000 PFU of rMd5, rMd5-MeqGCN, or rMd5ΔMeq. Three chickens were randomly selected at each time point, except at day 6, when two chickens were selected. On days 14 and 21 postinfection, blood samples were collected in heparin for reactivation assays (see below) and were subsequently euthanized for tissue sample collection. On days 6 and 21 postinfection, lymphoid organs and feather follicles were collected for IHC. On day 14 postinfection, lymphoid organs were collected to determine lymphoid organ/body weight ratios. Organ/body weight ratios between groups were analyzed by using the Kruskal-Wallis test with significance set at P < 0.05.
(ii) Experiment 2.
To study the role of the Meq homodimers on oncogenesis, SPF (Hy-Vac) day-old chicks (nine per group) were inoculated subcutaneously with 2,000 PFU of virus rMd5, rMd5-MeqGCN, or rMd5ΔMeq and reared in modified Horsfall-Brauer isolation units for 8 weeks. Weekly mortality was recorded, and all chickens were necropsied at time of death or at termination of the experiments and evaluated for MDV-specific lesions in the viscera and nerves. To confirm that the phenotypic changes observed in rMd5-MeqGCN were due to the exclusive homodimerization of Meq, a revertant virus (rMd5-MeqGCNR) was inoculated into three chickens (3,000 PFU) and evaluated for MDV specific lesions.
(ii) Experiment 3.
To study the role of Meq homodimers on horizontal transmission, six SPF (Charles River) day-old chicks were inoculated subcutaneously with 3,000 PFU with rMd5 or rMd5-MeqGCN; three additional uninoculated chicks were reared with each group and served as contacts to assess horizontal transmission. At 8 weeks postinfection, buffy coats were obtained from heparinized blood by centrifugation at 500 × g. DNA was extracted by using a Purelink Genomic DNA extraction kit (Invitrogen), and PCR was performed using the MDV-specific Meq primers SR1118 (5′-GAT CCC GGG GAG ATG TCT CAG GAG CCA GAG C-3′) and SR1135 (5′-GAT CCC GGG TCA GGG TCT CCC GTC ACC TGG AAA CC-3′) to detect the presence of MDV genome.
Reactivation assay.
Buffy coats were collected as described above at days 14 and 21 postinfection. DEF monolayers seeded in 35-mm plates were inoculated with 106 lymphocytes in duplicate, and viral plaques were counted at 7 days postinoculation.
RESULTS
Mutations in the leucine zipper of Meq are sufficient to confer homodimerization.
It has previously been shown that Meq has the ability to form homodimers and heterodimers like the Jun family of bZIP transcription factors, with which Meq shares considerable homology (21). It is well established that the specific amino acid residues within the leucine zipper of bZIP proteins determine their dimerization properties. Among these transcription factors, the yeast protein GCN4 is well characterized, and the leucine zipper region is known to allow for the exclusive formation of homodimers (16). As in other studies (10, 11, 14), we also utilized the leucine zipper region of GCN4 in place of the parental leucine zipper of Meq to study the function of Meq homodimers in MDV pathogenesis. The leucine zipper of Meq was successfully “swapped” with the leucine zipper of GCN4 using overlapping PCR, resulting in a Meq homodimer mutant, MeqGCN (Fig. 2). The open reading frames of Meq and MeqGCN, fused to the FLAG and HA epitope tags (HA-Meq, Flag-Meq, HA-MeqGCN, and Flag-MeqGCN) were expressed in DF-1 cells. Coimmunoprecipitation experiments were performed with anti-Flag antibody-conjugated beads to test whether the leucine zipper mutations in MeqGCN were sufficient to confer homodimerization (Fig. 3). First, Flag-Meq or Flag-MeqGCN was cotransfected with HA-Meq in DF-1 cells (Fig. 3a, left panel). Flag-Meq protein complexes were immunoprecipitated with anti-Flag antibody and evaluated for the presence of HA proteins by Western blot analysis. Flag-Meq, but not Flag-MeqGCN, effectively precipitated HA-Meq. Conversely, Flag-MeqGCN precipitated HA-MeqGCN but not HA-Meq (Fig. 3a, right panel). These results demonstrated the strong homodimerization property of MeqGCN.
FIG. 3.

Homo- and heterodimerization of Meq and MeqGCN. (a) Coimmunoprecipitation analysis of tagged Meq and MeqGCN proteins. The indicated plasmids were cotransfected in DF-1 cells. Twenty percent of the total cell lysates that were used for coimmunoprecipitation were also included as controls. Meq coprecipitates only with Meq (left panel), and MeqGCN coprecipitates only with MeqGCN (right panel), demonstrating homodimerization. Membranes were reprobed with anti-Flag antibody as a control. (b) MeqGCN had impaired ability to form heterodimer with c-Jun or c-Fos. The indicated plasmids were cotransfected in DF-1 cells, immunoprecipitated with either anti-Fos or anti-Jun antibody, and blotted with anti-Meq antibody. Ten percent of the total cell lysates that were used for immunoprecipitation were also included in the same gel as a control. Both c-Fos and c-Jun are effectively precipitated Meq but only weakly precipitated MeqGCN.
As a next step, the ability of Meq and MeqGCN to dimerize with c-Jun or c-Fos was examined (Fig. 3b). Meq or MeqGCN was coexpressed with c-Fos or c-Jun, and their ability to heterodimerize with other bZIP family protein assessed. As shown in the left panel of Fig. 3b, while Meq effectively pulled down c-Fos, very little, if any, c-Fos was found to associate with MeqGCN. Likewise, c-Jun interacted with Meq much more avidly than MeqGCN. This and the above experiment suggest that MeqGCN preferentially, if not exclusively, forms homodimer under physiological conditions.
Meq homodimer mutant (MeqGCN) retains DNA binding, transactivation, and repressive functions.
Previous characterization of Meq has shown that Meq homodimers and heterodimers differ in their DNA binding affinities. Although both Meq homodimers and heterodimers bind AP-1 sequences, Meq together with c-Jun bound AP-1 sequences with greater affinity than Meq alone. In addition, Meq alone bound the MDV origin of replication (MDV Ori) located in the pp38/14 bidirectional promoter (21). Luciferase reporter assays indicated functional differences between Meq and Meq plus c-Jun in that although Meq activated the AP-1 containing meq promoter, Meq plus c-Jun resulted in higher activation. Furthermore, Meq expression was shown to repress the MDV pp38/pp14 bidirectional promoter, which contains the MDV Ori (21). Therefore, EMSA and luciferase assays were performed to test whether the DNA binding and transactivation/repression functions of MeqGCN were still functional. Recombinant Flag-Meq and Flag-MeqGCN proteins were prepared from recombinant baculovirus-infected cells. Recombinant proteins were incubated with 32P-labeled consensus AP-1 oligonucleotide or an oligonucleotide encompassing a portion of the MDV Ori. Both Meq and MeqGCN proteins bound both AP-1 and MDV Ori oligonucleotides, and the intensity of band shifts for both Meq and MeqGCN decreased in the presence of specific competitor, indicating specific binding (Fig. 4). Luciferase assays were then performed to test the transactivation and repressive functions of MeqGCN compared to Meq. DF-1 cells were transfected with pcDNA empty vector, pcDNA-Meq, or pcDNA-MeqGCN, together with pGL3-meq promoter reporter vector. As shown in Fig. 5, both Meq and MeqGCN significantly activated the meq promoter, although MeqGCN activated the meq promoter to significantly lower levels than Meq (P < 0.05). Luciferase assays were also performed to test the ability of MeqGCN to repress the previously described pp38 and pp14 promoters. DF-1 cells were transfected with pcDNA, pcDNA-Meq, or pcDNA-MeqGCN and either the pGL3-pp38 or the pGL3-pp14 promoter reporter vectors. Although pcDNA-Meq and pcDNA-MeqGCN significantly repressed the pp38 promoter, only pcDNA repressed the pp14 promoter (P < 0.05) (Fig. 5). Nonetheless, both pcDNA-Meq and pcDNA-MeqGCN significantly repressed the pGL2-3X-Ori, which contains three copies of the sequence element found in the pp38/14 bidirectional promoter that Meq was previously reported to bind (21, 29).
FIG. 4.
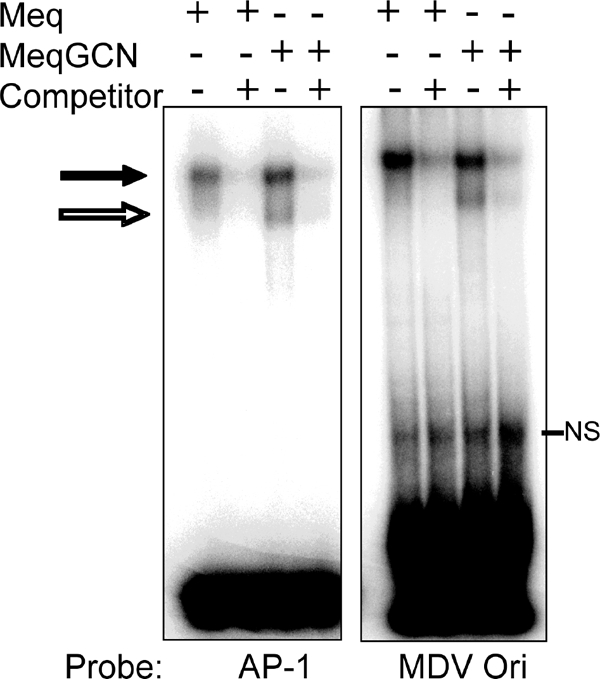
EMSAs of Meq and MeqGCN protein were performed to test the DNA binding capacity of MeqGCN. Purified baculovirus-expressed proteins, Meq and MeqGCN, were incubated with radiolabeled oligonucleotide probes, followed by gel retardation analysis. Two different probes were used: a consensus AP-1 oligonucleotide and an ACACA probe derived from the MDV origin of replication (MDV Ori). Band shifts are observed by Meq and MeqGCN (black arrow), the intensity of which decreases in the presence of unlabeled competitor. NS, nonspecific bands.
FIG. 5.
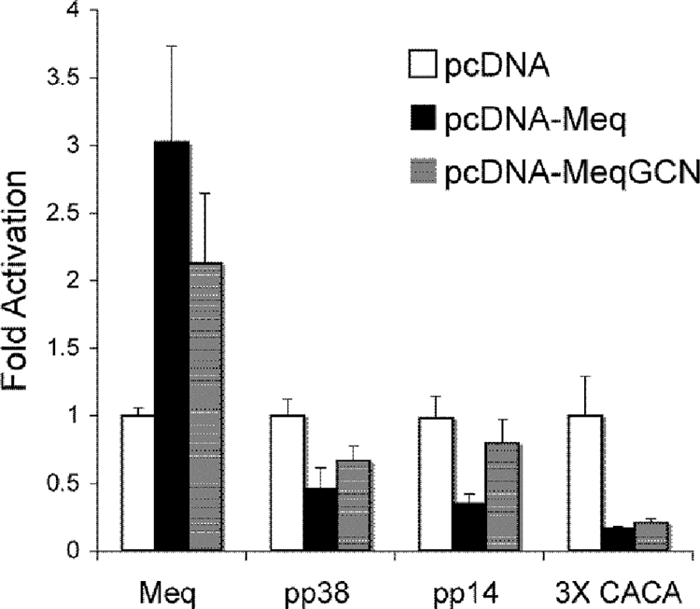
Luciferase assays demonstrate that MeqGCN has functional transactivation and repression activities. DF-1 cells were transfected with meq promoter, pp14 promoter, pp38 promoter, or 3X-Ori luciferase reporter plasmids and pcDNA (empty vector), pcDNA-Meq, or pcDNA-MeqGCN. Both Meq and MeqGCN activate the meq promoter and repress the pp38/14 bidirectional promoters and 3X-Ori, indicating MeqGCN maintains transactivation/repressive functions. Luciferase values are expressed as the fold difference relative to the pcDNA vector. Error bars indicate the standard deviation.
Colony formation in soft agar.
Colony formation in soft agar is a marker of transformation, and Meq expression in fibroblasts has been shown to promote anchorage-independent growth (20). Soft-agar assay was used to test the ability of Meq homodimers to promote colony formation in soft agar. Meq and MeqGCN expression in selected DF-1 cells was confirmed by IFA (Fig. 6a, top panel). As expected, selected DF-1 cells expressing the pcDNA-Meq construct formed large colonies in soft agar compared to control cells selected with pcDNA empty vector (Fig. 6a, bottom panel). Furthermore, cells expressing pcDNA-Meq had 23-fold more colonies >100 μm in size compared to pcDNA control cells. In contrast, DF-1 cells expressing pcDNA-MeqGCN only had 3.5-fold as many colonies >100 μm in size compared to empty vector control cells (Fig. 6b), indicating that Meq homodimers have a reduced potential for transformation in vitro.
FIG. 6.

In vitro soft-agar assay demonstrating that MeqGCN has reduced transformation potential. (a) The top row shows immunofluorescence analysis of Meq expression from pools of selected DF-1 cells transfected with pcDNA, pcDNA-Meq, and pcDNA-MeqGCN. In the bottom panels, a soft-agar assay was performed using pcDNA, pcDNA-Meq, and pcDNA-MeqGCN selected DF-1 cells to assess anchorage-independent growth. (b) Number of colonies >100 μm observed in cells expressing pcDNA, pcDNA-Meq, or pcDNA-MeqGCN. The average numbers of colonies counted from three random fields are shown. Error bars indicate the standard deviation.
Construction of Meq homodimer mutant virus rMd5-MeqGCN.
A recombinant Md5 mutant virus in which the leucine zipper region of meq was replaced with the corresponding region of GCN4 (rMd5-MeqGCN) was successfully constructed. The leucine zipper region of the meq gene was replaced with the leucine zipper region of GCN4 by overlapping PCR, and the EcoQ fragment containing the chimeric meqGCN gene was cloned into the A6Δmeq and SN5Δmeq cosmids. The resultant cosmids A6-MeqGCN and SN5-MeqGCN, together with cosmids P89, B40, and SN16, were cotransfected into DEF by the calcium phosphate method, and a recombinant virus was subsequently recovered by homologous recombination. Southern blot analyses were performed to assess the integrity of the rMd5-MeqGCN viral genome. Genomic DNA from rMd5-, rMd5-MeqGCN-, and rMd5ΔMeq-infected DEF was digested with EcoRI or PstI and probed with 32P-labeled Md5 cosmids or EcoQ fragment DNA, respectively. No differences were observed between rMd5 and rMd5-MeqGCN EcoRI digestion patterns, confirming the integrity of the recombinant genomes (Fig. 7). DNA from rMd5-, rMd5-MeqGCN-, and rMd5ΔMeq-infected DEF was also digested with PstI because there is a PstI site located within the leucine zipper region of the meq gene that is absent in the mutant MeqGCN gene. As expected, PstI-digested rMd5-MeqGCN DNA probed with 32P-labeled EcoQ fragment resulted in a single band compared to two bands observed for rMd5 (Fig. 7). Like rMd5-MeqGCN, rMd5ΔMeq, also lacks the PstI site due to the deletion of the meq gene; therefore, a single yet smaller band was detected by Southern blot (Fig. 7).
FIG. 7.

Southern blot analysis of rMd5, rMd5-MeqGCN, and rMd5ΔMeq. (A) DNA was digested with EcoRI and probed with total viral MDV DNA. The restriction profile of rMd5-MeqGCN is similar to rMd5, indicating that no gross genome rearrangements occurred. The arrow indicates the location of the EcoQ fragment. Due to the meq deletion in the EcoQ fragment of rMd5ΔMeq, this fragment migrates faster. (B) DNA was digested with PstI and probed with EcoQ fragment. The introduced LZ mutations in rMd5-MeqGCN resulted in the loss of a PstI site, and therefore a single band is observed, in contrast to the two bands for rMd5. Likewise, rMd5ΔMeq does not have a PstI site and, as a consequence of the meq deletion, results in a faster-migrating single band.
IFA was performed to evaluate Meq expression in virus-infected DEF. Meq expression was observed in both rMd5- and rMd5-MeqGCN-infected DEF but not in rMd5ΔMeq-infected cells. In addition, a magnification of ×1,000 clearly showed that expression of MeqGCN localized to the nucleus (Fig. 8). Although less expression was observed in rMd5-MeqGCN compared to rMd5, this correlates with luciferase data, which indicated that MeqGCN activated the meq promoter to a lesser extent than parental Meq (Fig. 5).
FIG. 8.

Immunofluorescence analysis of DEF cells infected with recombinant viruses. (a) rMd5ΔMeq-infected DEF (magnification, ×100); (b) rMd5-MeqGCN-infected DEF (magnification, ×100); (c) rMd5-infected DEF (magnification, ×100); (d) rMd5-MeqGCN-infected DEF (magnification, ×1,000). Meq expression is observed in the nucleus of rMd5- and rMd5-MeqGCN-infected DEF but not rMd5ΔMeq-infected DEF.
In vitro and in vivo replication of rMd5-MeqGCN.
The in vitro growth properties of rMd5-MeqGCN were tested to assess whether the leucine zipper mutations had any effect on virus replication in vitro. Our results indicate that rMd5 and rMd5-MeqGCN virus replicated similarly at the time points tested (days 0, 2, 4, and 6) (Fig. 9). To test the role of Meq homodimers in in vivo replication, 4-week-old SPF chickens were inoculated with 3,000 PFU of rMd5, rMd5-MeqGCN, or rMd5ΔMeq virus. At 6 days after inoculation, two randomly selected birds were euthanized, and lymphoid organs (thymus, spleen, and bursa of Fabricius) were collected and evaluated for viral lytic antigen (pp38) expression by IHC. Expression of pp38 was evident in the bursa of Fabricius (Fig. 10), thymus, and spleen (data not shown) with both rMd5- and rMd5-MeqGCN-infected chickens, indicating that the leucine zipper mutations in the rMd5-MeqGCN virus did not affect early cytolytic infection. To further assess the role of Meq homodimers in viral replication, lymphoid organ body weight ratios were determined from three birds from each group at day 14 postinfection to evaluate lymphoid organ atrophy. As expected, splenomegaly, as well as thymic and bursal atrophy, was observed in chickens inoculated with rMd5, indicating high levels of cytolytic infection. Although there were no significant differences between all groups, bursal and thymic atrophy were less evident in the rMd5-MeqGCN group compared to the rMd5 group (Fig. 11).
FIG. 9.

In vitro growth properties of rMd5 and rMd5-MeqGCN. DEFs were infected with the indicated viruses and harvested on days 2, 4, and 6 after infection, and titers were determined on fresh DEF. Day 0 indicates the titer of the virus in the inoculum. The experiment was performed in duplicate, and the titer (logarithm of the mean number of PFU per dish) is indicated.
FIG. 10.

Infection of lymphoid tissue and feather follicles. Immunohistochemistry of bursa and feather follicle at days 6 and 21 postinfection, respectively, with anti-pp38 monoclonal antibody (H19). Positive cells are indicated by double staining; counterstaining was performed with hematoxylin.
FIG. 11.
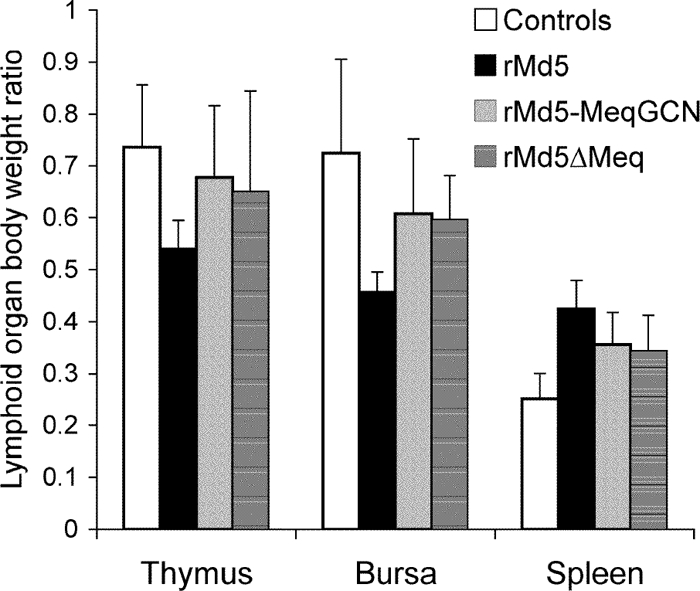
Lymphoid organ/body weight ratios of rMd5-, rMd5-MeqGCN-, and rMd5ΔMeq-infected chickens day 14 postinfection. Three birds from each group were euthanized, lymphoid organs were collected, and chickens and lymphoid organs were weighed. Statistical analysis was performed by using the Kruskal-Wallis test. Significance was set at P < 0.05. Although no significant differences were observed, less lymphoid organ atrophy was observed in chickens infected with rMd5-MeqGCN compared to birds infected with rMd5.
It is well documented that MDV switches from an active cytolytic infection to a latent infection approximately 7 to 8 days postinfection, and virus reactivation can be observed when latently infected lymphocytes are cocultured with fibroblasts (2). To examine whether Meq homodimers are involved in the establishment of latency and reactivation, peripheral blood lymphocytes (buffy coats) were obtained from three chickens in each group at days 14 and 21 postinfection and cocultured with DEF. To confirm viral infection, portions of collected lymphocytes were fixed on microscope slides, and IFAs were performed to the detect viral antigen, pp38. As shown in Table 1, few or no plaques were obtained from the peripheral blood mononuclear cells of chickens infected with rMd5-MeqGCN and rMd5ΔMeq. However, at day 21, latently infected lymphocytes were positive for viral antigen pp38 (Fig. 12), suggesting that these viruses are defective in reactivation.
TABLE 1.
Virus reactivation from peripheral blood lymphocytesa
| Group | Avg no. of PFU at:
|
|
|---|---|---|
| Day 14 | Day 21 | |
| rMd5 | 45 | 120 |
| rMd5-MeqGCN | 1 | 0 |
| rMd5ΔMeq | 2.5 | 0 |
Reactivation assays were performed on days 14 and 21 after inoculation. The numbers represent the average number of PFU observed when 106 peripheral blood lymphocytes were cocultured with DEF.
FIG. 12.

Detection of MDV pp38 protein in latently infected cells at 21 days postinfection. IFA was performed with anti-pp38 monoclonal antibody (H19) on peripheral blood mononuclear cells collected from rMd5- and rMd5-MeqGCN-infected chickens. Texas Red-conjugated secondary antibodies were used to detect pp38 (white arrows), and nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole).
Transmission of MDV occurs through replication of virus in the feather follicle epithelium (FFE) and release of dander containing infectious virus (4). Therefore, to determine whether the rMd5-MeqGCN homodimer mutant was able to replicate in the FFE, expression of the lytic viral antigen pp38 was evaluated in three chickens from each inoculated group by IHC. Tissues from all inoculated groups tested positive for pp38 antigen, indicating the ability of rMd5-MeqGCN, like rMd5, to replicate in the FFE (Fig. 10). To further assess the role of Meq homodimers in MDV transmission, day-old SPF chickens were inoculated with 3,000 PFU of rMd5 or rMd5-MeqGCN and reared with three uninfected contact chicks. Buffy coats were collected from contact birds at 8 weeks postinfection, DNA was extracted, and PCR for Meq was performed. In both groups, two out of three contact chickens tested positive for Meq or MeqGCN, respectively. Altogether, these results confirm that rMd5-MeqGCN retains the ability to transmit horizontally by shedding through the feather follicle epithelium.
Oncogenicity of rMd5-MeqGCN.
To determine whether Meq homodimers are involved in transformation of T cells, groups of nine SPF chickens were inoculated at day of age with 2,000 PFU of rMd5, rMd5-MeqGCN, or rMd5ΔMeq and then observed for mortality for a period of 8 weeks. All chickens that died during the experiment or were euthanized at the termination of the experiment were evaluated for MDV-specific lesions. MDV-associated mortality was observed in rMd5-infected chickens starting at week 4 postinfection, and none of the chickens survived the end of the experiment. In contrast, all of the rMd5-MeqGCN-infected chickens survived the 8-week experiment, and microscopic examination of the vagus, brachial, and sciatic nerves showed no signs of Marek's disease. Similarly, as expected, the rMd5ΔMeq-infected chickens also survived the duration of the experiment and were free of MDV- specific lesions (Fig. 13 and Table 2).
FIG. 13.

Mortality in chickens inoculated with rMd5, rMd5-MeqGCN, and rMd5ΔMeq. Chickens were inoculated with 2,000 PFU of the indicated viruses at 1 day of age and maintained in isolation for 8 weeks, and weekly mortality was recorded. Uninoculated chickens served as negative controls. Chickens that died during the experiment were evaluated for MDV-specific gross lesions.
TABLE 2.
Pathogenicity of rMd5, rMd5-MeqGCN, rMd5ΔMeq, and rMd5-MeqGCNR in SPF MDV maternal-antibody-negative chickens
| Virus testeda | No. of chickens affected/total no. tested
|
|
|---|---|---|
| Mortality (%) | Tumor incidence (%)b | |
| rMd5 | 9/9 (100) | 9/9 (100) |
| rMd5-MeqGCN | 0/9 (0) | 0/9 (0) |
| rMd5ΔMeq | 0/9 (0) | 0/9 (0) |
| rMd5-MeqGCNRc | 3/3 (100) | 3/3 (100) |
| No virus | 0/9 (0) | 0/9 (0) |
All chickens were inoculated with 3,000 PFU of the indicated viruses.
Chickens that were not positive for gross tumors were further evaluated for microscopic tumors.
Performed as an independent experiment.
Construction and biological properties of revertant virus rMd5-MeqGCNR.
To verify that the phenotypic changes observed with rMd5-MeqGCN were due to changes introduced in the leucine zipper region, a revertant virus was constructed. DNA obtained from rMd5-MeqGCN-infected CEF was cotransfected with purified parental MDV EcoQ DNA, which contains the meq gene. A revertant virus, rMd5-MeqGCNR, was selected by plaque purification and screened for the presence of both parental and MeqGCN-mutant meq genes by PCR. PCR revealed that of the 130 plaques tested, 3 contained one copy of the parental meq gene restored in the viral genome. Three chickens were infected with 3,000 PFU of rMd5-MeqGCNR, and by week 5 postinfection all three were positive for MDV gross tumors, confirming that the attenuated phenotype of rMd5-MeqGCN was due to the mutations introduced in the leucine zipper of Meq. In addition, PCR analysis of virus isolated from the rMd5-MeqGCNR-infected chickens revealed that both copies of the parental meq gene had been restored.
DISCUSSION
MDV is a ubiquitous, highly contagious, potent oncogenic virus, which is controlled by vaccination to prevent economic losses otherwise caused by Marek's disease. Due to widespread use of vaccines, MDV continues to evolve toward greater virulence and, therefore, a better understanding of the molecular mechanisms of viral pathogenesis is warranted to control this economically important disease of poultry.
A number of genes unique to MDV are encoded within the repeat long region or at its junction with the unique long region and have been associated with pathogenesis. For example, disruption of pp38, vIL8, or LORF11 genes from oncogenic MDV resulted in reduced tumor incidence as a consequence of impaired lytic replication in chickens (6, 9, 19, 27). On the other hand, disruption of vTR (viral telomerase RNA) or meq gene did not appear to have an effect on lytic replication in vivo but resulted in 60 and 100% reductions in tumor incidence, respectively (25, 36).
MDV-1 (GaHV-2)-encoded Meq protein is consistently detected in MDV-induced tumors and is a potentially multifaceted transcription factor that has been shown to directly interact with cell cycle regulator, cyclin-dependent kinase 2 (CDK2), and the corepressor C-terminal binding protein (CtBP) (24). Interestingly, abrogation of the interaction of Meq with CtBP in an MDV-1 recombinant virus (pRB1B-Ct20) resulted in loss of transformation; however, the molecular mechanisms associated with this phenotype remain to be elucidated (1), although it can be speculated that Meq may interfere with the ability of CtBP to function as a corepressor, as has been described for adenovirus early region 1A (9a).
Meq shares homology with the Jun/Fos bZIP family of transcription factors, of which viral counterparts v-Jun and v-Fos are oncogenic, therefore implying a similar oncogenic potential (35). In vitro transformation studies and the loss of transformation observed in vivo with rMd5ΔMeq provides convincing proof that Meq is essential for transformation by MDV. However, the mechanisms of Meq transformation remain unknown. In vitro characterization of Meq DNA binding and transactivation or repression activities have provided support for the hypothesis that Meq homodimers and heterodimers have distinct roles in MDV-1 pathogenesis (13, 20, 21, 29); however, the role of Meq homodimers and heterodimers in MDV-1 pathogenesis has not been characterized.
In the present study, a recombinant virus in which the leucine zipper of the Meq protein was replaced with the leucine zipper of the yeast GCN4 protein was successfully generated. It has been well established that the leucine zipper region of bZIP proteins determines their dimerization specificity, which in turn affects their DNA binding properties (16, 38). The mutant Meq protein we generated containing the dimerization domain of GCN4 retained both DNA binding and transactivation/repression functions in vitro. However, rMd5-MeqGCN expressed MeqGCN to a lesser extent than parental rMd5 virus expressed Meq in infected DEF (Fig. 8). This correlates with in vitro luciferase data, which showed that parental Meq activates the meq promoter at significantly greater levels than MeqGCN (Fig. 5), supporting a previous report that Meq, together with c-Jun, has a greater binding affinity for AP-1 sequences and higher transactivation activity on the meq promoter than Meq alone (21). Since MeqGCN transactivates the Meq promoter to a lesser extent than parental Meq, it is possible that MeqGCN may also have decreased the transactivation of other AP-1-containing promoters. This decrease in transactivation potential could contribute to the decreased transformation observed by MeqGCN in the soft-agar assay (Fig. 6). However, transactivation activity does not always correlate with transformation, as has been observed in studies with v-Jun mutants that are strong transactivators and poor transformers and vice versa (10). We also found that MeqGCN homodimers had a reduced ability to transrepress the pp38/14 promoter, although MeqGCN repressed MDVOri sequences found within the pp38/14 promoter at levels comparable to parental Meq. It is possible that MeqGCN homodimers in the context of the full pp38/14 promoter are not able to bind the pp38/14 promoter, as well as parental Meq or bind other elements in the promoter with different affinities, therefore decreasing transrepression. It has been shown that v-Jun-GCN4 leucine zipper chimeric proteins are more stable than parental v-Jun homodimers, and it is therefore possible that the stability of MeqGCN homodimers be different than that of parental Meq homodimers.
The contribution of other MDV proteins or cellular factors to MDV transformation cannot be dismissed either. Disruption of Meq contact with CtBP resulted in a loss of oncogenicity showing the importance of Meq and CtPB interactions (1). In addition, Meq has been shown to associate with cell cycle regulators, CDK2, and p53 (17, 24), although the contributions of these interactions in MDV transformation have not been defined. It remains possible that MeqGCN homodimers have altered interactions with these or other unknown cellular factors. However, taken together, our data demonstrate that MeqGCN homodimers are not sufficient to induce transformation of T cells in chickens. Further investigations with Meq heterodimer mutants might aid in further defining the role of Meq in MDV transformation.
The role of Meq homodimers in MDV-1 pathogenesis was determined by evaluating both cytolytic infection and transformation in vivo. In vitro, rMd5-MeqGCN replicated similar to the parental virus and, in vivo, cytolytic infection was evident in lymphoid organs and feather follicles, as assessed by the expression of MDV-1 lytic protein pp38 (Fig. 10). However, evaluation of effects of MeqGCN on cytolytic infection is difficult because Meq expression is not essential for early cytolytic infection (25). Meq is not always detected early in infection, and expression of Meq in vivo is variable and has been shown to depend on the virus strain and chicken line used (8, 26). Since the FFE is the site of fully productive infection and source of horizontal transmission (4), we evaluated the role of Meq homodimers on transmission by IHC (Fig. 10) and contact transmission. Our results show that Meq homodimerization did not interfere with either replication in the FFE or transmission to contact chickens.
Atrophy of thymus and bursa of Fabricius and splenomegaly is a marker of MDV-1 lytic infection and is a good indicator of the virulence of MDV-1 pathotypes. Atrophy is evident as early as 8 days postinfection with no significant differences observed among MDV-1 pathotypes. However, at day 14 postinfection, although lymphoid organ atrophy is less than at day 8 postinfection, significant differences between MDV-1 pathotypes are evident. By day 14 postinfection, less atrophy is observed with less-virulent pathotypes, indicating the affected tissues are able to recover. On the other hand, a greater degree of atrophy at day 14 postinfection is observed with the more virulent pathotypes. However, it is still unknown whether more-virulent strains are able to infect a greater number of cell types or whether they maintain a prolonged cytolytic infection, therefore causing more damage to tissue than is caused by less-virulent strains (5). To assess the virulence of rMd5-MeqGCN, lymphoid organ/body weight ratios were determined for rMd5-MeqGCN-, rMd5-, and rMd5ΔMeq-infected chickens. As expected, lymphoid organ atrophy and splenomegaly were observed at day 14 postinfection in the rMd5-infected group, whereas rMd5-MeqGCN-infected chickens displayed less lymphoid organ atrophy and were more similar to uninfected controls (Fig. 11). Although no significant differences were observed at day 14 among all groups, the statistical power of the time point was limited due to the small number of chickens examined and may not accurately reflect the biological differences between these viruses. However, these results suggest that rMd5-MeqGCN is less virulent than parental rMd5.
One of the goals of the present study was to dissect the Meq functions in the hope of separating its transforming potentials from the in vivo replication/latency functions. A perhaps oversimplified model is that Meq/Jun heterodimers, like v-Jun, impart transforming function, whereas Meq/Meq homodimers are involved in latency entry and/or reactivation. To test this model, the replicative properties of rMd5-MeqGCN virus were assessed in vivo by evaluating early cytolytic infection and reactivation from latency. Although early cytolytic infection was not impaired (Fig. 10), the virus appeared to be defective in reactivation (Table 1). A switch from lytic to latent infection in MDV-1 normally occurs at approximately 7 days postinfection, and virus from latently infected cells can be reactivated when cocultured with fibroblasts in vitro (2). In our study, few viral plaques were detected at day 14, and none were detected at day 21 for both the rMd5-MeqGCN and the rMd5ΔMeq groups. However, although not quantitative, the presence of latently infected cells in the chickens was confirmed by the detection of viral antigen pp38 (Fig. 12) and PCR amplification of the MDV-1 genome (data not shown) in all infected groups. Since both rMd5-MeqGCN and rMd5ΔMeq viruses appear to be defective in reactivation, it may be argued that Meq heterodimers might play an important role in virus reactivation from latency.
Inoculation of chickens with rMd5-MeqGCN also showed that this recombinant virus is apathogenic. During the 8-week experiment, none of the chickens infected with rMd5-MeqGCN developed gross or microscopic tumors, whereas in the rMd5 group all of the chickens that died or were euthanized suffered from Marek's disease (Fig. 13). Like rMd5ΔMeq, rMd5-MeqGCN did not transform T cells, further supporting in vitro data and implicating Meq heterodimers in MDV-1 transformation. In order to rule out the possibility that tumor formation was inhibited by the production of antibodies directed toward the GCN leucine zipper, we tested convalescent chicken serum on cells expressing Meq and MeqGCN by IFA. None of the sera obtained from rMd5- or rMd5-MeqGCN-infected chickens reacted positively with cells expressing either Meq or MeqGCN (data not shown). These results are further supported by previous work with retrovirus constructs expressing v-Jun GCN4 leucine zipper mutants, which remained oncogenic in infected chickens (10, 14). This suggests that immune responses to MeqGCN did not contribute to the lack of oncogenicity observed in rMd5-MeqGCN-infected chickens. Importantly, the phenotypic differences observed in the present study for rMd5-MeqGCN can be attributed to the leucine zipper mutations, since the Md5 phenotype was fully restored in the revertant virus rMd5-MeqGCNR.
In summary, the present study provides the first in vivo evidence that Meq homodimers are not sufficient for MDV-1 transformation, reinforcing the notion that the participation of Meq/Jun may be crucial to its transforming ability. In order to better characterize the Meq transcriptional pathways involved in MDV-1 pathogenesis, studies with recombinant Meq heterodimer virus are under way to investigate the role of Meq-Jun heterodimers in MDV transformation in vivo.
Acknowledgments
This study was supported by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service, grants 2004-35204-14840 (to S.M.R. and B.L.) and 2006-01604 (to H.-J.K.). P.F.S. was supported by NIH predoctoral training grant T32-AI052072.
We thank Shailbala Singh, Vinayak Brahmakshatriya, Xiali Liu, and Kathy Rector for help with animal studies. We are grateful to Stephen Safe and Junn-lin Liu for providing valuable reagents.
Footnotes
Published ahead of print on 29 October 2008.
REFERENCES
- 1.Brown, A. C., S. J. Baigent, L. P. Smith, J. P. Chattoo, L. J. Petherbridge, P. Hawes, M. J. Allday, and V. Nair. 2006. Interaction of MEQ protein and C-terminal-binding protein is critical for induction of lymphomas by Marek's disease virus. Proc. Natl. Acad. Sci. USA 1031687-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calnek, B. W. 1986. Marek's disease: a model for herpesvirus oncology. Crit. Rev. Microbiol. 12293-320. [DOI] [PubMed] [Google Scholar]
- 3.Calnek, B. W. 2001. Pathogenesis of Marek's disease virus infection. Curr. Top. Microbiol. Immunol. 25525-55. [DOI] [PubMed] [Google Scholar]
- 4.Calnek, B. W., H. K. Adldinger, and D. E. Kahn. 1970. Feather follicle epithelium: a source of enveloped and infectious cell-free herpesvirus from Marek's disease. Avian Dis. 14219-233. [PubMed] [Google Scholar]
- 5.Calnek, B. W., R. W. Harris, C. Buscaglia, K. A. Schat, and B. Lucio. 1998. Relationship between the immunosuppressive potential and the pathotype of Marek's disease virus isolates. Avian Dis. 42124-132. [PubMed] [Google Scholar]
- 6.Cui, X., L. F. Lee, H. D. Hunt, W. M. Reed, B. Lupiani, and S. M. Reddy. 2005. A Marek's disease virus vIL-8 deletion mutant has attenuated virulence and confers protection against challenge with a very virulent plus strain. Avian Dis. 49199-206. [DOI] [PubMed] [Google Scholar]
- 7.Ferrin, L. J., and R. D. Camerini-Otero. 1991. Selective cleavage of human DNA: RecA-assisted restriction endonuclease (RARE) cleavage. Science 2541494-1497. [DOI] [PubMed] [Google Scholar]
- 8.Gimeno, I. M., R. L. Witter, A. M. Fadly, and R. F. Silva. 2005. Novel criteria for the diagnosis of Marek's disease virus-induced lymphomas. Avian Pathol. 34332-340. [DOI] [PubMed] [Google Scholar]
- 9.Gimeno, I. M., R. L. Witter, H. D. Hunt, S. M. Reddy, L. F. Lee, and R. F. Silva. 2005. The pp38 gene of Marek's disease virus (MDV) is necessary for cytolytic infection of B cells and maintenance of the transformed state but not for cytolytic infection of the feather follicle epithelium and horizontal spread of MDV. J. Virol. 794545-4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Grand, R.J.A., C. Baker, P. M. Barral, R. K. Bruton, J. Parkhill, T. Szestak, and P.H. Gallimore. 2007. The significance of the CtBP-Ade1A interaction during viral infection and transformation, p. 44-60. In G. Chinnadurai (ed.), CtBP family proteins. Landes Bioscience, New York, NY.
- 10.Hartl, M., and P. K. Vogt. 1992. Oncogenic transformation by Jun: role of transactivation and homodimerization. Cell Growth Differ. 3899-908. [PubMed] [Google Scholar]
- 11.Hughes, M., A. Sehgal, M. Hadman, and T. Bos. 1992. Heterodimerization with c-Fos is not required for cell transformation of chicken embryo fibroblasts by Jun. Cell Growth Differ. 3889-897. [PubMed] [Google Scholar]
- 12.Izumiya, Y., S. F. Lin, T. J. Ellison, A. M. Levy, G. L. Mayeur, C. Izumiya, and H. J. Kung. 2003. Cell cycle regulation by Kaposi's sarcoma-associated herpesvirus K-bZIP: direct interaction with cyclin-CDK2 and induction of G1 growth arrest. J. Virol. 779652-9661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones, D., L. Lee, J. L. Liu, H. J. Kung, and J. K. Tillotson. 1992. Marek disease virus encodes a basic-leucine zipper gene resembling the fos/jun oncogenes that is highly expressed in lymphoblastoid tumors. Proc. Natl. Acad. Sci. USA 894042-4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jurdic, P., I. Treilleux, L. Vandel, E. Tabone, S. Huguier, A. Sergeant, and M. Castellazzi. 1995. Tumor induction by v-Jun homodimers in chickens. Oncogene 111699-1709. [PubMed] [Google Scholar]
- 15.Kim, H., S. You, I. J. Kim, J. Farris, L. K. Foster, and D. N. Foster. 2001. Increased mitochondrial-encoded gene transcription in immortal DF-1 cells. Exp. Cell Res. 265339-347. [DOI] [PubMed] [Google Scholar]
- 16.Kouzarides, T., and E. Ziff. 1989. Leucine zippers of fos, jun and GCN4 dictate dimerization specificity and thereby control DNA binding. Nature 340568-571. [DOI] [PubMed] [Google Scholar]
- 17.Kung, H. J., L. Xia, P. Brunovskis, D. Li, J. L. Liu, and L. F. Lee. 2001. Meq: an MDV-specific bZIP transactivator with transforming properties. Curr. Top. Microbiol. Immunol. 255245-260. [DOI] [PubMed] [Google Scholar]
- 18.Lee, L. F., B. Lupiani, R. F. Silva, H. J. Kung, and S. M. Reddy. 2008. Recombinant Marek's disease virus (MDV) lacking the Meq oncogene confers protection against challenge with a very virulent plus strain of MDV. Vaccine 261887-1892. [DOI] [PubMed] [Google Scholar]
- 19.Lee, L. F., R. F. Silva, X. Cui, H. Zhang, M. Heidari, and S. M. Reddy. 2007. Characterization of LORF11, a unique gene common to the three Marek's disease virus serotypes. Avian Dis. 51851-857. [DOI] [PubMed] [Google Scholar]
- 20.Levy, A. M., O. Gilad, L. Xia, Y. Izumiya, J. Choi, A. Tsalenko, Z. Yakhini, R. Witter, L. Lee, C. J. Cardona, and H. J. Kung. 2005. Marek's disease virus Meq transforms chicken cells via the v-Jun transcriptional cascade: a converging transforming pathway for avian oncoviruses. Proc. Natl. Acad. Sci. USA 10214831-14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy, A. M., Y. Izumiya, P. Brunovskis, L. Xia, M. S. Parcells, S. M. Reddy, L. Lee, H. W. Chen, and H. J. Kung. 2003. Characterization of the chromosomal binding sites and dimerization partners of the viral oncoprotein Meq in Marek's disease virus-transformed T cells. J. Virol. 7712841-12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu, J. L., L. F. Lee, Y. Ye, Z. Qian, and H. J. Kung. 1997. Nucleolar and nuclear localization properties of a herpesvirus bZIP oncoprotein, MEQ. J. Virol. 713188-3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu, J. L., Y. Ye, L. F. Lee, and H. J. Kung. 1998. Transforming potential of the herpesvirus oncoprotein MEQ: morphological transformation, serum-independent growth, and inhibition of apoptosis. J. Virol. 72388-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu, J. L., Y. Ye, Z. Qian, Y. Qian, D. J. Templeton, L. F. Lee, and H. J. Kung. 1999. Functional interactions between herpesvirus oncoprotein MEQ and cell cycle regulator CDK2. J. Virol. 734208-4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lupiani, B., L. F. Lee, X. Cui, I. Gimeno, A. Anderson, R. W. Morgan, R. F. Silva, R. L. Witter, H. J. Kung, and S. M. Reddy. 2004. Marek's disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc. Natl. Acad. Sci. USA 10111815-11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parcells, M. S., V. Arumugaswami, J. T. Prigge, K. Pandya, and R. L. Dienglewicz. 2003. Marek's disease virus reactivation from latency: changes in gene expression at the origin of replication. Poult. Sci. 82893-898. [DOI] [PubMed] [Google Scholar]
- 27.Parcells, M. S., S. F. Lin, R. L. Dienglewicz, V. Majerciak, D. R. Robinson, H. C. Chen, Z. Wu, G. R. Dubyak, P. Brunovskis, H. D. Hunt, L. F. Lee, and H. J. Kung. 2001. Marek's disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J. Virol. 755159-5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng, F., G. Bradley, A. Tanaka, G. Lancz, and M. Nonoyama. 1992. Isolation and characterization of cDNAs from BamHI-H gene family RNAs associated with the tumorigenicity of Marek's disease virus. J. Virol. 667389-7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian, Z., P. Brunovskis, L. Lee, P. K. Vogt, and H. J. Kung. 1996. Novel DNA binding specificities of a putative herpesvirus bZIP oncoprotein. J. Virol. 707161-7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy, S. M., B. Lupiani, I. M. Gimeno, R. F. Silva, L. F. Lee, and R. L. Witter. 2002. Rescue of a pathogenic Marek's disease virus with overlapping cosmid DNAs: use of a pp38 mutant to validate the technology for the study of gene function. Proc. Natl. Acad. Sci. USA 997054-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reference deleted.
- 32.Ross, N., G. O'Sullivan, C. Rothwell, G. Smith, S. C. Burgess, M. Rennie, L. F. Lee, and T. F. Davison. 1997. Marek's disease virus EcoRI-Q gene (meq) and a small RNA antisense to ICP4 are abundantly expressed in CD4+ cells and cells carrying a novel lymphoid marker, AV37, in Marek's disease lymphomas. J. Gen. Virol. 78(Pt. 9)2191-2198. [DOI] [PubMed] [Google Scholar]
- 33.Ross, N. L. 1999. T-cell transformation by Marek's disease virus. Trends Microbiol. 722-29. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook, J., et al. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 35.Shaulian, E., and M. Karin. 2001. AP-1 in cell proliferation and survival. Oncogene 202390-2400. [DOI] [PubMed] [Google Scholar]
- 36.Trapp, S., M. S. Parcells, J. P. Kamil, D. Schumacher, B. K. Tischer, P. M. Kumar, V. K. Nair, and N. Osterrieder. 2006. A virus-encoded telomerase RNA promotes malignant T-cell lymphomagenesis. J. Exp. Med. 2031307-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tulman, E. R., C. L. Afonso, Z. Lu, L. Zsak, D. L. Rock, and G. F. Kutish. 2000. The genome of a very virulent Marek's disease virus. J. Virol. 747980-7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogt, P. K. 2001. Jun, the oncoprotein. Oncogene 202365-2377. [DOI] [PubMed] [Google Scholar]
- 39.Witter, R. L. 1997. Increased virulence of Marek's disease virus field isolates. Avian Dis. 41149-163. [PubMed] [Google Scholar]