

Abstract
We have described a novel essential replicative DNA helicase from Bacillus anthracis, the identification of its gene, and the elucidation of its enzymatic characteristics. Anthrax DnaB helicase (DnaBBA) is a 453-amino-acid, 50-kDa polypeptide with ATPase and DNA helicase activities. DnaBBA displayed distinct enzymatic and kinetic properties. DnaBBA has low single-stranded DNA (ssDNA)-dependent ATPase activity but possesses a strong 5′→3′ DNA helicase activity. The stimulation of ATPase activity appeared to be a function of the length of the ssDNA template rather than of ssDNA binding alone. The highest specific activity was observed with M13mp19 ssDNA. The results presented here indicated that the ATPase activity of DnaBBA was coupled to its migration on an ssDNA template rather than to DNA binding alone. It did not require nucleotide to bind ssDNA. DnaBBA demonstrated a strong DNA helicase activity that required ATP or dATP. Therefore, DnaBBA has an attenuated ATPase activity and a highly active DNA helicase activity. Based on the ratio of DNA helicase and ATPase activities, DnaBBA is highly efficient in DNA unwinding and its coupling to ATP consumption.
Bacillus anthracis is a gram-positive bacterium and the etiological agent of the disease anthrax (29, 30). B. anthracis and other gram-positive bacteria pose a serious threats to human health. Thus, considerable efforts have been placed on understanding the physiology and pathology of these microorganisms. Currently, the molecular and cellular biology of B. anthracis is poorly understood. A detailed understanding of proteins involved in fundamental cellular processes such as DNA replication is critical to combating diseases caused by this organism.
Chromosomal DNA replication requires the concerted actions of many different proteins in stable and transient complexes (35). Extensive studies of the process chromosomal DNA replication in Escherichia coli, as well as its plasmids and phages, have led to it serving as a model system for the study of DNA replication in both prokaryotes and eukaryotes (1, 2, 7, 15, 18, 19, 23-25, 35, 40, 41, 44, 45). DnaB protein appears to be involved in all stages of DNA replication from initiation to termination (7, 21, 23). The DnaB helicase is a multifunctional enzyme that is involved in the formation and translocation of the replication machinery in E. coli and λ bacteriophage and thus plays a pivotal role (4, 6, 9, 11, 15, 22, 35, 38, 40, 42). Due to its ability to physically interact with a variety of other replication proteins, the DnaB protein plays a key role in the assembly of the primosome and subsequent movement of the replication apparatus (2, 3, 5, 10, 14, 19, 20, 22, 26, 37, 37, 40, 43). DnaB unwinds the DNA duplex into two single parental DNA template strands, which are protected by single-stranded DNA (ssDNA) binding protein (SSB). The process proceeds unidirectionally (5′→3′) in a forklike manner (12, 38). Replication of the “leading strand” by DNA polymerase III holoenzyme, consisting of DNA polymerase III core and associated proteins, is continuous. However, DNA synthesis on the “lagging” strand is necessarily discontinuous. DnaG primase, acting in concert with DnaB helicase, initiates the template-dependent synthesis of short RNA primers, which are extended by the DNA polymerase III holoenzyme.
The DnaB helicase of E. coli (DnaBEC) has three distinct functional domains, which are as follows: N-terminal domain α, amino acid residues 1 to 156; domain β, amino acid residues 157 to 302; and C-terminal domain γ, amino acid residues 303 to 471. These domains appear to be present in all bacterial replicative DNA helicases (10, 16). Previous studies involving functional analysis of DnaBEC indicate that domain β contains both the ATP binding and the ATPase active sites, and domain γ likely includes the ssDNA binding site and one of the two sites for hexamer formation. Recombinant purified domain β polypeptide hydrolyzes ATP, albeit at a slower rate. Partial proteolysis of DnaBEC with trypsin allows removal of the α domain from DnaBEC and formation of βγ hexamer. The βγ polypeptide retains the hexameric property, as well as ssDNA-dependent ATPase activity. However, it lacks DNA helicase activity completely. Thus, domain α is not required for ATPase activity, or ssDNA binding, but it is essential for the DNA helicase activity of DnaBEC. Sequence analysis indicates that domain α does not contain any known enzymatic motif and is unlikely to have an enzymatic function. Among these three domains, domain α is the least-conserved domain of DnaBEC. Therefore, the role(s) of the α domain in DNA unwinding remains unclear at the present time.
The replicative DNA helicase and primase interact as a transient complex in DNA replication (40). Chemical cross-linking has confirmed the existence of the complex and established a 6:3 helicase/primase ratio for the E. coli enzymes (43). The interaction plays a crucial role in DNA replication because it serves to stimulate and regulate the relevant activities of the two enzymes (8, 31). For example, the primase-helicase interaction recruits primase to the replication fork, significantly enhances primase and helicase activities, and regulates the length and sequence specificity of primer synthesis and/or initiation. The susceptibility of primase action to dilution suggests that primase enters and exits the replication fork interacting with the helicase transiently. This finding is consistent with the necessity for primase to synthesize RNA primers distal to helicase action and then reinitiate primer synthesis on newly generated single strands (50, 51).
Although significant progress has been made in identifying the major components of the DNA replication machinery in E. coli, studies on select agents, such as B. anthracis, remain nonexistent. Many pathogenic bacteria have physiology that is quite different from that observed with E. coli. Thus, it is likely that the mechanisms of DNA replication in these organisms are different. Development of more targeted drugs or antibiotics against these organisms has been hampered due to the lack of knowledge of chromosomal DNA replication and the overall molecular biology of these bacterial pathogens. A major area of progress has been in the area of genome sequencing. The genomes of a large number of these pathogenic organisms have been completely sequenced. Even though the growth of these bacteria in large scale and purification of their replication proteins remain daunting, PCR-based cloning and expression of individual replication proteins for critical biochemical analysis has become feasible.
We describe here the identification of the gene and characterization of the B. anthracis homolog of the replicative DNA helicase, DnaB protein. We have also carried out a comprehensive analysis of its enzymatic characteristics, including ATPase, DNA helicase, and the mechanism of ssDNA binding (35).
MATERIALS AND METHODS
Nucleic acids and other reagents.
All chemicals used in the present study were reagent or spectroscopy grade and obtained from Sigma-Aldrich Chemical Co. (Milwaukee, WI). High-pressure liquid chromatography-grade water was obtained from Fisher Chemicals (Pittsburgh, PA). Oligonucleotides and nucleotides were purchased from Sigma-Genosys and Fisher Chemicals, Inc. (Pittsburgh, PA), respectively.
Buffers.
Lysis buffer was composed of 25 mM Tris-HCl (pH 7.9), 10% sucrose, and 250 mM NaCl. Buffer A was composed of 25 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 10% glycerol, 5 mM dithiothreitol, and NaCl as indicated. Buffer B, used for anisotropy studies, contained 20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, and 10% glycerol, and KCl as indicated.
In vivo deletion analysis of putative helicase genes BAS0880 and BAS5321.
Approximately 1 kb of sequence flanking each locus to be deleted was amplified by PCR and cloned on either side of the omega kanamycin (Omega-Km) resistance element in pMR1 (27). The upstream flanking region was cloned between the SacI and SmaI sites in pMR1, and the downstream region was cloned between the SalI and StuI sites in pMR1. By using primers carrying SacI and SalI restriction sites as tails, each flanking region contained one cohesive end and one blunt end to provide directionality in the ligations. Finally, the cloned upstream Omega-Km downstream constructions were amplified with the SacI and SalI tailed primers and ligated into SacI- and SalI-digested pKS1, which provides a temperature-sensitive Lactococcus lactis subsp. cremoris pWV01 replicon (49). The pKS1-ΔBAS-Kmr clones were verified by PCR to consist of the upstream and downstream loci flanking the kanamycin resistance element in place of the gene to be deleted. The constructs were introduced into E. coli GM2163 by electroporation. Plasmid DNA was prepared and used to electroporate B. anthracis Sterne cells to kanamycin resistance (LB plus 100 μg of kanamycin/ml). Transformants were also confirmed to be resistant to the vector marker (10 μg of tetracycline/ml for pMR1 or 3 μg of erythromycin/ml for pKS1). Cells were grown for over 20 generations in the absence of drug selection to allow for the two recombination events, resulting in replacement of each gene with the kanamycin resistance marker. The occurrence of this event was monitored by detecting the loss of the vector marker resistance but retention of kanamycin resistance and was confirmed by PCR amplification with primers annealing to the kanamycin resistance element and to flanking genomic regions (i.e., outside the region manipulated) (Tables 1 and 2).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotypea | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| GM2163 | F−dam13::Tn9 dcm-6 hsdR2 ara-14 leuB6 fhuA31 lacY1 tsx78 glnV44 galK2 galT22 mcrA hisG4 rfbD1 rpsL146 xylA5 mtl-1 thi-1 mcrB1 | E. coli Genetic Stock Center (CGSC 6581) |
| BL21(DE3)RIL | E. coli B F−ompT hsdS(rB− mB−) dcm+ Tetrgal λ(DE3) endA Hte [argU ileY leuW Camr] | Stratagene |
| B. anthracis | ||
| Sterne 34F2 | pXO2− | Colorado Serum Co. |
| MDM808 | Sterne 34F2 ΔBAS0880 Kmr, isolate 1 | This study |
| MDM809 | Sterne 34F2 ΔBAS0880 Kmr; isolate 2 | This study |
| MDM801 | Sterne 34F2 BAS0880::pKS1-ΔBAS0880-Kmr Emr | This study |
| Plasmids | ||
| pMR1 | Modified pUTE29 (48) to contain an Ω-Kmr element; Apr in E. coli, Tcr and Kmr in B. anthracis, with extensive restriction enzyme sites flanking the Ω-Kmr gene | 27 |
| pKS1 | Vector with temperature-sensitive replicon from L. cremoris pWV01 replicon for allele replacement in B. anthracis; Kmr and Emr in E. coli and B. anthracis | 49 |
| pET30b | T7 RNA polymerase-based expression vector, Kmr | Novagen |
Apr, Tcr, Kmr, and Emr: ampicillin, tetracycline, kanamycin, and erythromycin resistance, respectively.
TABLE 2.
Oligonucleotides and primers used in this study
| Primer | Sequence |
|---|---|
| BAS5321-Up-F+Sac | ACTGACTTGAGCTCTCGTACCCCTCCGCATAATA |
| BAS5321-Up-R | AAAATGCCGTTTTCCCTACC |
| BAS5321-Dwn-F | CGGGATACCAACTGGGTTTA |
| BAS5321-Dwn-R+SalI | ACGCTTGTCGACCATCTTCGAAGCGACGTTCT |
| BAS5321-out-F | CGTTAAGCAAGGAGGATTGC |
| BAS5321-out-R | AAGTCAACCAAAAAGCGAACA |
| BAS0880-Up-F+Sac | ACGCTTGAGCTCGGAGACGGGAGGAATTAGGA |
| BAS0880-Up-R | ACCGGCATCGAAAAATACTG |
| BAS0880-Dwn-F | AGATTCATGTGGCGAAGCAT |
| BAS0880-Dwn-R+Sal | ACGCTTGTCGACCCGCCAACTTTTGCTTTACT |
| BAS0880-out-F | CCCTCCTAAGCCCCTTACAA |
| BAS0880-out-R | CGGAAAATCCCCATTAATCC |
| Kan-F | GTTTCAAAATCGGCTCCGTCGATACTATGT |
| Kan-R | GTAGGCGCTCGGGACCCCTATCTAGCGAAC |
| HCASE45-5′ | ATCTCCATGGTCACGACGTTGTAAAACGACGGCCAGTGAATTCGA |
| HCASE45-3′ | TCACGACGTTGTAAAACGACGGCCAGTGAATTCGAATCTCCATGG |
| 60-mer | GGGGGTCTCACGACGTTGTAAAACGACGGCCAGTGAATTCGAGCTCGGTACCCGGGGTAGGA |
B. anthracis DnaB helicase expression and purification.
The DnaBBA gene was amplified by PCR using genomic DNA from B. anthracis strain 9131, obtained as a gift from Theresa M Koehler of the University of Texas Houston Health Science Center, Houston (33, 34). The amplified gene was inserted in a pET30 vector (Novagen, Inc., Madison, WI) under the control of a T7 promoter (pET30-DnaBBA recombinant plasmid) and confirmed by DNA sequencing. DnaBBA was overexpressed in E. coli strain BL21(DE3)RIL (Stratagene, Inc.) harboring pET30-DnaBBA plasmid. Cells harboring the recombinant plasmid were grown in 2×YT media containing 50 μg of kanamycin/ml, 20 μg of tetracycline/ml, and 12 μg of chloramphenicol/ml with shaking at 37°C to an optical density at 600 nm of 0.4. IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a final concentration of 0.25 mM. The cells were shaken for an additional 12 h at 12°C and then harvested by centrifugation for 10 min at 5,000 × g. The cells were resuspended in 2.5% of the original culture volume of lysis buffer at 4°C and stored at −80°C until further use.
Extraction of the induced cells was as previously described for DnaBEC (13). DnaBBA protein was precipitated from the cell extract by the addition of 0.25 g of ammonium sulfate/ml, followed by chilling on ice overnight. The precipitate was collected by centrifugation. The precipitate was resuspended in buffer A containing 0.2 g of ammonium sulfate/ml. The suspension was stirred for 60 min at 0°C, followed by centrifugation. The protein pellet was resuspended in buffer A (fraction II).
DnaBBA protein was first fractionated by Q-Sepharose chromatography (GE Health Sciences, Piscataway, NJ). The salt concentration of DnaBBA fraction II was adjusted to the conductivity of buffer A100 (buffer A containing 100 mM NaCl) by dilution with buffer A0. The protein was loaded onto a 25-ml Q-Sepharose column equilibrated with buffer A100. At 100 mM NaCl, DnaBBA protein passes through the Q-Sepharose column without binding and is found in the flowthrough fractions. The flowthrough fractions (fraction III) were pooled and loaded onto to a 6-ml S-Sepharose column equilibrated with buffer A100. DnaBBA protein was eluted with a 10-column volume gradient of buffers A100 and A500. The peak fractions were identified by ssDNA-dependent ATPase and DNA helicase activities in conjunction with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The active fractions were pooled and concentrated by ultrafiltration using a YM-30 membrane (fraction IV). The purified DnaBBA protein was essentially homogeneous and >98% pure, as analyzed by SDS-PAGE. The protein concentration was determined by the method of Bradford (17).
ATPase assay.
ATPase assays were carried out based on previously described methods (16). The amount of enzyme used in the assays was selected such that the rate of hydrolysis would be linear in the time range examined. A standard 10-μl reaction mixture contained 10 mM MgCl2, 200 pmol of M13mp19 ssDNA, 500 μM [α-32P]ATP (1-2 × 103 cpm/pmol), and DnaBBA protein. Reactions were incubated at 37°C for 30 min and terminated by addition of 2 μl of 200 mM EDTA, followed by chilling on ice. Aliquots (2 μl) were applied to polyethyleneimine cellulose strips, prespotted with ADP-ATP marker. The strips were developed with 1 M formic acid-0.5 M LiCl and dried. The ADP-ATP spots were located by using 254-nm UV fluorescence. The portions containing ATP and ADP were excised and counted in a liquid scintillation counter using a toluene-based scintillator.
DNA helicase assay.
The helicase assays were based on the methods previously described by Biswas and Biswas (10). Unless otherwise indicated, DNA helicase activity was determined utilizing a M13mp19 partial duplex substrate hybridized to a radiolabeled 60-mer oligonucleotide (Table 2), possessing five-nucleotide forks at both the 5′ and the 3′ ends. A standard 20-μl reaction volume contained 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 10% glycerol, 5 mM dithiothreitol, 0.1 mM ATP, 17 fmol (1 × 104 to 2 × 104 cpm/μl) of substrate, and the indicated amount of DnaBBA protein. The mixtures were incubated at 30°C for 15 min, and the reactions were terminated by the addition of 4 μl of 2.5% SDS, 60 mM EDTA, and 1% bromophenol blue. Displacement was monitored by PAGE, followed by autoradiography.
Equilibrium ssDNA binding analysis.
Fluorescence experiments were performed on a steady-state photon-counting spectrofluorometer (PC1; ISS Instruments, Champaign, IL) equipped with a Hamamatsu R928P photomultiplier tube, and the measurements were made in L-format. Excitation and emission slits were adjusted at 8 and 4 nm, respectively (32).
5′-Fluorescein-labeled oligo(dT)25 [Fl-(dT)25] was used as a fluorescence anisotropy probe. The oligonucleotide was diluted in buffer B to a concentration of 3 nM and titrated with DnaBBA in the concentration range of 0.1 nM to 1 μM. The samples were excited at 488 nm, and the fluorescence anisotropy was measured at 540 nm (36), at which minimal variation in the total fluorescence intensity was observed. Fluorescence intensities were measured three times for 10 s each time and averaged. Anisotropy values were expressed as millianisotropy or mA, which is equal to the anisotropy divided by 1,000. The standard deviation for the anisotropy values was <2 mA. The total fluorescence intensity did not change significantly with increases in the protein concentration. Therefore, fluorescence lifetime changes or the scattered excitation light did not affect anisotropy measurements.
Analysis of DNA binding by fluorescence anisotropy.
The interaction of DnaBBA with labeled oligonucleotide can be represented as follows:
 |
(1) |
where R is the ligand, i.e., labeled oligonucleotide, and P is the protein or DnaBBA. At equilibrium, Ka, the equilibrium association constant can be given as:
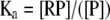 |
(2) |
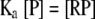 |
(3) |
The fraction of the binding sites occupied can be represented as:
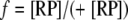 |
(4) |
Substituting for [RP] and rearranging the equation we get:
 |
(5) |
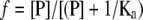 |
(6) |
Similarly, the equilibrium dissociation constant Kd (Kd = 1/Ka) can be expressed as:
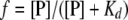 |
(7) |
At half-maximal binding, f = 0.5, and:
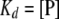 |
(8) |
Thus, Kd can be further defined as the DnaBBA concentration at which half of the sites are occupied. Nonlinear regression analysis of the anisotropy data was carried out using Prism 3.01 software (GraphPad Software, Inc., San Diego, CA). The Kd values, i.e., the concentrations of DnaBBA required to bind 50% of the oligonucleotides, were computed using the following equation:
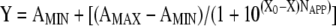 |
where AMIN and AMAX are the anisotropy values at the bottom and top plateaus, respectively; X represents the log of the DnaBBA concentration; X0 is the X value when the response is halfway between the top and the bottom; and NAPP is the Hill coefficient.
RESULTS
DnaB Helicase from B. anthracis.
The B. anthracis genome has been completely sequenced. An analysis of the genome indicates two genes that are homologous to the DnaB gene of E. coli: BAS0880 and BAS5321. Both of these genes are listed as “replicative DNA helicase” in GenBank. Therefore, we have analyzed these genes by in vivo gene disruption (Fig. 1).
FIG. 1.

Homology Alignment of DnaB sequences from B. anthracis (DnaBBA), E. coli (DnaBEC), and M. tuberculosis (DnaBTB). Alignment was carried out by CLUSTALW2 online using InterProScan. Amino acid residues displaying 100% identity are highlighted in red, and those displaying similarity are highlighted in blue.
Allele replacement mutagenesis of the B. anthracis replicative DNA helicase genes.
In order to establish whether either or both gene products (BAS0880 and BAS5321) play essential roles in replication, we used allele exchange methodology in an attempt to delete each gene (37, 38, 43). Deletion constructs for each locus were built in pMR1 (37) and pKS1 (38) and introduced into B. anthracis (Sterne strain) cells (see Materials and Methods). The resulting transformants were grown for over 20 generations and examined for loss of the vector marker but retention of the allele replacement marker. Deletions were only obtained for cells carrying the BAS0880 constructs. PCR amplification from primers outside the region cloned in the deletion construct and from the allele-replacement marker (Kmr) confirmed that BAS0880 had been replaced with the kanamycin resistance element (Fig. 1). These results establish that the putative replicative helicase locus, BAS0880, is not essential for growth and thus is unlikely to function as the replicative DNA helicase. Although we have not tested whether dnaB (BAS5321) deletions could be generated in the presence of a complementing copy of the gene on a plasmid, the inability to obtain uncomplemented deletions suggests that the dnaB locus is essential for growth or the viability of B. anthracis Sterne cells.
Homology with heterologous DnaB helicases.
The BAS5321 open reading frame (ORF) is 1,359 bp and codes for the DnaBBA polypeptide of 453 amino acids. The polypeptide sequence of DnaBBA revealed several important structural motifs: (i) a Walker type I nucleotide-binding motif and (ii) a DNA-binding motif (RAKCRR). The amino acid sequence of DnaBBA was compared to DnaB proteins of E. coli and Mycobacterium tuberculosis (DnaBTB). The sequence alignment is presented in Fig. 2. DnaBBA appears to have extensive sequence homology with these two DnaB proteins, DnaBEC and DnaBTB. In addition, the sequence alignment demonstrated that the DnaBBA lacked 17 N-terminal amino acid residues that are present in E. coli. Even though, the amino acid sequence of DnaBBA exhibits strong homology with DnaBEC and DnaBTB, it has a lower degree of homology in the N terminus and significantly higher degree of homology at the C terminus amino acid residues. As mentioned earlier, the N-terminal domain α does not have any enzymatic activity, despite its absolute requirement for DNA helicase activity. Among the three domains of DnaB helicase, domain α is the least conserved (∼19%), and domain γ is the most conserved (>60%) compared to other prokaryotic DnaB helicase, as is the case with DnaBBA. Computation was carried out based on similarity and identity. The alignment of multiple DnaB helicase sequences also indicated that the first 20 amino acid residues are probably not essential for DNA helicase activity (data not shown).
FIG. 2.

Allele replacement mutagenesis of the putative replicative DNA helicase gene BAS0880 in B. anthracis. PCR confirmation of B. anthracis Sterne clones with insertion of pKS1-ΔBAS0880-Kmr or deletion of BAS0880 was performed. (A) Analysis of the drug resistance marker phenotype of two colonies of each of strains MDM801 (row 1), MDM808 (row 2), and MDM809 (row 3), which were patched on LB agar medium containing 50 μg of kanamycin/ml (LB+Km; left side) or kanamycin plus 3 μg of erythromycin/ml (LB+Km+Em; right side). (B) Analysis of one colony of each of the three strains by PCR with primer pairs BAS0880-out-F + Kan-R (lanes a) and Kan-F + BAS0880-out-R (lanes b). The PCR products and drug resistance phenotypes are consistent with clone 1 (MDM801) representing a single crossover insertion of pKS1-ΔBAS0880-Kmr in the downstream region and with clones 2 and 3 (strains MDM808 and MDM809) carrying deletions of BAS0880 resulting from a second crossover to eliminate the pKS1 vector. (C) Schematic display of expected PCR products from cells with BAS0880 deleted. Up and Down indicate the flanking regions of gene BAS0880 amplified by PCR and cloned in pKS1; Omega-Kmr indicates the kanamycin resistance element cloned in place of gene BAS0880.
Expression and purification of DnaBBA.
We have cloned DnaBBA (BAS5321 ORF) and sequenced and expressed the ORF in E. coli by using a T7 expression system. SDS-PAGE analysis demonstrated that the recombinant protein migrated with a mass of ∼50 kDa as expected of a 453-amino-acid polypeptide. We have developed a purification protocol for recombinant DnaBBA. The DnaBBA was purified extensively to homogeneity (Fig. 3) using ammonium sulfate fractionation, followed by purification on Q-Sepharose and S-Sepharose ion-exchange chromatography steps. DnaBBA did not bind to Q-Sepharose at low ionic strength. This step helped remove contaminating endogenous DnaBEC, which bound to Q-Sepharose (6, 10). S-Sepharose chromatography removed other contaminating proteins. S-Sepharose fractions were assayed for DNA helicase and ATPase activities. The active fractions were pooled and concentrated.
FIG. 3.

SDS-PAGE analysis of purified DnaBBA. purified (fraction IV) DnaBBA was analyzed by a 5→18% gradient polyacrylamide gel followed by Coomassie blue staining with bovine serum albumin (68 kDa) and DnaBEC (52 kDa) as protein standards.
ssDNA-dependent ATPase activity of DnaBBA.
DnaBBA demonstrated a modest ssDNA-independent ATPase activity. Analysis of ATPase and dATPase activities showed that DnaBBA was able to hydrolyze either nucleotide with comparable efficiency (data not shown). The ATPase activity was stimulated by ssDNA cofactors, a property common to all DnaB helicases. We have analyzed synthetic oligonucleotides ranging in size from 15 to 60 nucleotides and M13mp19 bacteriophage ssDNA as cofactors (Fig. 4). We did not observe any significant stimulation below 15 nucleotides (data not shown). ATPase activity was slightly stimulated by ssDNA, and only M13mp19 ssDNA stimulated it significantly (Fig. 4B). The KdssDNA for oligonucleotide binding, determined by analysis of the ATPase plots in Fig. 4, indicated that the ssDNA binding did not appear to depend on the size of the DNA cofactor (Fig. 4B). Detailed kinetic analyses were carried out with the 60-mer oligonucleotide and M13mp19 ssDNA in order to determine the kinetic differences in rate and affinity (Fig. 5). The Km for ATP (KmATP) with the 60-mer oligonucleotide as DNA cofactor was 152 μM (Fig. 5A). The KmATP for DnaBBA with M13mp19 as DNA cofactor was 220 μM and was comparable to that observed with 60-mer (Fig. 5B). Therefore, ATP binding affinity did not change with ssDNA cofactors. However, the Vmax of DnaBBA with 60-mer was 7.2 × 105 pmol/min/mg, which increased to 2.7 × 106 pmol/min/mg with M13mp19 as the DNA cofactor. Thus, Vmax was enhanced by M13mp19 ssDNA ∼3-fold over that observed with the 60-mer cofactor and 6-fold over that observed with the 15-mer oligo(dT)15. Therefore, the Vmax increased proportionally with the size of the ssDNA cofactor. It likely indicated that with increasing length of the ssDNA, DnaBBA had more ssDNA to migrate or translocate, which could be responsible for the enhancement of the ATPase activity observed here.
FIG. 4.

ssDNA-dependent ATPase activity of DnaBBA. The ATPase activity of DnaBBA was measured in the presence of ssDNA cofactors. Reactions were carried out in a standard ATPase assay (see Materials and Methods) with 25 ng of DnaBBA and the indicated amounts of ssDNA and/or oligonucleotides. (A) Plots of ATPase activities with ssDNA cofactors. The data were analyzed by nonlinear regression analysis using Prism 6.0 (GraphPad Software). (B) Vmax and Kd values with each oligonucleotide or ssDNA as determined from the ATPase plots.
FIG. 5.

Kinetic analysis of ATPase activities of DnaBBA with ssDNA cofactors. ATPase activity of DnaBBA was analyzed by using the oligonucleotides 60-mer (A) and M13mp19 ssDNA (B). Reactions were carried out in a standard ATPase assay (see Materials and Methods) with 25 ng of DnaBBA and 200 ng of ssDNA and/or oligonucleotides and 25 to 500 μM [α-32P]ATP. The data were analyzed by nonlinear regression analysis using Prism 6.0. Insets represent Lineweaver-Burk (1/V versus 1/[S]) plots with linear regression of the corresponding ATPase data.
DNA helicase activity of DnaBBA.
We have examined the DNA helicase activities of DnaBBA. A protein titration of DnaBBA in DNA unwinding is shown in Fig. 6A. The standard assay contained 17 fmol of the 50-bp partial duplex substrate. Overall, 50% unwinding of the input substrate was observed with 20 ng of DnaBBA in 15 min at 30°C. In order to determine the rate of DNA unwinding, we carried out a time course analysis of the DNA unwinding in a 0- to 30-min range using 20 ng of DnaBBA. The initial rate of DNA unwinding was 10% per min or 1.7 fmol/min (Fig. 7B). Therefore, the rate of DNA unwinding was ∼85 pmol/min/mg.
FIG. 6.

DNA Helicase activities of DnaBBA and DnaBEC. Autoradiograph analyses of DNA helicase assays were carried out in a standard DNA helicase assay (see Materials and Methods) using the indicated amounts. Titration of purified DnaBBA was performed with the indicated amounts per 20-μl assay. The reaction products were analyzed in a 20-by-20-cm 8→12% gradient polyacrylamide gel and electrophoresed for 60 min at 190 V in 1× Tris-borate-EDTA containing 0.1% SDS. The gel was dried and autoradiographed at −80°C. The positions of the 32P-labeled partial duplex substrate and the unwound 60-mer substrates are indicated in the figure.
FIG. 7.

Kinetics of DNA unwinding by DnaBBA. (A) DNA helicase assays were carried out over 0 to 30 min with 20 ng of DnaBBA. Assays were carried out as described in Materials and Methods. (B) Kinetic plot of helicase activity following quantitation of unwinding by densitometry. The data were analyzed by nonlinear regression using Prism 6.0.
Nucleotide requirement of DNA helicase activity of DnaBBA.
The DNA helicase activity of DnaBBA is strictly ATP dependent. An ATP titration from 0.0.5 mM, in a standard DNA helicase assay of DnaBBA, is shown in Fig. 8A. The DNA helicase activity increases with ATP concentration. DNA helicase activity is not detectable below a 50 μM concentration. A quantitative analysis of the ATP dependence indicates that half-maximal DNA helicase activity is observed at 108 μM (Fig. 8B). The Km for ATPase (KmATP) activity, as determined from a Michaelis-Menten plot, was 150 μM with the 60-mer oligonucleotide and 220 μM with M13mp19 ssDNA as cofactor. Therefore, the KmATP determined from the ATPase activity correlated well with the Kd of ATP in the DNA helicase reactions.
FIG. 8.

Analysis of precise ATP requirement in the DNA helicase activity of DnaBBA. (A) DNA helicase assays were carried out with titration of ATP over a 0 to 500 μM concentration range. Assays were carried out as described in Materials and Methods. (B) Plot of helicase activity as a function of ATP concentration after quantitation of unwinding by densitometry of the corresponding autoradiograph. The data were analyzed by nonlinear regression using Prism 6.0.
We have examined other nucleotides in addition to ATP in the stimulation of DNA helicase activity of DnaBBA. Only adenosine nucleotides appeared capable of stimulating the DNA helicase activity (Fig. 9). A direct comparison of the two nucleotides indicated that dATP actually functions as a better cofactor than ATP. At both 100 and 500 μM, dATP produced comparable or slightly higher levels of unwinding than ATP. On the other hand, nonadenine nucleotides such as GTP, CTP, and UTP are not capable of replacing ATP/dATP in the DNA helicase activity. These nucleotides either do not bind or DnaBBA cannot hydrolyze them.
FIG. 9.

Analysis of nucleotide triphosphates in DNA helicase activity of DnaBBA. DNA helicase assays were carried out with ATP, GTP, CTP, UTP, and dATP at 100 and 500 μM concentrations. Assays were carried out as described in Materials and Methods.
Polarity of migration on ssDNA.
The polarity of translocation by DnaBBA was examined by using two analogous substrates with 5′ or 3′ forks constructed from M13mp19 circular ssDNA and two 45-bp oligonucleotides: one with a 5′ 10-nucleotide overhang and the other with a 3′ 10-nucleotide overhang. Each of these substrates contained an identical 35-bp duplex region. Thus, the energy required for unwinding these substrates remained constant. The substrate with 5′ overhang required a 5′→3′ migration of the DnaBBA helicase on the ssDNA template, whereas the substrate with 3′ overhang required a 3′→5′ migration of DnaBBA on ssDNA. The results presented in Fig. 10, indicated that DnaBBA unwound the substrate with the 3′ overhang at an ∼4-fold-higher rate than the substrate with 5′ overhang. The results in Fig. 10 indicated that DnaBBA has a 5′→3′ directionality of migration on ssDNA.
FIG. 10.

Polarity of translocation of DnaBBA on ssDNA template. (A) The polarity of translocation by DnaBBA was examined using two identical substrates with 5′ or 3′ forks constructed from M13mp19 circular ssDNA and two 45-bp oligonucleotides: one with a 5′ 10-nucleotide overhang and the other with a 3′ 10-nucleotide overhang. Each of these substrates contained a 35-bp identical duplex region. The kinetics with each substrate was measured over 0 to 30 min. Assays were carried out as described in Materials and Methods. (B) Plots of DNA helicase activities with each substrate after quantitation by densitometry. The data were analyzed by nonlinear regression using Prism 6.0.
ssDNA binding by DnaBBA.
The first step in DNA helicase action is the formation of helicase-ssDNA complex before DNA unwinding is initiated. All DNA helicases eventually form a ternary complex, helicase-dsDNA-NTP, before unwinding of the first base pair. Therefore, we have analyzed ssDNA binding by DnaBBA and the role nucleotide cofactors in ssDNA binding. We have analyzed the mechanism of DNA binding by DnaBBA using oligonucleotides labeled with a 5′-fluorescein moiety, and fluorescence anisotropy measurement was used to analyze DNA-protein complex formation. The binding constant for the interaction of DNA with DnaBBA was determined by using Fl-(dT)25. Fluorescence anisotropy was measured at increasing concentrations of DnaBBA until saturation in anisotropy was observed in the presence or absence of ATPγS. A semi-log plot of the anisotropy values at various DnaBBA concentrations in the presence of ATPγS generated the binding isotherm shown in Fig. 11A. With the addition of DnaBBA, the anisotropy value increased, which was due to an increase in the concentration of DnaBBA-Fl-(dT)25 complex as shown in Fig. 11A. A sigmoidal binding isotherm with a plateau at 230 mA at high DnaBBA concentration was observed (Fig. 11A). The Kd values, i.e., the concentrations of DnaBBA required to bind 50% of the oligonucleotides were computed. The Kd for DnaBBA-Fl-(dT)25 complex in the presence of ATPγS was (5.3 ± 1.0) × 10−8 M. The ssDNA binding isotherm without nucleotides is presented in Fig. 11B. The Kd determined from the binding isotherm was (4.0 ± 1.0) × 10−8 M. Therefore, the ssDNA binding by DnaBBA did not appear to depend on ATP/ATPγS.
FIG. 11.

ssDNA binding by DnaBBA and DnaBEC and modulation by ATPγS. ssDNA binding was measured using 3 nM Fl(dT)25 oligonucleotide probe. Titration was carried out with DnaBBA, and fluorescence anisotropy was measured as described in Materials and Methods. Anisotropy values were plotted against log of DnaBBA concentration, and the plots were analyzed by nonlinear regression using Prism 6.0. (A) DnaBBA binding in the presence of 1 mM ATPγS; (B) DnaBBA binding in the absence of nucleotides.
DISCUSSION
The replicative DNA helicases are important components of the cellular replication machinery for all organisms (35). Therefore, in order to understand the mechanism of DNA replication of anthrax genome, it was necessary to clone and express the replicative DNA helicase gene of anthrax. The anthrax genome has been sequenced recently (46). A search of the annotated anthrax genome sequence yielded two genes with homology to DnaBEC, BAS0880 and BAS5321. Allele replacement mutagenesis of anthrax replicative DNA helicase genes indicated that the BAS0880 ORF could be deleted without any phenotypical change of mutant anthrax cells unlike what was observed with the BAS5321 ORF. Attempts to delete the BAS5321 ORF did not result in the isolation of a viable deletion mutant, suggesting that this gene is essential. Therefore, BAS5321 gene is the only essential DNA helicase gene, and the gene product is likely the true replicative DNA helicase of anthrax. We have cloned and expressed BAS5321 ORF in E. coli. The gene product is a 50-kDa polypeptide (Fig. 3). It has significantly different chromatographic properties that allow its complete separation from endogenous DnaBEC in the host E. coli cells.
DNA-dependent ATPase activity of DnaBBA is likely coupled to translocation.
The ATPase activity of DnaBBA was strongly ssDNA dependent (Fig. 4). The Vmax increased proportionally with the size of the ssDNA cofactor. With increasing length of the ssDNA, DnaBBA appeared to have more ssDNA template for migration or translocation that in turn led to the enhancement of the ATPase activity observed here. In Fig. 4, ATPase activity increased with increasing lengths of the oligonucleotides; the maximum activity was observed with very long ssDNA template, M13mp19 ssDNA. The total amount of ssDNA in each point remained constant for all ssDNA templates. Consequently, the length of the ssDNA cofactor in the ATPase assay regulated the ATPase activity of DnaBBA. It appeared that DnaBBA hydrolyzed ATP predominantly during translocation on ssDNA templates. Binding ssDNA was not sufficient for stimulating ATPase activity of DnaBBA. Even with a long ssDNA template, ATPase activity of DnaBBA appeared significantly lower than that of its E. coli homolog. Thus, ATPase activity of DnaBBA was coupled directly to translocation on ssDNA, which is likely important in its ability to find an open replication fork and ATPase activity was minimized during inactivity. In summary, ATP utilization by DnaBBA was tightly regulated and wastage of ATP was minimized.
DnaBBA is highly active as a DNA helicase.
DnaBBA displayed robust DNA helicase activity in contrast to its attenuated ATPase activity. A titration of DnaBBA in a DNA helicase assay indicated that as little as 0.5 ng of DnaBBA exhibited detectable DNA helicase activity. Kinetic analysis of the DNA helicase activity indicated that initial rate of DNA unwinding was ∼10% of input substrate per min at 30°C. We also observed products of complete unwinding of a 50-bp duplex in ≤2 min, which indicated a rate of ≥25 bp/min, presumably, with one DnaBBA hexamer. It is perhaps possible that duplex DNA unwinding is not the rate-limiting step and rather a facile one under our analysis conditions. With DnaBEC, our earlier studies indicated that a full-length product of DNA helicase action is observed in approximately 5 min, and thus it could attain a rate of ≥10 bp/min under closely comparable reaction conditions (9). In vivo rates of replication fork movement for prokaryotes such as E. coli or B. anthracis could be as high as 1,000 bp/s. However, such high rates of fork movement require involvement of a number of proteins in the replisome in addition to DnaB. Further systematic mechanistic studies of DnaB helicases are required to identify the contributions of various steps in the DNA helicase activity.
It is also interesting that DnaBBA utilized ATP or dATP and not other ribo- or deoxynucleotides as a cofactor for DNA helicase activity, and it could have a slight preference for dATP. The ATP requirement (Fig. 8) for the DNA helicase appeared to parallel ATPase activity. Half-maximal DNA helicase activity (i.e., the 50% effective concentration) was observed with 108 μM ATP, a result comparable to the KmATP of its ATPase activity (152 to 220 μM). DnaBBA exhibited a preference for the substrate containing a 3′ tail, which indicated a 5′→3′ directionality of translocation on ssDNA. A directionality of 5′→3′ is common to many replicative DNA helicases. Therefore, in conjunction with our genetic analyses, 5′→3′ directionality of DnaBBA is in conformity with its role as the replicative DNA helicase of anthrax.
ssDNA binding by DnaBBA was nucleotide independent.
DnaBBA has ssDNA-dependent ATPase and DNA helicase activities that require it to bind ssDNA. True equilibrium DNA binding can be accurately measured by measuring changes in the fluorescence anisotropy of the ssDNA while titrating with DNA-binding protein, and it provides detailed quantitative information about the thermodynamics of protein-DNA interaction (14, 28, 32, 36, 39, 47). Titration of Fl-(dT)25 with an increasing DnaBBA concentration produced sigmoidal binding isotherms as shown in Fig. 11A. Nonlinear regression analysis of the binding isotherms produced an equilibrium binding constant. An inherent difficulty associated with these studies is the hydrolysis of ATP by the DNA helicases such as DnaBBA. Due to the rapid conversion of ATP to ADP, measurement of ssDNA binding in the presence of ATP is not possible. Therefore, the measurements had to be carried out in the presence of a nonhydrolyzable analogue of ATP, ATPγS. In the presence of ATPγS, saturable binding was observed with DnaBBA (Fig. 11). The dissociation constant with ATPγS was (5.3 ± 1.0) × 10−8 M. Without ATPγS or other nucleotides, the DnaBBA and ssDNA interaction was measured, and a saturable binding isotherm was produced with a Kd of 4.0 × 10−8 M (Fig. 11B).
DnaBBA bound ssDNA in the complete absence of nucleotides, and the DnaBBA-ssDNA complex then bound ATP. Translocation was then initiated, and ATP was hydrolyzed to provide energy for translocation. DnaBEC, on the other hand, requires nucleotide for binding ssDNA but not nucleotide hydrolysis; however, once it is bound to ssDNA, it becomes a stimulated nucleotidase as shown in Fig. 12.
FIG. 12.

DnaB translocation and unwinding reactions for B. anthracis (A) and E. coli (B).
Under our assay conditions as well as the fact that the initial DNA-binding step is nucleotide independent, this step could be rate limiting, which may also explain the kinetics of DNA helicase as discussed earlier. In addition, even after ssDNA binding, the ATPase activity is effectively a function of the size of the ssDNA (Fig. 4), which likely indicates that small oligonucleotides do not provide enough space for DnaBBA to translocate or move and, as space increases with the size of the ssDNA, so does the ATPase activity of DnaBBA. Thus, ATP hydrolysis by DnaBBA is tightly coupled to its movement on the ssDNA, leading to low ATP consumption and its high DNA helicase output.
In summary, DnaBBA is mechanistically distinct from its E. coli homolog DnaBEC in many different ways. DnaBBA appears to use ATP only during translocation on ssDNA, presumably, to find the target replication fork and its unwinding. ATPase activity without DNA is highly attenuated. Its high ssDNA-dependent ATPase and DNA helicase activities could be tied to its physiology and growth conditions inside mammalian tissue, with possible restriction of ATP availability.
Acknowledgments
We gratefully acknowledge grants from the UMDNJ Foundation and the National Institutes of Health (AI064974) to D.T.M. and S.B.B.
We thank all of our collaborators for helpful advice and comments during the course of this work. We thank Catherine A. Royer from the Centre de Biochimie Structurale, INSERM, Montpellier, Cedex 02, France, for the gift of BIOEQS software and training involving data analysis. We also thank members of the Biswas and Moir laboratories for a variety of technical assistance with B. anthracis genetic studies, protein purification, and assays.
Footnotes
Published ahead of print on 17 October 2008.
REFERENCES
- 1.Abhyankar, M. M., S. Zzaman, and D. Bastia. 2003. Reconstitution of R6K DNA replication in vitro using 22 purified proteins. J. Biol. Chem. 27845476-45484. [DOI] [PubMed] [Google Scholar]
- 2.Alfano, C., and R. McMacken. 1989. Ordered assembly of nucleoprotein structures at the bacteriophage lambda replication origin during the initiation of DNA replication. J. Biol. Chem. 26410699-10708. [PubMed] [Google Scholar]
- 3.Alfano, C., and R. McMacken. 1988. The role of template superhelicity in the initiation of bacteriophage lambda DNA replication. Nucleic Acids Res. 169611-9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arai, K., and A. Kornberg. 1981. Mechanism of dnaB protein action. II. ATP hydrolysis by dnaB protein dependent on single- or double-stranded DNA. J. Biol. Chem. 2565253-5259. [PubMed] [Google Scholar]
- 5.Arai, K., R. Low, J. Kobori, J. Shlomai, and A. Kornberg. 1981. Mechanism of dnaB protein action. V. Association of dnaB protein, protein n′, and other repriming proteins in the primosome of DNA replication. J. Biol. Chem. 2565273-5280. [PubMed] [Google Scholar]
- 6.Arai, K., S. Yasuda, and A. Kornberg. 1981. Mechanism of dnaB protein action. I. Crystallization and properties of dnaB protein, an essential replication protein in Escherichia coli. J. Biol. Chem. 2565247-5252. [PubMed] [Google Scholar]
- 7.Bastia, D., S. Zzaman, G. Krings, M. Saxena, X. Peng, and M. M. Greenberg. 2008. Replication termination mechanism as revealed by Tus-mediated polar arrest of a sliding helicase. Proc. Natl. Acad. Sci. USA 10510831-10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharyya, S., and M. A. Griep. 2000. DnaB helicase affects the initiation specificity of Escherichia coli primase on single-stranded DNA templates. Biochemistry 39745-752. [DOI] [PubMed] [Google Scholar]
- 9.Biswas, E. E., and S. B. Biswas. 1999. Mechanism of DNA binding by the DnaB helicase of Escherichia coli: analysis of the roles of domain gamma in DNA binding. Biochemistry 3810929-10939. [DOI] [PubMed] [Google Scholar]
- 10.Biswas, E. E., and S. B. Biswas. 1999. Mechanism of DnaB helicase of Escherichia coli: structural domains involved in ATP hydrolysis, DNA binding, and oligomerization. Biochemistry 3810919-10928. [DOI] [PubMed] [Google Scholar]
- 11.Biswas, E. E., S. B. Biswas, and J. E. Bishop. 1986. The dnaB protein of Escherichia coli: mechanism of nucleotide binding, hydrolysis, and modulation by dnaC protein. Biochemistry 257368-7374. [DOI] [PubMed] [Google Scholar]
- 12.Biswas, E. E., P. H. Chen, and S. B. Biswas. 2002. Modulation of enzymatic activities of Escherichia coli DnaB helicase by single-stranded DNA-binding proteins. Nucleic Acids Res. 302809-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biswas, E. E., P. H. Chen, and S. B. Biswas. 1995. Overexpression and rapid purification of biologically active yeast proliferating cell nuclear antigen. Protein Expr. Purif. 6763-770. [DOI] [PubMed] [Google Scholar]
- 14.Biswas, S. B., and E. E. Biswas-Fiss. 2006. Quantitative analysis of binding of single-stranded DNA by Escherichia coli DnaB helicase and the DnaB x DnaC complex. Biochemistry 4511505-11513. [DOI] [PubMed] [Google Scholar]
- 15.Biswas, S. B., and E. E. Biswas. 1987. Regulation of dnaB function in DNA replication in Escherichia coli by dnaC and lambda P gene products. J. Biol. Chem. 2627831-7838. [PubMed] [Google Scholar]
- 16.Biswas, S. B., P. H. Chen, and E. E. Biswas. 1994. Structure and function of Escherichia coli DnaB protein: role of the N-terminal domain in helicase activity. Biochemistry 3311307-11314. [DOI] [PubMed] [Google Scholar]
- 17.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72248-254. [DOI] [PubMed] [Google Scholar]
- 18.Bramhill, D., and A. Kornberg. 1988. Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the Escherichia coli chromosome. Cell 52743-755. [DOI] [PubMed] [Google Scholar]
- 19.Bramhill, D., and A. Kornberg. 1988. A model for initiation at origins of DNA replication. Cell 54915-918. [DOI] [PubMed] [Google Scholar]
- 20.Bruand, C., M. Farache, S. McGovern, S. D. Ehrlich, and P. Polard. 2001. DnaB, DnaD, and DnaI proteins are components of the Bacillus subtilis replication restart primosome. Mol. Microbiol. 42245-255. [DOI] [PubMed] [Google Scholar]
- 21.Bussiere, D. E., and D. Bastia. 1999. Termination of DNA replication of bacterial and plasmid chromosomes. Mol. Microbiol. 311611-1618. [DOI] [PubMed] [Google Scholar]
- 22.Dallmann, H. G., S. Kim, A. E. Pritchard, K. J. Marians, and C. S. McHenry. 2000. Characterization of the unique C terminus of the Escherichia coli tau DnaX protein: monomeric C-tau binds alpha and DnaB and can partially replace tau in reconstituted replication forks. J. Biol. Chem. 27515512-15519. [DOI] [PubMed] [Google Scholar]
- 23.Datta, H. J., G. S. Khatri, and D. Bastia. 1999. Mechanism of recruitment of DnaB helicase to the replication origin of the plasmid pSC101. Proc. Natl. Acad. Sci. USA 9673-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dodson, M., H. Echols, S. Wickner, C. Alfano, K. Mensa-Wilmot, B. Gomes, J. LeBowitz, J. D. Roberts, and R. McMacken. 1986. Specialized nucleoprotein structures at the origin of replication of bacteriophage lambda: localized unwinding of duplex DNA by a six-protein reaction. Proc. Natl. Acad. Sci. USA 837638-7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dodson, M., J. Roberts, R. McMacken, and H. Echols. 1985. Specialized nucleoprotein structures at the origin of replication of bacteriophage lambda: complexes with lambda O protein and with lambda O, lambda P, and Escherichia coli DnaB proteins. Proc. Natl. Acad. Sci. USA 824678-4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao, D., and C. S. McHenry. 2001. tau binds and organizes Escherichia coli replication proteins through distinct domains: domain IV, located within the unique C terminus of tau, binds the replication fork, helicase, DnaB. J. Biol. Chem. 2764441-4446. [DOI] [PubMed] [Google Scholar]
- 27.Giorno, R., J. Bozue, C. Cote, T. Wenzel, K. S. Moody, M. Mallozzi, M. Ryan, R. Wang, R. Zielke, J. R. Maddock, A. Friedlander, S. Welkos, and A. Driks. 2007. Morphogenesis of the Bacillus anthracis spore. J. Bacteriol. 189691-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gryczynski, I., R. F. Steiner, and J. R. Lakowicz. 1991. Intensity and anisotropy decays of the tyrosine calmodulin proteolytic fragments, as studied by GHz frequency-domain fluorescence. Biophys. Chem. 3969-78. [DOI] [PubMed] [Google Scholar]
- 29.Inglesby, T. V., D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. M. Friedlander, J. Hauer, J. McDade, M. T. Osterholm, T. O'Toole, G. Parker, T. M. Perl, P. K. Russell, K. Tonat, et al. 1999. Anthrax as a biological weapon: medical and public health management. JAMA 2811735-1745. [DOI] [PubMed] [Google Scholar]
- 30.Inglesby, T. V., T. O'Toole, D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. M. Friedlander, J. Gerberding, J. Hauer, J. Hughes, J. McDade, M. T. Osterholm, G. Parker, T. M. Perl, P. K. Russell, and K. Tonat. 2002. Anthrax as a biological weapon, 2002: updated recommendations for management. JAMA 2872236-2252. [DOI] [PubMed] [Google Scholar]
- 31.Johnson, S. K., S. Bhattacharyya, and M. A. Griep. 2000. DnaB helicase stimulates primer synthesis activity on short oligonucleotide templates. Biochemistry 39736-744. [DOI] [PubMed] [Google Scholar]
- 32.Khopde, S., E. Biswas, and S. Biswas. 2002. Affinity and sequence specificity of DNA binding and site selection for primer synthesis by Escherichia coli primase. Biochemistry 4114820-14830. [DOI] [PubMed] [Google Scholar]
- 33.Koehler, T. M. 2002. Bacillus anthracis genetics and virulence gene regulation. Curr. Top. Microbiol. Immunol. 271143-164. [DOI] [PubMed] [Google Scholar]
- 34.Koehler, T. M., Z. Dai, and M. Kaufman-Yarbray. 1994. Regulation of the Bacillus anthracis protective antigen gene: CO2 and a trans-acting element activate transcription from one of two promoters. J. Bacteriol. 176586-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornberg, A., and T. A. Baker. 1992. DNA replication. W. H. Freeman & Co., New York, NY.
- 36.Lakowicz, J. R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Plenum Publishers, New York, NY.
- 37.LeBowitz, J. H., and R. McMacken. 1984. The bacteriophage lambda O and P protein initiators promote the replication of single-stranded DNA. Nucleic Acids Res. 123069-3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LeBowitz, J. H., and R. McMacken. 1986. The Escherichia coli dnaB replication protein is a DNA helicase. J. Biol. Chem. 2614738-4748. [PubMed] [Google Scholar]
- 39.LeTilly, V., and C. A. Royer. 1993. Fluorescence anisotropy assays implicate protein-protein interactions in regulating trp repressor DNA binding. Biochemistry 327753-7758. [DOI] [PubMed] [Google Scholar]
- 40.Lu, Y. B., P. V. Ratnakar, B. K. Mohanty, and D. Bastia. 1996. Direct physical interaction between DnaG primase and DnaB helicase of Escherichia coli is necessary for optimal synthesis of primer RNA. Proc. Natl. Acad. Sci. USA 9312902-12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markowitz, V. M., F. Korzeniewski, K. Palaniappan, E. Szeto, G. Werner, A. Padki, X. Zhao, I. Dubchak, P. Hugenholtz, I. Anderson, A. Lykidis, K. Mavromatis, N. Ivanova, and N. C. Kyrpides. 2006. The integrated microbial genomes (IMG) system. Nucleic Acids Res. 34D344-D348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McMacken, R., K. Ueda, and A. Kornberg. 1977. Migration of Escherichia coli dnaB protein on the template DNA strand as a mechanism in initiating DNA replication. Proc. Natl. Acad. Sci. USA 744190-4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitkova, A., S. Khopde, and S. Biswas. 2003. Mechanism and stoichiometry of interaction of DnaG primase and DnaB helicase of Escherichia coli in RNA primer synthesis. J. Biol. Chem. 27852253-52261. [DOI] [PubMed] [Google Scholar]
- 44.Mukhopadhyay, G., K. M. Carr, J. M. Kaguni, and D. K. Chattoraj. 1993. Open-complex formation by the host initiator, DnaA, at the origin of P1 plasmid replication. EMBO J. 124547-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Odegrip, R., S. Schoen, E. Haggard-Ljungquist, K. Park, and D. K. Chattoraj. 2000. The interaction of bacteriophage P2 B protein with Escherichia coli DnaB helicase. J. Virol. 744057-4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Read, T. D., S. N. Peterson, N. Tourasse, L. W. Baillie, I. T. Paulsen, K. E. Nelson, H. Tettelin, D. E. Fouts, J. A. Eisen, S. R. Gill, E. K. Holtzapple, O. A. Okstad, E. Helgason, J. Rilstone, M. Wu, J. F. Kolonay, M. J. Beanan, R. J. Dodson, L. M. Brinkac, M. Gwinn, R. T. DeBoy, R. Madpu, S. C. Daugherty, A. S. Durkin, D. H. Haft, W. C. Nelson, J. D. Peterson, M. Pop, H. M. Khouri, D. Radune, J. L. Benton, Y. Mahamoud, L. Jiang, I. R. Hance, J. F. Weidman, K. J. Berry, R. D. Plaut, A. M. Wolf, K. L. Watkins, W. C. Nierman, A. Hazen, R. Cline, C. Redmond, J. E. Thwaite, O. White, S. L. Salzberg, B. Thomason, A. M. Friedlander, T. M. Koehler, P. C. Hanna, A. B. Kolsto, and C. M. Fraser. 2003. The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature 42381-86. [DOI] [PubMed] [Google Scholar]
- 47.Royer, C. A., W. R. Smith, and J. M. Beechem. 1990. Analysis of binding in macromolecular complexes: a generalized numerical approach. Anal. Biochem. 191287-294. [DOI] [PubMed] [Google Scholar]
- 48.Saile, E., and T. M. Koehler. 2002. Control of anthrax toxin gene expression by the transition state regulator abrB. J. Bacteriol. 184370-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shatalin, K. Y., and A. A. Neyfakh. 2005. Efficient gene inactivation in Bacillus anthracis. FEMS Microbiol. Lett. 245315-319. [DOI] [PubMed] [Google Scholar]
- 50.Stayton, M. M., L. Bertsch, S. Biswas, P. Burgers, N. Dixon, J. E. Flynn, Jr., R. Fuller, J. Kaguni, J. Kobori, M. Kodaira, R. Low, and A. Kornberg. 1983. Enzymatic recognition of DNA replication origins. Cold Spring Harbor Symp. Quant. Biol. 47(Pt. 2)693-700. [DOI] [PubMed] [Google Scholar]
- 51.Stayton, M. M., and A. Kornberg. 1983. Complexes of Escherichia coli primase with the replication origin of G4 phage DNA. J. Biol. Chem. 25813205-13212. [PubMed] [Google Scholar]