

Abstract
The genome sequence of the oral pathogen Streptococcus mutans predicts the presence of two putative polysaccharide deacetylases. The first, designated PgdA in this paper, shows homology to the catalytic domains of peptidoglycan deacetylases from Streptococcus pneumoniae and Listeria monocytogenes, which are both thought to be involved in the bacterial defense mechanism against human mucosal lysozyme and are part of the CAZY family 4 carbohydrate esterases. S. mutans cells in which the pgdA gene was deleted displayed a different colony texture and a slightly increased cell surface hydrophobicity and yet did not become hypersensitive to lysozyme as shown previously for S. pneumoniae. To understand this apparent lack of activity, the high-resolution X-ray structure of S. mutans PgdA was determined; it showed the typical carbohydrate esterase 4 fold, with metal bound in a His-His-Asp triad. Analysis of the protein surface showed that an extended groove lined with aromatic residues is orientated toward the active-site residues. The protein exhibited metal-dependent de-N-acetylase activity toward a hexamer of N-acetylglucosamine. No activity was observed toward shorter chitooligosaccharides or a synthetic peptidoglycan tetrasaccharide. In agreement with the lysozyme data this would suggest that S. mutans PgdA does not act on peptidoglycan but on an as-yet-unidentified polysaccharide within the bacterial cell surface. Strikingly, the pgdA-knockout strain showed a significant increase in aggregation/agglutination by salivary agglutinin, in agreement with this gene acting as a deacetylase of a cell surface glycan.
Streptococcus mutans is a pathogenic bacterium that is implicated in dental caries, infective endocarditis, and alpha-streptococcal shock syndrome in neutropenic patients (35). Its natural habitat is dental plaque, which is a well-known example of a naturally formed biofilm. In the oral cavity, S. mutans is considered among the primary etiological agents of dental caries because of its combined abilities to rapidly degrade carbohydrates, produce abundant acid, induce a tolerance to low-pH environments, and synthesize adherent glucans from sucrose (27).
In order to successfully colonize, S. mutans needs to avoid being eliminated by the various components of the innate immune system that can be found in the oral cavity. Specifically, saliva contains many defensive components/systems such as antimicrobial peptides, mucins, proline-rich glycoprotein, immunoglobulins, lactoferrin, cystatins, lysozyme, and (other) salivary (glyco)proteins (28, 38). For instance, bacterial aggregation through salivary agglutinin (SAG) prevents the adherence of microorganisms to the different oral surfaces (39).
One of the strategies that microorganisms use to evade the host innate immune system is de-N-acetylation of their cell surface glycans. A crucial role for exopolysaccharide deacetylation was shown in biofilm formation, colonization, and resistance to neutrophil phagocytosis and human antibacterial peptides in Staphylococcus epidermidis (42). Protection against host defenses by polysaccharide deacetylases has recently been reported for Listeria monocytogenes (9) and Streptococcus pneumoniae (40, 41). In both cases a peptidoglycan deacetylase modifies the cell wall to significantly increase its resistance to lysozyme hydrolysis.
Both of these bacterial peptidoglycan deacetylases are members of the carbohydrate esterase 4 (CE4) family (CAZY database, http://www.cazy.org/CAZY/). CE4 esterases are metal-dependent enzymes that deacetylate polysaccharides such as peptidoglycan, chitin, and acetylxylan. For instance, bacterial peptidoglycan deacetylases de-N-acetylate GlcNAc and N-acetylmuramic acid (MurNAc) sugars in the disaccharide repeat unit present in cell surface peptidoglycan (8, 17, 40, 41). Structural and biochemical characterization of the S. pneumoniae PgdA enzyme (PgdASp) showed that it coordinated an essential divalent metal cation at the active site (7). The metal ion coordinates a water molecule that performs a nucleophilic attack on the carbonyl carbon of the acetate moiety of GlcNAc. An aspartate residue acts as the catalytic base by activating the nucleophilic water, and a histidine, which protonates the leaving group, completes the general acid base catalysis mechanism (6, 7). Peptidoglycan deacetylases, chitin deacetylases, and acetylxylan esterases are able to deacetylate oligomeric GlcNAc in vitro, which is a pseudosubstrate for these enzymes (2, 3, 7, 37).
The genome sequence of S. mutans UA159 (1) displays two open reading frames that are annotated as putative polysaccharide deacetylases, one between positions 582168 and 581236 (GenBank locus tag SMU.623c, designated pgdA) and one between positions 912330 and 911434 (SMU.963c, pgdB). The predicted amino acid sequence of S. mutans PgdA (PgdASm) displays clear homology with the catalytic domains of the peptidoglycan GlcNAc de-N-acetylase of S. pneumoniae (PgdASp).
This study is aimed at determining the function of the pgdA gene product in S. mutans and understanding its possible role in host defense mechanisms. Through generation of a pgdA knockout, characterization of the recombinant enzyme, and determination of its structure, we demonstrate that PgdA is an active, metal-dependent CE4 esterase that plays a role in tuning cell surface properties and in interactions with (salivary) agglutinin, an essential component of the innate immune system, most likely through deacetylation of an as-yet-unidentified polysaccharide.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
Escherichia coli strains were grown either in liquid or on solid (1.5% agar) Luria-Bertani (LB) medium at 37°C. S. mutans UA159 (wild type) and its derivatives were grown in Todd-Hewitt broth (TH broth) or on 1.5% Todd-Hewitt agar containing 0.3% yeast extract, anaerobically at 37°C. Erythromycin was included where indicated at 200 μg/ml for E. coli and 10 μg/ml for S. mutans; ampicillin was used at 100 μg/ml for E. coli.
Construction of the S. mutans deacetylase-knockout strain.
Chromosomal DNA of S. mutans was isolated according to the method of Hanna et al. (18) and used as a template for PCR. To delete the pgdA (SMU.623c) gene in S. mutans UA159, we used a precise deletion method (13, 26). A crossover PCR deletion product was constructed in two steps. (i) Two fragments were generated with primer combinations pgdAuf/pgdAur and pgdAdf/pgdAdr, respectively. One fragment includes 554 bp upstream and 6 bp of the start of the gene; the other fragment includes 692 bp downstream and 6 bp of the end of the gene. (ii) The fragments were annealed at their overlapping region and PCR amplified as a single fragment using the outer primers (pgdAuf and pgdAdr). This fragment was digested with EcoRI and SphI and ligated into the suicide vector pORI280 (23), resulting in pLJ2. After the correct sequence was verified (BaseClear, Leiden, The Netherlands), pLJ2 was transformed into S. mutans UA159 (24). Selection for gene replacement was performed according to the method of Leenhouts et al. (23). pgdA gene deletion was confirmed by PCR using primers pgdAR and pgdAur. Furthermore, using quantitative PCR, pgdA gene expression was tested on the mRNA of wild-type and mutant strains using primers rtpgdAF and rtpgdAR. Also the expression of SMU.622c was monitored with primers rtSMU_622F and rtSMU_622R to exclude polar effects. All primer sequences and strain information are available in the supplemental material.
Construction of the S. mutans deacetylase overexpression plasmid.
When the protein sequence of PgdASm was analyzed with Signal-P (14), a signal sequence was predicted at the N terminus of the protein. The cleavage site was predicted between residues 41 and 42. Therefore, the DNA coding for amino acid residues 42 to 311 of the deacetylase was amplified by PCR from S. mutans UA159 genomic DNA by using primers SMU.623cF/R and cloned into the pGEX-6P-1 expression vector (Amersham) using the BamHI/EcoRI restriction sites; the sequence of the insert was verified by DNA sequencing.
Aggregation assay.
Overnight cultures of S. mutans were diluted 1:20 in fresh TH broth. At various time points a 2-ml sample from each culture was washed and resuspended in phosphate-buffered saline (PBS), pH 7.4. Aggregation indices (AIs) were subsequently determined according to the method of Malik and Kakii (29). One milliliter of an S. mutans suspension in PBS was rigorously vortexed to destroy aggregates, and the optical density at 600 nm (OD600 or ODtotal) of this suspension was determined. From another 1 ml of suspension, aggregates were removed by mild centrifugation (650 × g, 2 min), and the ODsupernatant was measured. AI was defined as (ODtotal − ODsupernatant)/ODtotal. The experiment was performed in triplicate.
Hydrophobicity assay.
The hydrophobicity of S. mutans cells was determined by measuring the adherence of the cells to xylene (Merck), as described by Rosenberg et al. (33). Cells from an overnight culture were harvested, washed once, and resuspended in PBS (pH 7.4). The OD600 of this sample was measured (A0). A 1.5-ml portion of this suspension was mixed with 0 or 1 ml of xylene (90 s; vortex). The mixture was allowed to stand for 15 min to ensure complete separation of the two phases. A 1-ml sample was carefully removed from the aqueous phase and left standing for 1 h to evaporate residual xylene, and subsequently the OD600 of the sample was measured (A). The percentage of bacterial adhesion to solvent was calculated by % adherence = (1 − A/A0) × 100. The experiment was performed in triplicate.
Lysozyme susceptibility.
Overnight cultures of S. mutans were diluted 1:20 in fresh TH broth and grown at 37°C. At indicated time points, during the exponential growth phase, the cultures were divided in two. To one part 40 μg/ml hen egg lysozyme was added; no lysozyme was added to the control culture. Subsequent growth was monitored by following the OD600 in a spectrophotometer (Molecular Devices, California) for 3 h.
In a second experiment, cells were grown until the start of the stationary phase, and the cultures were centrifuged (15 min, 4,000 × g, 4°C), resuspended in Tris-EDTA (TE) buffer (pH 8.0), and again divided and treated as described above. Changes in OD600 were subsequently recorded for 5 h. Four replicates were tested for each condition, and the experiment was performed in duplicate.
Agglutinin adhesion.
The susceptibility of S. mutans cells to aggregation by SAG was examined by the adhesion assay described previously (4, 5, 25). In brief, human parotid saliva was collected from one volunteer with a Lashley cup, and 10 ml of parotid saliva was cooled on ice water for 30 min to promote the formation of a precipitate and subsequently centrifuged at 16,000 × g. The resulting pellet (approximately 10-fold enriched in SAG) was dissolved in 1 ml PBS and used for binding studies. Overnight cultures of S. mutans were washed and resuspended in Tris-HCl buffer (pH 7.0) to a set density of OD600 = 1.0. Subsequently, 100 μl of this bacterial suspension was added to a microtiter plate the wells of which had been coated with 1 to 5 μg/ml (twofold serially diluted) crude SAG. After 2 h of incubation at 37°C the plates were washed three times with buffer. Adherent bacteria were detected after staining with 2.5 μM SYTO-13 (Invitrogen; 100 μl/well) in a Fluostar Galaxy microtiter plate fluorescence reader (Molecular Devices, California). The experiment was performed in triplicate.
Binding of wheat germ agglutinin (WGA) to S. mutans cells was quantified with an Alexa Fluor 488-WGA conjugate (Invitrogen). Overnight cultures were washed and resuspended in bovine serum albumin (BSA)-saline solution (0.25% BSA, 0.15 M NaCl; OD600 of 0.55). Alexa Fluor 488-WGA was subsequently added to final concentrations of 0, 1, 5, and 10 μg/ml. After 60 min of incubation (37°C), the cells were centrifuged, washed, and resuspended in BSA-saline. Bound Alexa Fluor 488-WGA was quantified in a fluorimeter (excitation, 495 nm; emission, 519 nm).
Production and purification of S. mutans PgdA.
E. coli BL21(DE3)pLysS cells containing the pGEX-6P-1 expression plasmid encoding residues 42 to 311 of the PgdA protein were grown at 37°C in LB medium containing ampicillin until they reached an OD600 of 0.6. Gene expression was induced by the addition of a final concentration of 0.25 mM isopropyl-β-d-thiogalactopyranoside followed by a further incubation for 4 h. Cells were harvested by centrifugation and resuspended in 50 ml of 25 mM Tris, 250 mM NaCl, pH 7.5, per liter culture volume. After lysis by sonication, the insoluble material was removed by centrifugation (22,500 × g, 30 min, 4°C). The soluble fraction was incubated with glutathione-Sepharose beads (Amersham) at 4°C for 3 h. Cleavage of the glutathione S-transferase tag from PgdASm was achieved by incubation at 4°C with PreScission protease. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis showed good expression of the protein and confirmed successful cleavage of 95 to 100% of the fusion protein. PgdA protein separated from glutathione beads was purified further by gel filtration in lysis buffer containing 2 mM EDTA. A single peak that corresponded to the expected size of the monomeric protein (30.5 kDa) was observed, and sodium dodecyl sulfate-polyacrylamide gel electrophoresis showed that these fractions contained no contaminating proteins.
Crystallization and structure solution.
Gel filtration fractions were pooled and concentrated to 21 mg/ml using a 20-ml VivaSpin 10,000-molecular-weight-cutoff spin concentrator. Diffraction-quality cubic crystals were grown by vapor diffusion, using equal volumes of protein solution and mother liquor consisting of 2.4 M Na/K phosphate, pH 5.6. Crystals were soaked with 0.1 M ZnCl2 for 5 min followed by washing in a cryoprotectant solution (2.4 M Na/K phosphate, pH 5.6, 12% [wt/vol] polyethylene glycol 400 containing no ZnCl2) for 10 s. These crystals were tested on beamline BM14 at the European Synchrotron Radiation Facility (Grenoble, France), and a fluorescence scan of the crystals indicated the presence of Zn. A single anomalous dispersion data set was collected to 1.45-Å resolution at the Zn K edge (λ1 = 1.28 Å). Images were scaled using the HKL suite (31). The data between 15 and 1.45 Å were scaled in I213 with unit cell dimensions a = b = c = 128.57, one molecule per asymmetric unit, Rmerge of 0.045 (0.291 for the last shell), 99.1% completeness (98.7% for the last shell), and 3.6-fold anomalous redundancy (3.3-fold for the last shell). Analysis of the Harker section (u, v, w = 0.5) in the anomalous Patterson map revealed a single 70-σ peak. A single zinc site was identified using the ShelxD program (36), and the obtained phases were used to generate electron density maps in which WarpNtrace (32) built 238 out of 270 residues. Refinement was performed with CNS (10) interspersed with model building in O (21) and COOT (15); this resulted in a final model with an R factor of 0.165 (Rfree = 0.185) and good geometry (root mean square deviation from ideal bond lengths and angles is 0.012 Å and 1.7°, respectively). Density for a single zinc molecule within the active site of the protein was observed within the calculated electron density maps. Figures were generated using the PyMOL Molecular Graphics System, DeLano Scientific (http://www.pymol.org).
Fluorescamine-based de-N-acetylase activity assay.
Purified PgdASm was tested for de-N-acetylase activity using a 96-well plate assay as previously reported (7). Standard reaction mixtures consisted of 1 μM PgdASm, 50 mM Bis-Tris (pH 7.0), and 1 mM oligosaccharide substrate (Sigma) in a total volume of 50 μl and were incubated for 12 h or 16 h at 37°C. The reactions were stopped by the addition of 20 μl of 2 mg/ml fluorescamine in acetonitrile and the subsequent addition of 50 μl 0.4 M borate buffer, pH 9.0. Fluorescence was quantified using a FLX 800 Microplate fluorescence reader (Bio-Tek, Burlington, VT), with excitation and emission wavelengths of 360 and 460 nm, respectively. A calibration curve using glucosamine showed that the free amine labeling reaction was linear up to concentrations of 600 μM glucosamine. Measurements are shown as averages of three or four replicates.
Protein structure accession number.
The coordinates of the PgdASm structure have been deposited at the Protein Data Bank, entry code 2XXX.
RESULTS
S. mutans possesses a putative polysaccharide deacetylase.
The genome sequence of S. mutans UA159 displays two open reading frames that can be identified as polysaccharide deacetylases. Reading frame SMU.623c (pgdA) displays 51% similarity to the polysaccharide deacetylase of S. pneumoniae and 50% similarity to the polysaccharide deacetylase of L. monocytogenes (7, 9). An alignment of S. mutans PgdA with enzymatically and structurally characterized CE4 esterases including a chitin deacetylase, peptidoglycan deacetylases PgdASp and PdaABs (Bacillus subtilis PdaA), and an acetylxylan esterase (Fig. 1) shows that the S. mutans PgdA protein contains all of the catalytic and zinc binding residues. Structural studies of PgdASp and Colletotrichum lindemuthianum chitin de-N-acetylase (CDACl) have shown that a divalent metal cation is coordinated by an aspartic acid, in motif 1, and two histidine residues within motif 2 (6, 7). Treatment of PgdASp with the metal chelator EDTA caused complete loss of activity (7), confirming that the metal is essential for catalysis. PgdASm retains this classical Asp-His-His arrangement within the sequence alignment, suggesting that the protein could bind a metal cation within the active site. In the CE4 esterase catalytic mechanism, another aspartic acid in motif 1 (PgdASm numbering Asp114) acts as a catalytic base by activating the nucleophilic water (6, 7). A conserved arginine (Arg211) at the start of motif 3 that forms a hydrogen bond with the catalytic acid is also essential for catalysis. The catalytic machinery is completed by a histidine (His281) in motif 5 which acts as the catalytic acid and a conserved aspartate (Asp244 in motif 4) that alters the pKa on the catalytic histidine (6, 7). Thus, it appears that PgdASm contains all residues required for de-N-acetylation of a suitable substrate.
FIG. 1.

Structure-based sequence alignment of CE4 esterases. The sequences of three known de-N-acetylases, a de-O-acetylase, and PgdASm from the CE4 family are shown: S. mutans PgdA, C. lindemuthianum chitin de-N-acetylase, B. subtilis PdaA, Streptomyces lividans xylan de-O-acetylase, and S. pneumoniae PgdA. The secondary structures of PgdASm and PgdASp are indicated above and below the alignment, respectively. The secondary structure is highlighted as red helices, and blue strands represent the CE4 esterase domain. Secondary structure not present in the canonical CE4 fold is shown in green. The five CE4 active-site motifs (MT1 to MT5, yellow) are indicated below the alignment. The metal coordinating residues are colored cyan, and the catalytic residues are colored magenta. Residues highlighted in orange show large shifts in surface-exposed loops. The alignment was performed using the Aline program (written and kindly provided by Charlie Bond, University of Western Australia, and Alexander Schüttelkopf, Dundee University).
Characterization of the ΔpgdA strain.
Quantitative PCR showed expression of pgdA in the wild type but not in the mutant strain. Expression levels of the downstream SMU.622c gene were similar in the two strains. Together these expression profiles indicate the successful deletion of the pgdA gene in S. mutans without polar effects.
The ΔpgdA strain did not display any obvious differences in growth characteristics from the wild type. There was no difference in growth rate (see Fig. 3) or chain length (data not shown). Biofilm formation of the ΔpgdA strain was examined as described previously (30). No differences were found between the knockout and wild-type strains (data not shown). However, when the cells were grown on brain heart infusion agar plates, close inspection showed that the colony morphology of the ΔpgdA strain was clearly different from that of the wild type (Fig. 2). We decided to determine the hydrophobicity of both cell types by measuring the adherence of the cells to xylene. When the volume of xylene applied was two-thirds of that of the cell culture, the fraction of cells adhering to xylene was 0.55 (±0.08) for the wild type and 0.63 (±0.04) for the ΔpgdA strain. Hence, deletion of the pgdA gene slightly increased the cell surface hydrophobicity (Student's t test, P = 0.07).
FIG. 3.

AI of S. mutans UA159 wild type and the pgdA-knockout strain during growth. AIs of the wild-type strain are presented as gray bars; those of the pgdA-knockout strain are presented as white bars. OD600 values of the cultures at time points are given in squares for the wild type and triangles for the pgdA knockout. Both AI and OD600 values shown are means of three independent samples with standard deviations.
FIG. 2.

Images of S. mutans UA159 wild type and the pgdA-knockout strain. Closeup images of a single colony of S. mutans UA159 (wild type; left) and the ΔpgdA strain (right). Bacteria were grown anaerobically on brain heart infusion agar plates at 37°C for 7 days. Images were taken with a digital Zeiss camera installed on a Zeiss stereomicroscope (Stemi SV6; Hallbergmoos, Germany) at ×32 magnification.
Further differences between the cell surface of wild-type and ΔpgdA strains became apparent in an aggregation assay. Figure 3 shows that the AI of the pgdA-knockout strain is significantly higher than that of the wild type, throughout the growth phase. The AI of the wild-type strain did not seem to depend on the growth phase, while the AI of the knockout strain was significantly higher at the late exponential phase than at the earlier growth phases. One-way analysis of variance was used to analyze the data.
The ΔpgdA strain is not hypersensitive to lysozyme.
When lysozyme (40 μg/ml) was added during the exponential growth phase (Fig. 4A and B), no effects were observed in either the wild type or the ΔpgdA strain. In TH broth, in the stationary phase, the pH of the culture is 5.5. To exclude the effect of pH on the activity of lysozyme, the cells were washed with TE buffer (pH 8.0) prior to treatment. The addition of lysozyme resulted in a small decrease in the OD of both strains, 21% ± 6.1% for the wild-type strain and 18% ± 1.8% for the knockout strain (Fig. 4C and D). Increased concentrations of lysozyme (up to 1.28 mg/ml) did not result in any further inhibitory effects on either strain (data not shown).
FIG. 4.

Susceptibility of S. mutans to lysozyme. S. mutans wild type and the pgdA knockout were grown in TH broth. In the early exponential phase or at the beginning of the stationary phase (as indicated by the arrows), lysozyme (40 μg/ml) was added to the experimental cultures. (A and C) Wild-type strain; (B and D) pgdA knockout. Solid symbols indicate the control cultures without the addition of lysozyme. Open symbols indicate the cultures treated with lysozyme.
Therefore, while PgdASm appears to possess all of the residues required for de-N-acetylase activity, it seems not to play a role in lysozyme resistance of S. mutans, in contrast to what has been reported for PgdASp. The mechanism of lysozyme resistance in S. mutans might be different from that in S. pneumoniae. There might also be differences in substrate specificity between the two proteins.
Agglutinin susceptibility.
The prediction of signal sequence in PgdASm indicates that the protein is an extracellular protein which is secreted or bound to the cell surface. It may modify cell surface glycans at the interface between the bacteria and the host. Further investigations focused on the possibility that PgdASm helps S. mutans evade other components of the innate immune response in the oral cavity. SAGs are known to adhere to bacterial cells and modulate clearance and colonization of the oral cavity (39). As a further investigation of the activity of PgdASm, we studied the susceptibility of the wild-type and knockout strains of S. mutans toward agglutination with SAG. Figure 5A shows the adherence of S. mutans cells to SAG as indicated by SYTO-13 fluorescence. In assays of both the S. mutans wild-type and ΔpgdA strains, the number of S. mutans cells that adhered to SAG increased with increasing concentrations of SAG. Strikingly, at higher SAG concentrations used, adherence of the pgdA-knockout strain was much more pronounced, indicating an increased susceptibility to binding SAG. Since we used crude SAG in this test, we investigated the ability of PgdASm to bind to Alexa Fluor 488-labeled WGA, a lectin that specifically binds to N-acetylglucosamine and sialic acid residues. Figure 5B shows that adherence of S. mutans cells to WGA is significantly higher for the knockout strain than for the wild-type strain, suggesting that there is a greater number of GlcNAc-containing saccharides on the cell surface. Interestingly, agglutinins are known to bind GlcNAc but not glucosamine residues, a finding which is compatible with PgdASm acting as a GlcNAc de-N-acetylase that may help the bacteria evade the host innate immune response by disruption of interactions with SAG.
FIG. 5.
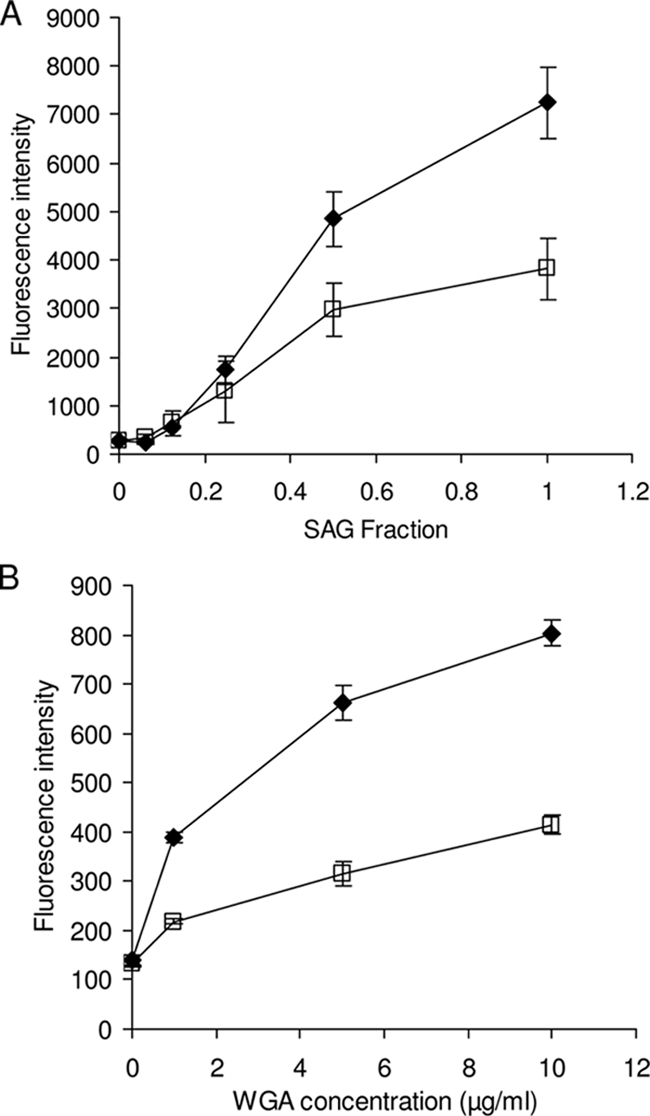
Susceptibility of S. mutans toward aggregation by agglutinin. Overnight cultures of S. mutans UA159 wild type (blank squares) and the pgdA knockout (solid diamonds) were washed with buffer and tested for adherence to increasing concentrations of SAG (A) using the fluorescence of SYTO-13 and WGA (B) using the fluorescence of the Alexa Fluor 488 conjugate, as described in Materials and Methods. The values shown are the means of three independent samples with standard deviations.
PgdASm possesses de-N-acetylase activity toward the chitooligosaccharide GlcNAc6.
To study the potential de-N-acetylase activity of PgdASm, the enzyme was overexpressed as a glutathione S-transferase fusion protein in E. coli and purified to yield 10 mg of pure PgdASm per liter of bacterial culture. An assay system for de-N-acetylases, based on the labeling of free amines with fluorescamine (7), has previously been used to determine the activity of de-N-acetylases. A screen of enzyme activity was performed using four different substrates, GlcNAc3, GlcNAc4, GlcNAc6, and a chemically synthesized GlcNAc-MurNAc-GlcNAc-MurNAc tetrasaccharide repeat of peptidoglycan. In the presence of both cobalt and zinc the protein showed significant de-N-acetylase activity toward chitohexaose (Fig. 6A and B). No activity was observed with the other oligosaccharides (data not shown).
FIG. 6.

De-N-acetylase activity of S. mutans PgdA. (A) S. mutans PgdA exhibits metal-dependent de-N-acetylase activity. The de-N-acetylase activity of recombinant PgdASm protein purified in the absence of EDTA. Assay mixtures containing 1 mM chitohexaose and 1 μM PgdASm were preincubated for 5 min in solution with different EDTA concentrations. The addition of a 10-fold excess of CoCl2 and ZnCl2 was used to reactivate the protein after EDTA treatment. (B) S. mutans PgdA steady-state kinetics. PgdASm (1 μM) was incubated with various concentrations of chitohexaose. The experiments were performed in triplicate, and the mean arbitrary fluorescent units (afu) were converted to the molar concentration of product using a glucosamine calibration curve under identical conditions. The reaction maintained first-order kinetics for 16 h (data not shown), and initial velocities were measured after 12 h.
PgdASm is a metal-dependent deacetylase.
Previously, Blair et al. showed that the PgdASp enzyme is metal dependent and activity was lost after the addition of the metal-chelating agent EDTA (7). To determine if the ability of PgdASm to de-N-acetylate chitohexaose was metal dependent, PgdASm protein was purified as described in Materials and Methods but without EDTA in the gel filtration buffer. This EDTA-free protein exhibited activity similar to that of the EDTA-purified protein assayed in the presence of excess metal, suggesting that the enzyme is able to scavenge metal ions during growth and purification steps. The addition of a range of divalent metal cations did not increase activity of PgdASm purified in the absence of EDTA (data not shown), suggesting full occupancy of the metal binding site with the scavenged divalent metal cation. To further investigate the metal dependency of the reaction, the PgdASm protein stock was incubated with different concentrations of EDTA, or water as a control, for 5 min. The final concentrations in this assay were 1 μM protein and 1, 10, and 100 μM EDTA. Assays were started by the addition of the protein-EDTA mixture to the substrate in 96-well plates. Figure 6A shows that the addition of increasing amounts of EDTA caused significant reduction of the PgdASm de-N-acetylase activity. When the enzyme was preincubated with 10 μM and 100 μM EDTA, we observed the loss of 97% and >99% activity, respectively (Fig. 6A). These results suggest that, like other CE4 esterases, the de-N-acetylation of chitohexaose by PgdASm is a metal-dependent process. It is, however, possible that EDTA treatment removes structurally important metal ions, leading to loss of activity. To investigate this, assays were performed with enzyme preincubated with 10 μM EDTA and in the presence of 100 μM ZnCl2 or CoCl2. The EDTA-treated protein was reactivated in the presence of these divalent metal cations. In the presence of Co2+, 80% of the wild-type protein activity was observed. However, when Zn2+ was added, complete activity was reconstituted. This may suggest that the recombinant form of PgdASm binds a zinc ion within its active site and that in vivo the PgdASm protein may be fully activated when coordinating a zinc ion.
Both S. mutans and its human hosts lack the catalytic machinery required to synthesize chitin and chitooligosaccharides. Hence, chitohexaose is likely to represent only a “pseudosubstrate,” with the production of free amine fitting a first-order reaction rate (data not shown). Initial velocity measurements fitted Michaelis-Menten kinetics, and the Km was 2.4 mM ± 0.2 mM (kcat [s−1] = 0.017 ± 0.001) (Fig. 6B). The Km value is similar to those reported with the peptidoglycan deacetylase PgdASp and its GlcNAc3 pseudosubstrate (Km = 3.8 ± 0.5 mM; kcat [s−1] = 0.55 ± 0.03) (7), while the turnover is decreased 30-fold. The chitooligosaccharide used in this assay more accurately represents the natural substrate of the chitin deacetylase from the plant pathogen C. lindemuthianum, CDACl. This protein has a Km of 48 ± 3 μM and a kcat (s−1) of 5.4 ± 0.8 in assay mixtures containing GlcNAc6. The much lower Km and kcat values observed in PgdASm assays support the hypothesis that it is not a chitin deacetylase. In conclusion, these data suggest that PgdASm is capable of catalyzing the de-N-acetylation of an N-acetylglucosamine residue on a longer oligosaccharide.
PgdASm adopts the canonical CE4 fold with an ordered active-site zinc.
To gain insight into why PgdASm requires longer chitooligosaccharides for activity, S. mutans PgdA was crystallized from Na/K phosphate solutions, synchrotron diffraction data were collected to 1.45 Å, and the structure was solved. A single-wavelength anomalous dispersion experiment exploiting a zinc soaked into the active site yielded a high-quality experimental electron density map, which could be partially automatically interpreted with WarpNtrace (32). Refinement produced a final model with an R factor of 0.165 (Rfree = 0.185) and good geometry. Residues 42 to 67 and 100 to 105 did not have well-defined electron density and were not included in the model. The PgdASm structure consists of an extended N-terminal domain (amino acids 68 to 99) that incorporates two α-helices (α0′ and α0) (Fig. 7A). The catalytic domain (amino acids 106 to 311) adopts a distorted TIM barrel fold comprising eight parallel β-strands, with the C-terminal ends of five of these strands forming the solvent-exposed active-site region, surrounded by eight α-helices (Fig. 7A). This structural fold has been observed in other CE4 esterases, and despite topological differences described for other CE4 esterases, PgdASm shares a similar topology with the PgdASp protein (6, 7). Superposition on the catalytic domain of the PgdASp structure gives a root mean square deviation of 1.3 Å on 168 equivalent Cα atoms. Insertions between β3-α3 and α5-β7 in the PgdASm protein cause reorganization of some surface-exposed loops compared to PgdASp (Fig. 7A). An anomalous difference electron density map was generated, revealing a 99-σ peak that was modeled as a zinc ion coordinated octahedrally by Asp115, His166, His170, and a water molecule (Fig. 7B). These residues align with the highly conserved metal coordination residues in other CE4 esterases (6-8, 37) (Fig. 1). Additional tetrahedral electron density was observed close to the zinc ion and was modeled as a phosphate ion, presumably originating from the crystallization mother liquor that contained 2.4 M phosphate (Fig. 7B). This phosphate ion makes a bidentate interaction with the zinc ion via two of its oxygen atoms approximately 2.0 and 3.0 Å away, occupying a position similar to that of the ordered acetate (the product of the reaction) observed in the CDACl and PgdASp structures and forming comparable interactions (6, 7). Interestingly, the reaction catalyzed by CE4 enzymes has been proposed to proceed through a tetrahedral oxyanion intermediate, and this may, to some extent, be mimicked by the phosphate. Despite extensive soaking studies with substrate/product analogues, it appeared to be impossible to displace the ordered phosphate from the active site.
FIG. 7.

Overview of the S. mutans PgdA structure. (A) Comparison of the overall structures of the S. mutans PgdA and the S. pneumoniae PgdA. α-helices are colored red and β-strands are colored blue in the CE4 esterase domain. Secondary structure elements at the termini of the proteins, outside the typical CE4 fold, are shown in green. Exposed loop regions which differ significantly due to inserts in the S. mutans PgdA structure compared to PgdASp (2C1G) are shown in orange. Secondary structure elements are named in accordance with the sequence alignment in Fig. 1. (B) Stereo image of the active site of S. mutans PgdA. A phosphate ion (yellow) was observed coordinating with the zinc ion (magenta) and other ligands including a water molecule (shown in cyan) in an octahedral manner. The unbiased 1.45-Å |Fo| − |Fc|, φcalc electron density map is shown (blue) contoured at 2.5 σ. (C) S. mutans PgdA contains an extended surface groove containing exposed aromatic residues. PgdASm and PgdASp (2C1G) structures are shown in surface representation. All aromatic residues are represented as sticks and colored blue. A putative intermediate of PgdASp deacetylation of GlcNAc3, as described previously (7), is shown in stick representation and colored green. This potential tetrahedral intermediate was superposed onto the PgdASm structure using the PgdASp coordinates to generate a model of a PgdASm-chitooligosaccharide complex. Aromatic residues that line the active site or putative oligosaccharide binding site of PgdASm are labeled. Surface representations of the metal binding triad and the four active-site residues are colored pink.
The surface of the PgdASm protein contains an extended groove lined with aromatic residues.
Analysis of the PgdASm surface revealed a deep groove that extends from the active site toward the α0 helix at the N terminus of the protein (Fig. 7C). The groove is 36 Å long and as deep as 13 Å in some places. This surface feature extends away from the active site for 13 Å and then encompasses a 110° turn before continuing for a further 22 Å. It is formed by residues from the β5-β6 region, the α5 helix, and the β4-α4 loop and exits in a tunnel created by the α0 and α4 helices. A series of exposed aromatic residues line the groove and appear to be positioned approximately 10 Å from each other. To investigate the potential interactions with sugars, the PgdASm structure was superimposed on a PgdASp structure in which a chitotrioside carrying the previously proposed oxyanion reaction intermediate was docked (6, 7). Intriguingly, the 10-Å distance between aromatic residues is almost identical to that observed between the first and third N-acetylglucosamine sugars on the superimposed GlcNAc3 reaction intermediate (Fig. 7C). Tyr172 is positioned opposite the active site with its hydroxyl group pointing toward the superimposed sugar. At further distances from the active site there are three aromatic residues, Trp239, Phe262, and Trp236, which line the groove and are orientated so that they may participate in stacking interactions with longer polysaccharide substrates. Trp236 is found at the opening of the groove furthest away from the active site. In contrast, the PgdASp structure contains three solvent-exposed aromatic residues in close proximity to the active site and within 5 Å of the docked GlcNAc trimer (Fig. 7C). The discovery of the novel putative carbohydrate binding groove combined with the de-N-acetylase activity of PgdASm suggests a different substrate and function for this enzyme, in comparison with those of PgdASp. This result is consistent with the difference in lysozyme resistance found between the PgdASm- and the PgdASp-knockout strains.
DISCUSSION
GenBank locus tag SMU.623c in the genome sequence of S. mutans UA159 (1) reveals an open reading frame with clear homology to the catalytic domains of the peptidoglycan deacetylases of S. pneumoniae and L. monocytogenes. The SMU.623c open reading frame contains all of the catalytic residues required for de-N-acetylase activity in CE4 esterases (Fig. 1). In both L. monocytogenes (9) and S. pneumoniae (41), deletion of the homologous polysaccharide deacetylase resulted in an increased susceptibility of the cells to lysozyme. In the oral cavity, the natural habitat of S. mutans, lysozyme plays a crucial role as part of the innate immune system (39). To understand the function of the PgdASm, we generated a knockout of the pgdA gene in S. mutans.
Examination of the susceptibility to lysozyme in S. mutans UA159 and in the pgdA knockout showed that PgdASm is not involved in lysozyme resistance of S. mutans (Fig. 4). Both the wild type and the mutant are almost fully resistant to lysozyme. Nevertheless, deletion of pgdA had a clear effect on colony morphology and aggregation behavior of S. mutans (Fig. 2 and 3), illustrating the activity and functionality of the protein in modifying cell surface properties.
The 1.45-Å-resolution X-ray structure of the S. mutans pgdA gene product also exposes PgdA as a typical CE4 esterase (Fig. 7A to C), in which the active site appears intact and fully functional. Investigation of the possible enzymatic activity of the protein showed that it was capable of de-N-acetylating GlcNAc residues on the oligosaccharide chitohexaose in a divalent metal cation-dependent mechanism (Fig. 6). In contrast, no activity was observed toward shorter chitooligosaccharides or a peptidoglycan-derived tetrasaccharide. The inability of the enzyme to release acetate from the latter tetrasaccharide makes it unlikely that this PgdA is active against peptidoglycan. This complements the conclusion that the knockout is not lysozyme sensitive. A long and deep groove extends from the active site over the surface of the PgdASm structure (Fig. 7). It is lined with three evenly spaced aromatic residues that are generally observed in carbohydrate-processing/binding proteins. The 10-Å distances between these residues suggest that six sugars could bind within the groove with a penultimate sugar ideally positioned within the active site. Attempts to elucidate the mechanism of binding of chitohexaose to the protein and an investigation of the function of aromatic residues within the surface groove are ongoing.
Recently, a putative peptidoglycan deacetylase (EF_1843) of Enterococcus faecalis, a bacterium that is able to survive in host macrophages, was also shown not to be involved in lysozyme resistance (19). Accordingly, reversed-phase high-performance liquid chromatography and matrix-assisted laser desorption ionization-time of flight analysis of the E. faecalis peptidoglycan provided evidence that EF_1843 is not involved in deacetylation of cell wall saccharides, similar to what is reported here for PgdASm. Strikingly, strains with knockouts of the EF_1843 gene exhibited a significant decrease in the ability of the bacteria to survive in mouse peritoneal macrophages. An alignment of the two proteins shows that they share 49% sequence identity covering 75% of the protein, with the exception of the N terminus. It is a reasonable hypothesis that PgdASm may perform a similar role in S. mutans resistance to phagocytic killing.
In the absence of any involvement in lysozyme sensitivity, an alternative role for S. mutans PgdA in innate immune interactions may lie in modulation of interactions with SAG. The most prominent effect of knocking out pgdA is the increased susceptibility to agglutination via lectins. Both adherence to WGA and that to SAG increased significantly upon deletion of the polysaccharide deacetylase (Fig. 5). Particularly relevant is of course the increased interaction with SAG (Fig. 5A). These lectins (agglutinins) display a high affinity against acetyl groups, in agreement with a role of PgdASm in N deacetylation of a cell wall component that would result in a lower susceptibility to agglutination. The capability to protect itself against agglutination by the host defense system can be seen as an important virulence trait of S. mutans. Indeed, clinical studies have indicated that parotid saliva primarily affects the in vivo prevalence of S. mutans by clearing the bacteria from the mouth rather than promoting adherence to oral surfaces (11, 12). Furthermore, it was recently demonstrated that SAG (also called gp-340) can be regarded as a infection (caries) susceptibility protein (20). Adherence of S. mutans to SAG has already been studied extensively (16, 34). Antigen I/II, a surface receptor on streptococci, is believed to mediate bacterial SAG binding. Deletion of this antigen resulted in less SAG-mediated aggregation in S. mutans (22). The suggestion that PgdA could also be involved in a specific and more direct aggregation mechanism needs further study, but the higher agglutination susceptibility and the availability of an X-ray structure could facilitate exploitation of S. mutans PgdA as a potential antistreptococcal target.
Supplementary Material
Acknowledgments
D. M. F. van Aalten is supported by a Wellcome Trust Senior Research Fellowship.
We thank A. J. Ligtenberg and J. T. D. Leito for their assistance in the SAG adherence experiments and D. E. Blair for his useful advice and technical expertise. We also thank the European Synchrotron Radiation Facility, Grenoble, France, for the time at beamline BM14.
Footnotes
Published ahead of print on 31 October 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Ajdic, D., W. M. McShan, R. E. McLaughlin, G. Savic, J. Chang, M. B. Carson, C. Primeaux, R. Tian, S. Kenton, H. Jia, S. Lin, Y. Qian, S. Li, H. Zhu, F. Najar, H. Lai, J. White, B. A. Roe, and J. J. Ferretti. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. USA 9914434-14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker, L. G., C. A. Specht, M. J. Donlin, and J. K. Lodge. 2007. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot. Cell 6855-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banks, I. R., C. A. Specht, M. J. Donlin, K. J. Gerik, S. M. Levitz, and J. K. Lodge. 2005. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot. Cell 41902-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bikker, F. J., A. J. Ligtenberg, C. End, M. Renner, S. Blaich, S. Lyer, R. Wittig, W. van't Hof, E. C. Veerman, K. Nazmi, J. M. de Blieck-Hogervorst, P. Kioschis, A. V. Nieuw Amerongen, A. Poustka, and J. Mollenhauer. 2004. Bacteria binding by DMBT1/SAG/gp-340 is confined to the VEVLXXXXW motif in its scavenger receptor cysteine-rich domains. J. Biol. Chem. 27947699-47703. [DOI] [PubMed] [Google Scholar]
- 5.Bikker, F. J., A. J. Ligtenberg, K. Nazmi, E. C. Veerman, W. van't Hof, J. G. Bolscher, A. Poustka, A. V. Nieuw Amerongen, and J. Mollenhauer. 2002. Identification of the bacteria-binding peptide domain on salivary agglutinin (gp-340/DMBT1), a member of the scavenger receptor cysteine-rich superfamily. J. Biol. Chem. 27732109-32115. [DOI] [PubMed] [Google Scholar]
- 6.Blair, D. E., O. Hekmat, A. W. Schuttelkopf, B. Shrestha, K. Tokuyasu, S. G. Withers, and D. M. van Aalten. 2006. Structure and mechanism of chitin deacetylase from the fungal pathogen Colletotrichum lindemuthianum. Biochemistry 459416-9426. [DOI] [PubMed] [Google Scholar]
- 7.Blair, D. E., A. W. Schuttelkopf, J. I. MacRae, and D. M. van Aalten. 2005. Structure and metal-dependent mechanism of peptidoglycan deacetylase, a streptococcal virulence factor. Proc. Natl. Acad. Sci. USA 10215429-15434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blair, D. E., and D. M. van Aalten. 2004. Structures of Bacillus subtilis PdaA, a family 4 carbohydrate esterase, and a complex with N-acetyl-glucosamine. FEBS Lett. 57013-19. [DOI] [PubMed] [Google Scholar]
- 9.Boneca, I. G., O. Dussurget, D. Cabanes, M. A. Nahori, S. Sousa, M. Lecuit, E. Psylinakis, V. Bouriotis, J. P. Hugot, M. Giovannini, A. Coyle, J. Bertin, A. Namane, J. C. Rousselle, N. Cayet, M. C. Prevost, V. Balloy, M. Chignard, D. J. Philpott, P. Cossart, and S. E. Girardin. 2007. A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc. Natl. Acad. Sci. USA 104997-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54905-921. [DOI] [PubMed] [Google Scholar]
- 11.Carlen, A., J. Olsson, and A. C. Borjesson. 1996. Saliva-mediated binding in vitro and prevalence in vivo of Streptococcus mutans. Arch. Oral Biol. 4135-39. [DOI] [PubMed] [Google Scholar]
- 12.Carlen, A., J. Olsson, and P. Ramberg. 1996. Saliva mediated adherence, aggregation and prevalence in dental plaque of Streptococcus mutans, Streptococcus sanguis and Actinomyces spp. in young and elderly humans. Arch. Oral Biol. 411133-1140. [DOI] [PubMed] [Google Scholar]
- 13.Deng, D. M., M. J. Liu, J. M. ten Cate, and W. Crielaard. 2007. The VicRK system of Streptococcus mutans responds to oxidative stress. J. Dent. Res. 86606-610. [DOI] [PubMed] [Google Scholar]
- 14.Emanuelsson, O., S. Brunak, G. von Heijne, and H. Nielsen. 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2953-971. [DOI] [PubMed] [Google Scholar]
- 15.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 602126-2132. [DOI] [PubMed] [Google Scholar]
- 16.Ericson, T., and J. Rundegren. 1983. Characterization of a salivary agglutinin reacting with a serotype c strain of Streptococcus mutans. Eur. J. Biochem. 133255-261. [DOI] [PubMed] [Google Scholar]
- 17.Gilmore, M. E., D. Bandyopadhyay, A. M. Dean, S. D. Linnstaedt, and D. L. Popham. 2004. Production of muramic delta-lactam in Bacillus subtilis spore peptidoglycan. J. Bacteriol. 18680-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanna, M. N., R. J. Ferguson, Y. H. Li, and D. G. Cvitkovitch. 2001. uvrA is an acid-inducible gene involved in the adaptive response to low pH in Streptococcus mutans. J. Bacteriol. 1835964-5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hebert, L., P. Courtin, R. Torelli, M. Sanguinetti, M. P. Chapot-Chartier, Y. Auffray, and A. Benachour. 2007. Enterococcus faecalis constitutes an unusual bacterial model in lysozyme resistance. Infect. Immun. 755390-5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jonasson, A., C. Eriksson, H. F. Jenkinson, C. Kallestal, I. Johansson, and N. Stromberg. 2007. Innate immunity glycoprotein gp-340 variants may modulate human susceptibility to dental caries. BMC Infect. Dis. 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47110-119. [DOI] [PubMed] [Google Scholar]
- 22.Lee, S. F., A. Progulske-Fox, G. W. Erdos, D. A. Piacentini, G. Y. Ayakawa, P. J. Crowley, and A. S. Bleiweis. 1989. Construction and characterization of isogenic mutants of Streptococcus mutans deficient in major surface protein antigen P1 (I/II). Infect. Immun. 573306-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leenhouts, K., G. Buist, A. Bolhuis, A. ten Berge, J. Kiel, I. Mierau, M. Dabrowska, G. Venema, and J. Kok. 1996. A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol. Gen. Genet. 253217-224. [DOI] [PubMed] [Google Scholar]
- 24.Li, Y. H., N. Tang, M. B. Aspiras, P. C. Lau, J. H. Lee, R. P. Ellen, and D. G. Cvitkovitch. 2002. A quorum-sensing signaling system essential for genetic competence in Streptococcus mutans is involved in biofilm formation. J. Bacteriol. 1842699-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ligtenberg, A. J., E. C. Veerman, and A. V. Nieuw Amerongen. 2000. A role for Lewis a antigens on salivary agglutinin in binding to Streptococcus mutans. Antonie van Leeuwenhoek 7721-30. [DOI] [PubMed] [Google Scholar]
- 26.Link, A. J., D. Phillips, and G. M. Church. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 1796228-6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loesche, W. J. 1986. Role of Streptococcus mutans in human dental decay. Microbiol. Rev. 50353-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lumikari, M., and J. Tenovuo. 1991. Effects of lysozyme-thiocyanate combinations on the viability and lactic acid production of Streptococcus mutans and Streptococcus rattus. Acta Odontol. Scand. 49175-181. [DOI] [PubMed] [Google Scholar]
- 29.Malik, A., and K. Kakii. 2003. Pair-dependent co-aggregation behavior of non-flocculating sludge bacteria. Biotechnol. Lett. 25981-986. [DOI] [PubMed] [Google Scholar]
- 30.O'Toole, G. A., and R. Kolter. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28449-461. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. A 276307-326. [DOI] [PubMed] [Google Scholar]
- 32.Perrakis, A., R. Morris, and V. S. Lamzin. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6458-463. [DOI] [PubMed] [Google Scholar]
- 33.Rosenberg, M., D. Gutnick, and E. Rosenberg. 1980. Adherence of bacteria to hydrocarbons: a simple method for measuring cell-surface hydrophobicity. FEMS Microbiol. Lett. 929-33. [Google Scholar]
- 34.Rundegren, J. 1986. Calcium-dependent salivary agglutinin with reactivity to various oral bacterial species. Infect. Immun. 53173-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salam, M. A., R. Nakao, H. Yonezawa, H. Watanabe, and H. Senpuku. 2006. Human T-cell responses to oral streptococci in human PBMC-NOD/SCID mice. Oral Microbiol. Immunol. 21169-176. [DOI] [PubMed] [Google Scholar]
- 36.Schneider, T. R., and G. M. Sheldrick. 2002. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 581772-1779. [DOI] [PubMed] [Google Scholar]
- 37.Taylor, E. J., T. M. Gloster, J. P. Turkenburg, F. Vincent, A. M. Brzozowski, C. Dupont, F. Shareck, M. S. Centeno, J. A. Prates, V. Puchart, L. M. Ferreira, C. M. Fontes, P. Biely, and G. J. Davies. 2006. Structure and activity of two metal-ion dependent acetyl xylan esterases involved in plant cell wall degradation reveals a close similarity to peptidoglycan deacetylases. J. Biol. Chem. 28110968-10975. [DOI] [PubMed] [Google Scholar]
- 38.Tenovuo, J., M. Lumikari, and T. Soukka. 1991. Salivary lysozyme, lactoferrin and peroxidases: antibacterial effects on cariogenic bacteria and clinical applications in preventive dentistry. Proc. Finn. Dent. Soc. 87197-208. [PubMed] [Google Scholar]
- 39.Van Nieuw Amerongen, A., J. G. Bolscher, and E. C. Veerman. 2004. Salivary proteins: protective and diagnostic value in cariology? Caries Res. 38247-253. [DOI] [PubMed] [Google Scholar]
- 40.Vollmer, W., and A. Tomasz. 2002. Peptidoglycan N-acetylglucosamine deacetylase, a putative virulence factor in Streptococcus pneumoniae. Infect. Immun. 707176-7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vollmer, W., and A. Tomasz. 2000. The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J. Biol. Chem. 27520496-20501. [DOI] [PubMed] [Google Scholar]
- 42.Vuong, C., S. Kocianova, J. M. Voyich, Y. Yao, E. R. Fischer, F. R. DeLeo, and M. Otto. 2004. A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J. Biol. Chem. 27954881-54886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.