

Abstract
Quorum sensing (QS), or cell-cell communication in bacteria, is achieved through the production and subsequent response to the accumulation of extracellular signal molecules called autoinducers (AIs). To identify AI-regulated target genes in Vibrio cholerae El Tor (V. choleraeEl), the strain responsible for the current cholera pandemic, luciferase expression was assayed in an AI− strain carrying a random lux transcriptional reporter library in the presence and absence of exogenously added AIs. Twenty-three genes were identified and shown to require the QS transcription factor, HapR, for their regulation. Several of the QS-dependent target genes, annotated as encoding hypothetical proteins, in fact encode HD-GYP proteins, phosphodiesterases that degrade the intracellular second messenger cyclic dimeric GMP (c-di-GMP), which is important for controlling biofilm formation. Indeed, overexpression of a representative QS-activated HD-GYP protein in V. choleraeEl reduced the intracellular concentration of c-di-GMP, which in turn decreased exopolysaccharide production and biofilm formation. The V. cholerae classical biotype (V. choleraeCl), which caused previous cholera pandemics and is HapR−, controls c-di-GMP levels and biofilm formation by the VieA signaling pathway. We show that the VieA pathway is dispensable for biofilm formation in V. choleraeEl but that restoring HapR in V. choleraeCl reestablishes QS-dependent repression of exopolysaccharide production. Thus, different pandemic strains of V. cholerae modulate c-di-GMP levels and control biofilm formation in response to distinct sensory pathways.
Over the past century, pandemic outbreaks of the diarrheal disease cholera have been caused by only two Vibrio cholerae biotypes (11). The classical biotype (V. choleraeCl) was responsible for the sixth pandemic, which lasted from 1899 to 1923. The etiological agent of the current seventh pandemic that began in 1961 is V. cholerae El Tor (V. choleraeEl). V. cholerae is an aquatic organism and its life cycle includes only transient colonization of the human small intestine. In humans, virulence factors elicit diarrhea, which is often fatal to the host but is also critical for efficiently transmitting the bacterium back into the environment (12). In marine habitats, V. cholerae is often found attached to other organisms in microbial communities called biofilms (19, 20). Ingestion of contaminated water or food is the mode of transmission into the human gastrointestinal tract. Understanding the mechanisms used by these two V. cholerae biotypes to control virulence factor production and biofilm formation, as well as understanding the factors responsible for the emergence of V. choleraeEl as the dominant biotype, have been the focus of active research for decades.
A bacterial cell-cell communication process called quorum sensing (QS) is used to control virulence and biofilm formation in V. choleraeEl (14, 34, 56). QS is accomplished through the synthesis, release, and subsequent detection of signaling molecules called autoinducers (AIs). Extracellular AI levels increase proportionately to increasing cell density, so the detection of AIs allows bacteria to monitor their population numbers.
V. choleraeEl strain C6706 uses two AIs, called CAI-1 and AI-2, for QS (5, 16, 17, 34, 38, 39). At low cell density, in the absence of CAI-1 and AI-2, the cognate sensor kinase receptors (CqsS and LuxP/Q, respectively) autophosphorylate and transfer phosphate to the phosphotransfer protein LuxU (Fig. 1A). LuxU transfers the phosphate to the response regulator LuxO. Phosphorylated LuxO (LuxO∼P) in conjunction with σ54 activates the transcription of genes encoding four small RNAs (sRNAs) called Qrr1-4, for quorum-regulatory RNAs. The Qrr sRNAs, along with the sRNA chaperone Hfq, destabilize the mRNA encoding the transcription factor HapR (21, 31). Thus, under low-cell-density conditions, HapR protein is not produced, so the expression of HapR-repressed genes is permitted, while HapR-activated genes are not expressed. At high cell density, CqsS and LuxP/Q bind their cognate AIs, and this event switches the sensors from acting as kinases to acting as phosphatases. Phosphate is removed from LuxO via LuxU. Dephosphorylated LuxO no longer activates transcription of the qrr genes, and in the absence of the Qrr sRNAs, hapR mRNA is translated into HapR protein. Thus, under high-cell-density conditions, HapR binds target promoter DNAs and regulates gene expression (14, 28, 34, 51, 55). In contrast to V. choleraeEl strains, which are predominantly HapR+, V. choleraeCl strain O395 carries a frameshift mutation in hapR preventing QS regulation of HapR-controlled genes (22).
FIG. 1.

Multiple regulatory pathways control biofilm formation in V. cholerae. (A) In V. choleraeEl, sensory inputs from two QS AIs, CAI-1 (squares) and AI-2 (circles), regulate HapR, which in turn modulates the levels of c-di-GMP, vps transcription, biofilm formation, and virulence. (B) In V. choleraeCl, the VieSAB sensory system, in response to an unknown signal (triangles), controls c-di-GMP levels to control vps transcription, biofilm formation, and virulence in a HapR-independent manner.
In V. choleraeEl, at high cell density, HapR represses the expression of vpsT, which encodes VpsT, a transcriptional activator of the biosynthesis operons vpsA-K and vpsL-Q, leading to a reduction in biofilm formation (4, 14, 30, 52, 53). At high cell density, QS also controls the transcription of multiple genes encoding proteins involved in modulating the levels of the intracellular second messenger molecule bis-(3′-5′)-cyclic dimeric GMP (c-di-GMP) (51). These are diguanylate cyclases that produce and phosphodiesterases that degrade c-di-GMP, respectively (for a review of c-di-GMP, see reference 41). Diguanylate cyclases have a conserved domain that includes but is not limited to the sequence motif GGDEF. Phosphodiesterases contain either an EAL domain or an HD-GYP domain. V. cholerae is predicted to encode 41 GGDEF proteins, 22 EAL proteins, and 9 HD-GYP proteins (13). The cumulative effect of HapR control of this class of genes in V. choleraeEl is to promote high levels of c-di-GMP at low cell density, contributing to biofilm formation, and low levels of c-di-GMP at high cell density, causing a reduction in biofilms (Fig. 1A). By contrast, V. choleraeCl strain O395, which is naturally HapR−, uses the phosphodiesterase VieA, a component of the VieSAB system, to control c-di-GMP (Fig. 1B) (32, 48, 50). Recently, however, we showed that this V. choleraeCl strain responds to CAI-1 and AI-2 to control the levels of a GGDEF protein through a HapR-independent pathway (15).
The above observations suggest that V. choleraeEl and V. choleraeCl could use distinct sensory pathways to control c-di-GMP levels and biofilm formation. To investigate this idea, in the current study we identified four putative HD-GYP proteins that are activated at high cell density by QS in V. choleraeEl. Consistent with this assignment of their function, overexpression of one of these, the VCA0681 protein, reduced vps expression and, in turn, biofilm formation. Degradation of c-di-GMP by VCA0681 required the canonical HD-GYP motif because mutation of this sequence hindered its capacity to destroy c-di-GMP and prevented the repression of vps transcription and biofilm formation. V. choleraeCl encodes all of the HD-GYPs present in V. choleraeEl, and the introduction of a functional HapR in V. choleraeCl restored the QS-dependent modulation of c-di-GMP levels and exopolysaccharide production. Conversely, the elimination of VieA in V. choleraeEl did not alter c-di-GMP signaling. Therefore, we propose that the different V. cholerae biotypes utilize distinct signal transduction pathways, QS in V. choleraeEl and VieA in V. choleraeCl, to coordinate c-di-GMP production and biofilm formation.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
All V. cholerae strains used were derivatives of either wild-type (WT) V. choleraeEl C6706str (49) or WT V. choleraeCl O395str (10). These included V. choleraeEl ΔluxOEl (SLS349), luxO D47EEl (SLS340), ΔhapREl (BH1543), ΔhapR ΔvieAEl (BH2728), ΔvieAEl (JC1185), and ΔcqsA ΔluxS (BH1575) mutants as well as V. choleraeCl ΔvieACl (JC1176), HapR+ ΔvieACl (BH2744), and HapR+ WT V. choleraeCl (WTCl) (BH2683) mutants. Escherichia coli S17-1λpir (6) and ElectroMAX DH10B (Invitrogen) cells were used for cloning. Bacteria were grown at 37°C with shaking, except for in bioluminescence assays, which were performed on cultures grown at 30°C, and in biofilm assays, during which cultures were not shaken. The following antibiotics, purchased from Sigma, were used (concentrations are given in parentheses in μg/ml): chloramphenicol (10), kanamycin (100), ampicillin (100) streptomycin (5000), and tetracycline (10). Expression of VCA0681, VCA0681FLAG, and VCA0681FLAG(D229A) was induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma).
DNA manipulations.
Standard protocols were used for all DNA manipulations (45). Restriction enzymes and T4 DNA ligase for cloning were purchased from New England Biolabs (NEB), and Herculase enhanced DNA polymerase for PCR amplification was purchased from Stratagene. Standard protocols (46) were used to make deletion constructs, as well as the HapR+ V. choleraeCl strains, which contained a copy of the V. choleraeEl hapR gene under the control of its own promoter at the lacZ site. Luciferase-based reporter plasmids were made by amplifying the promoters of the genes encoding the VC1295, VC1348, VC2340, VCA0210, and VCA0931 proteins from WT V. choleraeEl and cloning them into the pBBRlux vector (described previously in reference 31) by insertion into the SpeI and BamHI restriction sites. The pVCA0681 and pVCA0681FLAG vectors were constructed by cloning vca0681, containing or lacking a C-terminal FLAG epitope tag, into the EcoRI and BamHI sites of pEVS143 (9). The pVCA0681FLAG(D289A) derivative was constructed using the QuikChange II XL site-directed mutagenesis kit (Stratagene) according the instructions of the manufacturer. Western blotting of total protein was performed with monoclonal anti-Flag M2-peroxidase (horseradish peroxidase) antibody (Sigma) as described by the manufacturer. The primer sequences used for plasmid construction are available upon request.
Construction of the random lux transcriptional reporter library.
The random lux transcriptional reporter library was constructed as follows. Briefly, genomic DNA from WT V. choleraeEl was partially digested with Sau3A1 (New England Biolabs), and fragments in the size range from 1 to 2 kb were isolated by agarose gel purification using a QiaEx II gel extraction kit (Qiagen) and subsequently ligated into the BamHI site of the promoterless lux transcriptional plasmid described previously (31). The ligation mixture was transformed into E. coli strain S17-1λpir, and the E. coli transformants were conjugated with an AI-deficient V. choleraeEl ΔcqsA ΔluxS strain (BH1575) to transfer the library.
Differential bioluminescence induction.
A total of ∼40,000 V. choleraeEl ΔcqsA ΔluxS exconjugants were arrayed in microtiter plates in LB medium with a Genetix QPix2 XT colony picker and incubated overnight. Approximately 10% of the clones produced light at a level 50-fold above background and were presumed to contain active promoters. These clones were rearrayed into new microtiter plates, incubated overnight, and then combined at a 1:1 ratio with an overnight culture of an AI-deficient strain (CAI-1−, AI-2−) and, in parallel in a separate microtiter plate, with an AI-proficient strain (CAI-1+, AI-2+). Cultures were diluted 1/300 and incubated with agitation at 30°C for 8 h. This time point was chosen to ensure that strains had reached high cell density. Bioluminescence and optical density at 600 nm (OD600) were measured for each culture by use of an EnVision XCite multilabel plate reader (Perkin Elmer), and candidate AI-regulated cultures that showed differential bioluminescence between the AI-deficient and AI-proficient cultures of ≥3-fold were selected. Candidate AI-regulated clones were identified as targets if the clone displayed a similar bioluminescence regulation pattern and scale in a Wallac 1409 liquid scintillation counter (Perkin Elmer) as described previously (34). Finally, the lux reporter plasmid from each AI-regulated target clone was isolated from the V. choleraeEl ΔcqsA ΔluxS strain and electroporated into E. coli ElectroMAX DH10B cells (Invitrogen) for conjugation into V. cholerae WTEl, ΔluxOEl, luxO D47EEl, and ΔhapREl strains. Bioluminescence/OD600 (LUX/OD) was also measured for these overnight cultures.
Bioluminescence assays.
In coculture experiments, donor strains were combined at a 1:1 ratio with recipients, diluted 1/300, and incubated for 8 h. Monocultures were incubated overnight. Absorbance and bioluminescence were quantified thereafter. Bioluminescence measurements were made on a Wallac 1409 counter, except for those shown below (see Fig. 3), which were obtained using an EnVision XCite reader and multiplied by 100 to adjust for differences in reader sensitivity. LUX/OD is defined as counts min−1 ml−1/OD600.
FIG. 3.
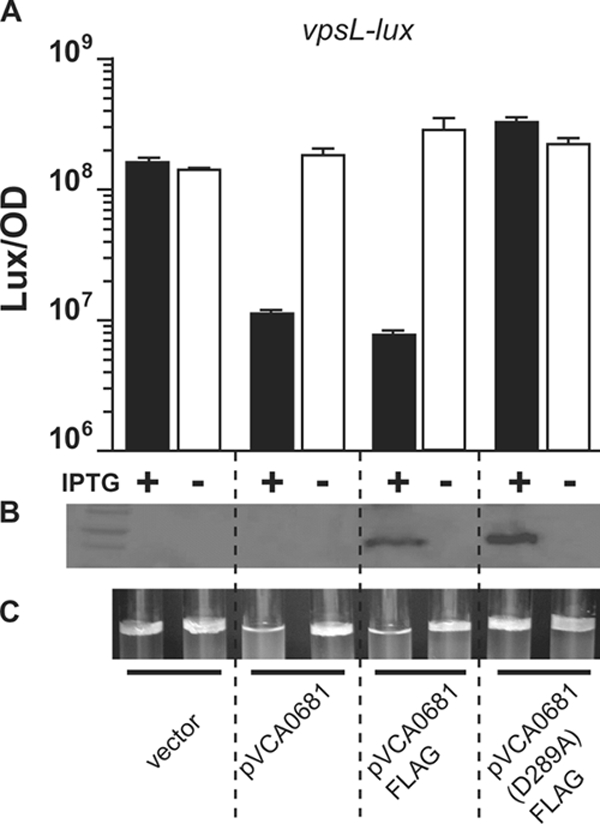
HD-GYP activity represses V. choleraeEl vpsL expression and biofilm formation. Expression of vpsL-lux in a V. choleraeEl low-cell-density mutant (luxO D47E) carrying a vector control (first pair of bars) or a vector carrying VCA0681 (second pair of bars), VCA0681-FLAG (third pair of bars), or VCA0681(D289A)-FLAG (fourth pair of bars) was measured in the presence (black bars) or absence (white bars) of IPTG. The mean LUX/OD and the standard deviation of triplicate cultures diluted 1/00 and subsequently grown for 8 h are shown. (B) Western blot of the strains shown in panel A visualized with anti-FLAG antibodies. (C) Biofilm formation of static cultures of the strains shown in panel A incubated for 48 h.
Quantification of intracellular c-di-GMP levels.
Relative levels of c-di-GMP/OD600 were determined for overnight cultures of V. cholerae WTEl, ΔluxOEl, and luxO D47EEl strains carrying the vector pEVS141, pVCA0681, or pVCA0681(D289A) as well as for V. cholerae WTCl, HapR+ WTCl, ΔvieACl, and HapR+ ΔvieACl strains and the HapR+ ΔvieACl strain carrying pVCA0681. Tandem high-pressure liquid chromatography-mass spectrometry was used to determine the relative levels of c-di-GMP, and the OD600 was also determined as described previously (51).
Biofilm formation.
Static 5-ml cultures of 1/100 dilutions of overnight cultures were incubated in LB containing chloramphenicol and IPTG where appropriate. The formation of a pellicle at the air-broth interface was assessed after ∼48 h.
RESULTS
Identification of AI-regulated genes in V. choleraeEl.
To identify QS-regulated genes in the V. choleraeEl strain C6706, we employed a strategy that simulated the gradual accumulation of AIs in cultures grown from low to high cell densities. A library of random V. choleraeEl genomic fragments fused to a promoterless luciferase cassette (Materials and Methods) was transformed into an AI-deficient V. choleraeEl strain. The transcriptional fusion library was then screened for genes controlled by AIs by combining overnight cultures of the candidates at a 1:1 ratio with an overnight culture of an AI-deficient V. choleraeEl strain (CA-1−, AI-2−) and, in parallel, with an AI-proficient V. choleraeEl strain (CAI-1+, AI-2+). Twenty-four unique candidate genes were identified that were differentially regulated ≥3-fold (Table 1).
TABLE 1.
AI-controlled target genes in V. cholerae
| Locus | Annotationa | Fold change (+AIs/−AIs) |
|---|---|---|
| VC0287 | Thermoresistant gluconokinase (gntV) | 0.11 |
| VC1185 | GGDEF family protein | 0.01 |
| VCA0025b | Transporter (nadC) | 0.3 |
| VCA0939 | Sensory box, GGDEF family protein | 0.04 |
| VCA1070 | Hypothetical protein | 0.1 |
| VC0098 | Methyl-accepting chemotaxis protein | 14 |
| VC0300b | Conserved hypothetical protein | 13 |
| VC0402 | MSHA biogenesis protein (mshJ) | 7 |
| VC1280b | Hypothetical protein | 4.6 |
| VC1362 | Amino acid ABC transporter, periplasmic amino acid-binding protein | 9.2 |
| VC1606 | Conserved hypothetical protein | 3.5 |
| VC1933 | Hypothetical protein | 8.8 |
| VC1967 | Methyl-accepting chemotaxis protein | 3.4 |
| VC2338 | Beta-galactosidase | 9.6 |
| VC2341 | Long-chain-fatty-acid-CoA ligase, putative | 3.9 |
| VCA0080 | GGDEF family protein | 10 |
| VCA0157 | NADH dehydrogenase, putative | 5.1 |
| VCA0681 | Conserved hypothetical protein | 8.4 |
| VCA0691 | Acetoacetyl-CoA reductase | 223 |
| VCA0731 | Hypothetical protein | 9.5 |
| VCA0877 | Hydrolase, putative | 4.3 |
| VCA0880 | Hypothetical protein | 85 |
| VCA0895 | Hypothetical protein | 6.9 |
| VCA0981b | Hypothetical protein | 50 |
MSHA, mannose-sensitive hemagglutinin; CoA, coenzyme A.
Isolated multiple times.
To verify QS regulation, each candidate reporter plasmid was transformed into WT V. choleraeEl and into various QS mutants that are “locked” either in the low-cell-density or in the high-cell-density mode. The “locked” low-cell-density mutant luxO D47EEl carries a constitutively active allele of luxO mimicking LuxO∼P and therefore overexpress the qrr genes, which repress hapR mRNA (Fig. 1A). By contrast, the ΔluxOEl mutant mimics high cell density because the qrr genes are not expressed, and HapR is produced constitutively. As anticipated, AI-activated targets were highly expressed in the WT and ΔluxO strains, while AI-repressed targets were highly expressed in the luxO D47E and ΔhapR strains (Table 1). Because each candidate exhibited a similar expression pattern in a luxO D47E strain and a ΔhapR strain that both simulate low cell density, our interpretation is that expression of each target gene is dependent on LuxO∼P as well as HapR. There was one exception. One QS target gene annotated VCA0939, encoding a GGDEF protein, was AI repressed and required LuxO∼P, but not HapR, for its control (15).
Because we and others are studying the link between QS and c-di-GMP in V. choleraeEl and V. choleraeCl (3, 15, 51), of particular interest to us were two of the target genes identified in this screen, vca0681 and vca0895 (AI activated ∼8-fold and ∼7-fold, respectively) (Table 1). vca0895 was previously identified as AI activated in a microarray study (56), and both vca0895 and vca0681 are predicted to encode proteins that are annotated as hypothetical. Our inspection of VCA0681 and VCA0895 show they encode proteins containing HD-GYP domains, presumably with c-di-GMP phosphodiesterase activity. Only one HD-GYP protein, RpfG, from Xanthomonas has been studied to date (8, 43, 44). We scanned the V. choleraeEl genome for homologs of RpfG. Seven proteins had high similarity; the two we identified (the VCA0681 and VCA0895 proteins) and five additional proteins (the VC1295, VC1348, VC2340, VCA0210, and VCA0931 proteins). The VC2497 protein also showed reasonable similarity to RpfG but it lacked several of the residues critical for phosphodiesterase activity and was not considered further.
Alignment of the seven putative HD-GYP proteins to RpfG shows that each contains the conserved HD and GYP residues as well as several other residues identified in the consensus sequence that are predicted to be important for phosphodiesterase activity (see Fig. S1 in the supplemental material) (13). The divergence in the N termini of the proteins is consistent with the modular domain architecture of other proteins involved in controlling c-di-GMP levels, with potential N-terminal signaling domains and C-terminal catalytic domains.
To determine whether any of the five genes not originally identified in our screen are controlled by QS, we constructed lux transcriptional fusions to each and measured expression by the above-described V. choleraeEl QS “locked” mutants. In addition to vca0681-lux and vca0895-lux, two other HD-GYP reporter fusions, vc2340-lux and vca0210-lux, are more highly expressed (∼32-fold and ∼9-fold, respectively) in WTEl and the locked high-cell-density mutant (the ΔluxOEl mutant) than in the low-cell-density mutant (the luxO D47EEl mutant) (Fig. 2). Thus, expression of genes encoding four of the seven HD-GYP proteins in V. choleraeEl is activated by QS at high cell density. Consistent with this finding, we note that vc2341, which is located adjacent to vc2340, and vca0211, which is located adjacent to vca0210, were previously identified as QS-activated genes (Table 1) (56). The remaining three fusions to genes encoding HD-GYP proteins (VC1295, VC1348, and VCA0931) either were not expressed or were not regulated by QS under our conditions, and they were not pursued further. Neither vc2340 nor vca0210 was identified in the genetic screen. We also did not identify vpsL, a known AI-controlled target. Thus, we suspect that the screen is not saturated in spite of several target genes having been identified multiple times (Table 1).
FIG. 2.
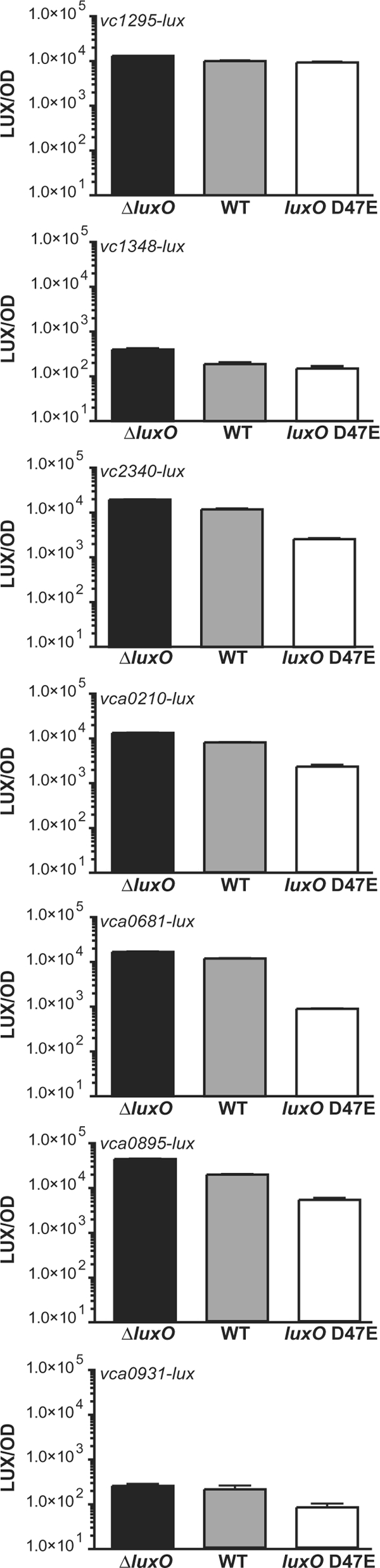
QS activates expression of four genes encoding HD-GYP proteins in V. cholerae. Expression of lux transcriptional fusions to the promoters of genes encoding predicted HD-GYP proteins was measured in the high-cell-density (WT and ΔluxO) and low-cell-density (luxO D47E) strains. Shown in each panel are the mean LUX/OD and the standard deviation from triplicate overnight cultures.
HD-GYP activity represses V. cholerae vps expression and biofilm formation.
QS control of multiple GGDEFs and EALs results in increased c-di-GMP at low cell densities and decreased c-di-GMP at high cell densities (51). c-di-GMP levels correlate with expression of the vps biosynthetic gene cluster. Thus, at high cell density, when c-di-GMP levels decline, vps transcription decreases (14, 51). Our present results suggest that if QS increases the production of four HD-GYP proteins at high cell density, because HD-GYP proteins degrade c-di-GMP, their activity could likewise contribute to the reduction in c-di-GMP and biofilm formation that occurs at high cell density. To test this idea, we examined whether the phosphodiesterase activity of the HD-GYP proteins identified in the screen, VCA0681 and VCA0895, influenced vps expression and biofilm formation. To do this, VCA0681 and VCA0895 were overexpressed in V. cholerae luxO D47EEl carrying either a vpsL-lux or a vpsT-lux transcriptional reporter. Overexpression of VCA0895 inhibited growth, which prevented our determination of its effects. Overexpression of VCA0681 reduced expression of both vpsL-lux (Fig. 3A) and vpsT-lux (data not shown). Deletion of either vca0681 or vca0895 from the V. choleraeEl genome did not alter vps expression or biofilm formation. This latter result was not unexpected, due to the extreme redundancy of phosphodiesterases.
Phosphodiesterase activity of Xanthomonas campestris RpfG requires the conserved His (H231) and Asp (D232) residues (43). We mutated the corresponding Asp in the VCA0681 protein to an Ala (D289A) and examined its effects (see Fig. S1 in the supplemental material). To monitor protein expression, a FLAG epitope tag was introduced at the C terminus. The FLAG tag that was added to the WT VCA0681 protein did not interfere with the repression of vpsL-lux (Fig. 3A). However, mutation of the active site (D289A) eliminated VCA0681 repression of vpsL-lux (Fig. 3A). Western blots showed that both the WT and mutant proteins were produced at similar levels (Fig. 3B). Thus, the putative active site of VCA0681 is required for its repression of vpsL.
To determine whether VCA0681 protein-mediated repression of vps transcription is sufficient to alter biofilm formation, we monitored biofilm pellicle formation in the V. cholerae luxO D47EEl strain carrying either VCA0681 or the active-site mutant. Consistent with the vps transcription results, biofilm formation was repressed by the WT VCA0681 protein but not the mutant protein (Fig. 3C). These results show that the phosphodiesterase activity of the representative protein we examined, VCA0681, is sufficient to reduce biofilm formation. At high cell density, AI activation of expression of vca0681 and genes encoding three other HD-GYP proteins likely contributes to the role QS plays in controlling biofilm formation (see Discussion).
The VCA0681 protein degrades c-di-GMP in V. choleraeEl.
To determine whether the decreased vps expression and reduced biofilm phenotype we observe as HD-GYP controlled (Fig. 3) are indeed a consequence of an HD-GYP-mediated reduction in c-di-GMP at high cell density, we measured c-di-GMP in V. choleraeEl expressing either VCA0681 or the active-site mutant (D289A). In high-cell-density overnight cultures, both the WT V. choleraeEl and ΔluxOEl strains had low c-di-GMP levels (Fig. 4). By contrast, the “locked” low-cell-density strain, luxO D47EEl, possessed ∼10-fold more c-di-GMP. When VCA0681 is overexpressed in the low-cell-density luxO D47EEl mutant, c-di-GMP is reduced to levels similar to those present in the WT V. choleraeEl and ΔluxOEl high-cell-density strains. Expression of the active-site mutant (D289A) in the luxO D47E strain has little effect on c-di-GMP levels, consistent with the impaired ability of this active-site mutant protein to repress vpsL expression or biofilm formation (Fig. 3).
FIG. 4.
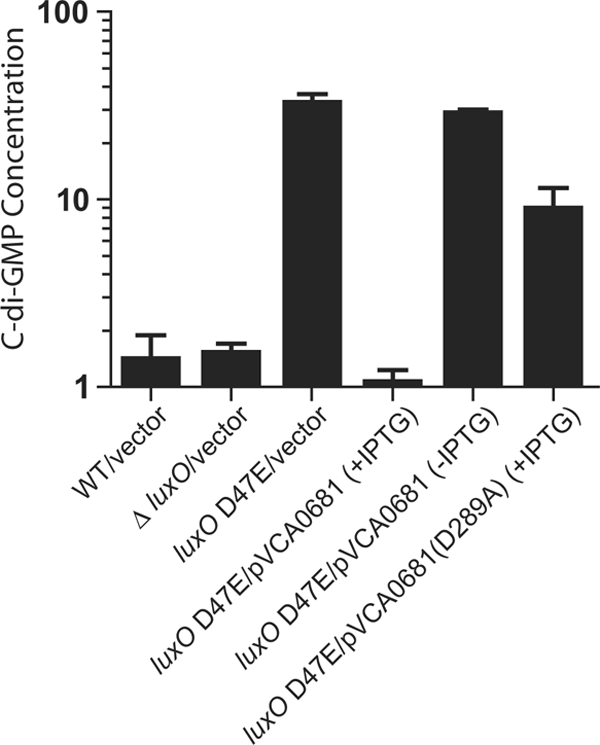
The VCA0681 protein controls c-di-GMP levels in V. choleraeEl. Relative c-di-GMP/1,000·OD600 (c-di-GMP concentration) in V. choleraeEl WT, ΔluxO, and luxO D47E strains and in the luxO D47E strain carrying IPTG-inducible vectors harboring VCA0681 or VCA0681(D289A). Shown are the mean values for triplicate cultures, with error bars indicating the standard deviation of the mean.
V. choleraeCl modulates c-di-GMP levels by a mechanism different from that of V. choleraeEl.
It has been proposed that the emergence of the V. choleraeEl biotype stems from its increased environmental fitness relative to the classical biotype (23). V. choleraeEl strain C6076 with a mutation in the hapR gene, because it is locked in the low-cell-density, low-c-di-GMP mode, is enhanced for vps production and produces robust biofilms (14, 56). By contrast, V. choleraeCl strain O395, which is naturally HapR−, does not produce strong biofilms. In this V. choleraeCl strain, deletion of the gene encoding the EAL protein VieA results in elevated vps expression and biofilm formation (50) The phosphodiesterase VieA is a component of the VieSAB signaling system that controls c-di-GMP levels and hence vps expression and biofilm formation in V. choleraeCl (32, 48, 50). While VieA controls >100 genes in V. choleraeCl, the role of VieA in V. choleraeEl appears to be minor (1). Because the ability to produce enhanced exopolysaccharide is correlated with enhanced environmental survival, we explored the relative importance of the QS and VieA signaling systems in biofilm formation in these two biotypes.
We constructed all possible combinations of V. choleraeEl and V. choleraeCl HapR+/− and VieA+/− strains. As a preliminary test for the function of HapR in each strain, we examined the canonical target luxCDABE, encoding luciferase, which is activated at high cell density in WT V. choleraeEl but not in an isogenic ΔhapR mutant (34). luxCDABE is not expressed in the V. choleraeCl parent; however, density-dependent activation of luxCDABE is restored in the HapR+ V. choleraeCl strain (see Fig. S2 in the supplemental material). Thus, the presence of HapR in V. choleraeCl restores HapR-dependent QS, at least with respect to luciferase.
To test whether the reintroduction of HapR in V. choleraeCl could likewise restore the HapR-dependent repression of vps transcription, we examined the expression of vpsL. As a reference, we measured vps transcription in the corresponding HapR+ and HapR− V. choleraeEl strains. Deletion of the hapR gene in V. choleraeEl enhanced vpsL expression 10-fold (Fig. 5A) (14). WT V. choleraeCl (HapR−) exhibits modest basal level expression of vps, and transcription is repressed ∼100-fold in HapR+ V. choleraeCl (Fig. 5A). These results show that the loss of hapR in either V. choleraeEl or V. choleraeCl relieves QS-dependent repression of vps transcription. Importantly, the absolute level of vps expression in the HapR− V. choleraeEl strain (Fig. 5A) is nearly 100 times higher than that of WT (HapR−) V. choleraeCl strain (Fig. 5A), consistent with the respective biofilm capabilities of the HapR− V. choleraeEl mutant and the naturally HapR− V. choleraeCl strain.
FIG. 5.

Expression of vpsL and levels of c-di-GMP are controlled differently in V. choleraeEl and V. choleraeCl. (A and B) Expression of vpsL-lux (A) and relative c-di-GMP levels (B) were measured for V. choleraeEl and V. choleraeCl strains containing or lacking hapR, and containing or lacking vieA. (C) Expression of vpsL-lux in V. choleraeCl ΔvieA and ΔvieA HapR+ strains carrying an IPTG-inducible vector expressing VCA0681 without or with IPTG. LUX/OD and c-di-GMP/1,000·OD600 (c-di-GMP concentration) are shown for triplicate cultures, with error bars indicating the standard deviation of the mean.
We carried out an analogous set of experiments to assess the role of VieA in both V. choleraeEl and V. choleraeCl. Mutation of vieA in either HapR+ or HapR− V. choleraeEl did not change vpsL expression (Fig. 5A). By contrast, mutation of vieA in V. choleraeCl enhanced vps transcription in both the HapR+ and HapR− strains (Fig. 5A) (50). Thus, VieA appears capable of controlling c-di-GMP and biofilms only in V. choleraeCl, whereas HapR can function in both biotypes.
In both V. choleraeEl and V. choleraeCl, elevated levels of vpsL and hence increased biofilms are a consequence of higher levels of c-di-GMP. Consistent with this, we found that the vpsL expression patterns we observed in the eight V. choleraeEl and V. choleraeCl strains (Fig. 5A) correlated with c-di-GMP levels. That is, high-level vps expression tracked with increased levels of c-di-GMP (compare Fig. 5A to 5B). The one exception to this pattern is that the extreme repression of vpsL in HapR+ V. choleraeCl is not mirrored by an equally drastic reduction in c-di-GMP. These results are consistent with our previous observations for V. choleraeEl that HapR represses vpsL transcription not only by a mechanism requiring c-di-GMP but also by repressing vpsT transcription directly through binding to the vpsT promoter (51). The HapR binding sites in the vpsT promoter region are identical in both V. choleraeEl and V. choleraeCl, and thus, the HapR-dependent repression of vpsL transcription we observed for V. choleraeCl may be due to this conserved c-di-GMP-independent regulatory mechanism. It remains possible, however, that this result occurred because we were at the lower limit of detection of c-di-GMP in V. choleraeCl.
We wondered whether we could override VieA control of vps in V. choleraeCl by artificially manipulating c-di-GMP levels through the induction of HD-GYP production in the VieA− V. choleraeCl strains. When VCA0681 expression was induced in a VieA− V. choleraeCl strain, vps levels decreased to the levels of the VieA+ V. choleraeCl strain (Fig. 5C). In HapR+ V. choleraeCl, expressing VCA0681 from a vector had less effect on vps expression than endogenous expression of vieA (compare results in Fig. 5C to those in Fig. 5A). Thus, VCA0681 cannot fully rescue a VieA defect in the HapR+ V. choleraeCl background. Nonetheless, the effect we did observe was mediated by c-di-GMP, because c-di-GMP levels were also reduced to background levels when VCA0681 is expressed (data not shown). We conclude that HD-GYP proteins, which are activated by HapR in V. choleraeEl strain C6706, may participate in both biotypes to control biofilm formation by altering c-di-GMP levels.
DISCUSSION
Multiple HapR-dependent GGDEFs and EALs participate in regulating the levels of c-di-GMP, which links QS to the control of biofilm formation in V. choleraeEl strain C6706 (14, 51). In the present work, we discovered that four AI-activated (i.e., HapR-activated) genes annotated in the genome as hypothetical are in fact genes for proteins with HD-GYP motifs predicted to encode phosphodiesterases that degrade c-di-GMP. The genes encoding these HD-GYPs are also present in the V. choleraeCl O395 genome, yet this strain is naturally HapR−. Thus, it was not obvious whether the HD-GYPs could impinge on c-di-GMP-mediated control of biofilm formation in V. choleraeCl. The difference in QS circuitry between the two V. cholerae biotypes prompted us to characterize the contribution of HapR, HD-GYP activity, c-di-GMP, and VieA to biofilm formation in V. choleraeEl and V. choleraeCl. Specifically, we showed that (i) proteins with an HD-GYP domain can alter biofilm formation in V. cholerae by reducing levels of c-di-GMP; (ii) an intact HD-GYP domain is required for this activity; (iii) HD-GYP proteins, although not activated by HapR in V. choleraeCl, when expressed are nonetheless capable of reducing c-di-GMP levels and exopolysaccharide production in this biotype; and (iv) HapR is capable of controlling c-di-GMP levels and biofilm formation in both biotypes, whereas a major input from VieA appears restricted to the V. choleraeCl biotype. Based on the results presented here, we suggest that c-di-GMP modulatory proteins play a role in biofilm formation in both V. choleraeEl and V. choleraeCl but that the signaling pathways controlling their activities differ in the two biotypes. The predominant sensory system governing the c-di-GMP/biofilm pathway in V. choleraeEl is the HapR-dependent QS circuit, while in V. choleraeCl, the VieA system is the primary sensory input (Fig. 1).
How the sensory information from the network of multiple diguanylate cyclases and phosphodiesterases in V. cholerae and other bacteria is integrated to produce a specific response mediated by c-di-GMP is poorly understood. Current theories include mechanisms for temporal control of diguanylate cyclase and phosphodiesterase gene expression, environmental control of the enzyme activities, discrete enzyme localization, and also feedback inhibition by c-di-GMP (42). V. choleraeEl was predicted by Galperin et al. (13) to encode 72 proteins that modulate c-di-GMP levels: 41 diguanylate cyclases (GGDEFs) and 31 phosphodiesterases (22 EALs and 9 HD-GYPs). Inactivation of a single diguanylate cyclase or phosphodiesterase rarely results in a dramatic phenotypic change, suggesting that phenotypes are observed only for strains in which multiple genes encoding c-di-GMP regulatory enzymes have been mutated. Alternatively, deletion of a single diguanylate cyclase or phosphodiesterase could produce a biofilm phenotype only under particular growth conditions or could alter a phenotype unrelated to biofilm formation. A few notable exceptions include genes encoding individual GGDEF proteins (CdgC, RocS, and MbaA) that, when mutated, produce an observable reduction in biofilm formation in V. choleraeEl grown under conditions similar to those in this study (2, 40). Even though many GGDEFs, EALs, and HD-GYPs are present in V choleraeCl, VieA appears to be the predominant EAL contributing to vps expression and biofilm formation, based on current studies (29, 50).
Our results suggest that the relative contributions of QS and VieA to the control of c-di-GMP vary between V. choleraeEl and V. choleraeCl (Fig. 1). In HapR+ V. choleraeEl, four HD-GYPs are expressed at high cell density. In conjunction with the GGDEFs and EALs described previously (51), the combined activities of these enzymes function to decrease c-di-GMP concentration, which reduces vps expression and terminates the formation of biofilms. HapR also represses toxin-coregulated pilin and cholera toxin production at high cell density in V. choleraeEl (34, 56). Together, these results are consistent with a model in which high-cell-density conditions in the host intestine promote decreased c-di-GMP levels, reduced biofilm formation, and the termination of toxin-coregulated pilin and cholera toxin production (14, 55). The synchronous termination of these factors enhances the transmission of V. choleraeEl from the intestine back into the environment. By contrast, c-di-GMP levels and biofilm formation are not repressed at high cell density in the naturally HapR− V. choleraeCl biotype. A possible role for QS in transmission predicts that strains of V. cholerae that are HapR+ (e.g., V. choleraeEl strain C6706) and HapR− (V. choleraeCl strain O395) should colonize with similar efficiencies at low cell density. However, upon the attainment of high cell density in the host intestine, QS in the HapR+ El Tor strains promotes detachment and transmission into the environment, while the lack of HapR in strains like V. choleraeCl hinders detachment and release from the host. Currently, however, there are no animal models to measure transmission differences between V. cholerae biotypes.
Environmental and clinical isolates of both V. choleraeEl and V. choleraeCl often contain nonfunctional HapR proteins (14, 22). We have previously proposed that such mutations could occur under conditions in which the cost incurred from the loss of HapR-mediated QS is outweighed by the gain provided by enhanced attachment via biofilm formation (14). This hypothesis would also predict that strains of V. choleraeCl should be isolated with nonfunctional VieA proteins, because they would produce increased exopolysaccharide and more-robust biofilms (Fig. 5A). However, we are aware of no incidences of natural VieA− V. choleraeCl isolates. Thus, we suspect that altered expression of some HapR-regulated target gene(s) in V. choleraeEl that is not involved in production of exopolysaccharide provides a selective advantage for HapR− V. cholerae. Based on these observations, we predict that such a putative target gene(s) is HapR regulated but not VieA regulated. We are currently measuring the selective advantage provided to QS+ compared to QS− strains under different growth conditions to identify such a target gene(s).
The history of cholera pandemics provides a model for studying the molecular mechanisms governing the emergence and reemergence of bacterial pathogens. In spite of the fact that clinical symptoms caused by V. choleraeEl are less severe than those caused by V. choleraeCl, V. choleraeEl appears to have outcompeted V. choleraeCl during the current pandemic (26). It has been proposed that this occurred because of the increased environmental fitness of V. choleraeEl (23), the differential susceptibility to lytic phages between the biotypes (47), or the ability of V. choleraeEl to more productively utilize carbohydrates such as chitin, which V. cholerae metabolizes when associated with zooplankton in the environment (33, 54). Many studies have examined the differences between these two important pandemic biotypes in terms of virulence genes and their expression (1, 7, 10, 18, 24, 25, 27, 36, 37), but less attention has been given to understanding the differences in biofilm formation between different V. cholerae biotypes (1, 35). Because the life cycle of V. cholerae includes only transient colonization of the human host, a better understanding of the selective pressures driving the evolution of this pathogen in aquatic habitats where vibrios are autochthonous members could contribute to our understanding of pandemic strains. Environmental attachment is proposed to be a crucial factor in the survival of V. cholerae in natural systems; therefore, understanding the signal transduction mechanisms controlling biofilm formation by V. cholerae in pathogenic and marine niches should shed light on the mechanisms underlying the emergence and reemergence of this infectious pathogen.
Supplementary Material
Acknowledgments
We thank J. Cong for strain construction and W. Ng for assistance with c-di-GMP quantification.
This work was supported by HHMI, by NIH grants 5R01GM065859 and 5R01A1054442, by NSF grant MCB-0343821 to B.L.B., and by an NIH postdoctoral fellowship (F32-AI054033-01) to B.K.H.
Footnotes
Published ahead of print on 24 October 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Beyhan, S., A. D. Tischler, A. Camilli, and F. H. Yildiz. 2006. Transcriptome and phenotypic responses of Vibrio cholerae to increased cyclic di-GMP level. J. Bacteriol. 1883600-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bomchil, N., P. Watnick, and R. Kolter. 2003. Identification and characterization of a Vibrio cholerae gene, mbaA, involved in maintenance of biofilm architecture. J. Bacteriol. 1851384-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camilli, A., and B. L. Bassler. 2006. Bacterial small-molecule signaling pathways. Science 3111113-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casper-Lindley, C., and F. H. Yildiz. 2004. VpsT is a transcriptional regulator required for expression of vps biosynthesis genes and the development of rugose colonial morphology in Vibrio cholerae O1 El Tor. J. Bacteriol. 1861574-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, X., S. Schauder, N. Potier, A. Van Dorsselaer, I. Pelczer, B. L. Bassler, and F. M. Hughson. 2002. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 415545-549. [DOI] [PubMed] [Google Scholar]
- 6.de Lorenzo, V., and K. N. Timmis. 1994. Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 235386-405. [DOI] [PubMed] [Google Scholar]
- 7.DiRita, V. J., M. Neely, R. K. Taylor, and P. M. Bruss. 1996. Differential expression of the ToxR regulon in classical and E1 Tor biotypes of Vibrio cholerae is due to biotype-specific control over toxT expression. Proc. Natl. Acad. Sci. USA 937991-7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dow, J. M., L. Crossman, K. Findlay, Y. Q. He, J. X. Feng, and J. L. Tang. 2003. Biofilm dispersal in Xanthomonas campestris is controlled by cell-cell signaling and is required for full virulence to plants. Proc. Natl. Acad. Sci. USA 10010995-11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn, A. K., D. S. Millikan, D. M. Adin, J. L. Bose, and E. V. Stabb. 2006. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl. Environ. Microbiol. 72802-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dziejman, M., E. Balon, D. Boyd, C. M. Fraser, J. F. Heidelberg, and J. J. Mekalanos. 2002. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc. Natl. Acad. Sci. USA 991556-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faruque, S. M., M. J. Albert, and J. J. Mekalanos. 1998. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 621301-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faruque, S. M., and G. B. Nair. 2002. Molecular ecology of toxigenic Vibrio cholerae. Microbiol. Immunol. 4659-66. [DOI] [PubMed] [Google Scholar]
- 13.Galperin, M. Y., A. N. Nikolskaya, and E. V. Koonin. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 20311-21. [DOI] [PubMed] [Google Scholar]
- 14.Hammer, B. K., and B. L. Bassler. 2003. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol. Microbiol. 50101-104. [DOI] [PubMed] [Google Scholar]
- 15.Hammer, B. K., and B. L. Bassler. 2007. Regulatory small RNAs circumvent the conventional quorum sensing pathway in pandemic Vibrio cholerae. Proc. Natl. Acad. Sci. USA 10411145-11149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henke, J. M., and B. L. Bassler. 2004. Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J. Bacteriol. 1866902-6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higgins, D. A., M. E. Pomianek, C. M. Kraml, R. K. Taylor, M. F. Semmelhack, and B. L. Bassler. 2007. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature 450883-886. [DOI] [PubMed] [Google Scholar]
- 18.Hung, D. T., J. Zhu, D. Sturtevant, and J. J. Mekalanos. 2006. Bile acids stimulate biofilm formation in Vibrio cholerae. Mol. Microbiol. 59193-201. [DOI] [PubMed] [Google Scholar]
- 19.Huq, A., E. B. Small, P. A. West, M. I. Huq, R. Rahman, and R. R. Colwell. 1983. Ecological relationships between Vibrio cholerae and planktonic crustacean copepods. Appl. Environ. Microbiol. 45275-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huq, A., P. A. West, E. B. Small, M. I. Huq, and R. R. Colwell. 1984. Influence of water temperature, salinity, and pH on survival and growth of toxigenic Vibrio cholerae serovar 01 associated with live copepods in laboratory microcosms. Appl. Environ. Microbiol. 48420-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jobling, M. G., and R. K. Holmes. 1997. Characterization of hapR, a positive regulator of the Vibrio cholerae HA/protease gene hap, and its identification as a functional homologue of the Vibrio harveyi luxR gene. Mol. Microbiol. 261023-1034. [DOI] [PubMed] [Google Scholar]
- 22.Joelsson, A., Z. Liu, and J. Zhu. 2006. Genetic and phenotypic diversity of quorum-sensing systems in clinical and environmental isolates of Vibrio cholerae. Infect. Immun. 741141-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaper, J. B., J. G. Morris, Jr., and M. M. Levine. 1995. Cholera. Clin. Microbiol. Rev. 848-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karaolis, D. K., R. Lan, J. B. Kaper, and P. R. Reeves. 2001. Comparison of Vibrio cholerae pathogenicity islands in sixth and seventh pandemic strains. Infect. Immun. 691947-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karaolis, D. K., R. Lan, and P. R. Reeves. 1995. The sixth and seventh cholera pandemics are due to independent clones separately derived from environmental, nontoxigenic, non-O1 Vibrio cholerae. J. Bacteriol. 1773191-3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolvin, J. L., and D. Roberts. 1982. Studies on the growth of Vibrio cholerae biotype eltor and biotype classical in foods. J. Hyg. (London) 89243-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovacikova, G., and K. Skorupski. 2000. Differential activation of the tcpPH promoter by AphB determines biotype specificity of virulence gene expression in Vibrio cholerae. J. Bacteriol. 1823228-3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovacikova, G., and K. Skorupski. 2002. Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol. Microbiol. 461135-1147. [DOI] [PubMed] [Google Scholar]
- 29.Lee, S. H., S. M. Butler, and A. Camilli. 2001. Selection for in vivo regulators of bacterial virulence. Proc. Natl. Acad. Sci. USA 986889-6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lenz, D. H., M. B. Miller, J. Zhu, R. V. Kulkarni, and B. L. Bassler. 2005. CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol. Microbiol. 581186-1202. [DOI] [PubMed] [Google Scholar]
- 31.Lenz, D. H., K. C. Mok, B. N. Lilley, R. V. Kulkarni, N. S. Wingreen, and B. L. Bassler. 2004. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 11869-82. [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Wilson, H. F., R. Tamayo, A. D. Tischler, D. W. Lazinski, and A. Camilli. 2008. The Vibrio cholerae hybrid sensor kinase VieS contributes to motility and biofilm regulation by altering the cyclic diguanylate level. J. Bacteriol. 1906439-6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meibom, K. L., M. Blokesch, N. A. Dolganov, C. Y. Wu, and G. K. Schoolnik. 2005. Chitin induces natural competence in Vibrio cholerae. Science 3101824-1827. [DOI] [PubMed] [Google Scholar]
- 34.Miller, M. B., K. Skorupski, D. H. Lenz, R. K. Taylor, and B. L. Bassler. 2002. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell 110303-314. [DOI] [PubMed] [Google Scholar]
- 35.Mueller, R. S., D. McDougald, D. Cusumano, N. Sodhi, S. Kjelleberg, F. Azam, and D. H. Bartlett. 2007. Vibrio cholerae strains possess multiple strategies for abiotic and biotic surface colonization. J. Bacteriol. 1895348-5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murley, Y. M., J. Behari, R. Griffin, and S. B. Calderwood. 2000. Classical and El Tor biotypes of Vibrio cholerae differ in timing of transcription of tcpPH during growth in inducing conditions. Infect. Immun. 683010-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murley, Y. M., P. A. Carroll, K. Skorupski, R. K. Taylor, and S. B. Calderwood. 1999. Differential transcription of the tcpPH operon confers biotype-specific control of the Vibrio cholerae ToxR virulence regulon. Infect. Immun. 675117-5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neiditch, M. B., M. J. Federle, S. T. Miller, B. L. Bassler, and F. M. Hughson. 2005. Regulation of LuxPQ receptor activity by the quorum-sensing signal autoinducer-2. Mol. Cell 18507-518. [DOI] [PubMed] [Google Scholar]
- 39.Neiditch, M. B., M. J. Federle, A. J. Pompeani, R. C. Kelly, D. L. Swem, P. D. Jeffrey, B. L. Bassler, and F. M. Hughson. 2006. Ligand-induced asymmetry in histidine sensor kinase complex regulates quorum sensing. Cell 1261095-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rashid, M. H., C. Rajanna, A. Ali, and D. K. Karaolis. 2003. Identification of genes involved in the switch between the smooth and rugose phenotypes of Vibrio cholerae. FEMS Microbiol. Lett. 227113-119. [DOI] [PubMed] [Google Scholar]
- 41.Romling, U., and D. Amikam. 2006. Cyclic di-GMP as a second messenger. Curr. Opin. Microbiol. 9218-228. [DOI] [PubMed] [Google Scholar]
- 42.Romling, U., M. Gomelsky, and M. Y. Galperin. 2005. c-di-GMP: the dawning of a novel bacterial signalling system. Mol. Microbiol. 57629-639. [DOI] [PubMed] [Google Scholar]
- 43.Ryan, R. P., Y. Fouhy, J. F. Lucey, L. C. Crossman, S. Spiro, Y. W. He, L. H. Zhang, S. Heeb, M. Camara, P. Williams, and J. M. Dow. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. USA 1036712-6717. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Ryan, R. P., Y. Fouhy, J. F. Lucey, B. L. Jiang, Y. Q. He, J. X. Feng, J. L. Tang, and J. M. Dow. 2007. Cyclic di-GMP signalling in the virulence and environmental adaptation of Xanthomonas campestris. Mol. Microbiol. 63429-442. [DOI] [PubMed] [Google Scholar]
- 45.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 46.Skorupski, K., and R. K. Taylor. 1996. Positive selection vectors for allelic exchange. Gene 16947-52. [DOI] [PubMed] [Google Scholar]
- 47.Takeya, K., T. Otohuji, and H. Tokiwa. 1981. FK phage for differentiating the classical and El Tor groups of Vibrio cholerae. J. Clin. Microbiol. 14222-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamayo, R., A. D. Tischler, and A. Camilli. 2005. The EAL domain protein VieA is a cyclic diguanylate phosphodiesterase. J. Biol. Chem. 28033324-33330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thelin, K. H., and R. K. Taylor. 1996. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. 642853-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tischler, A. D., and A. Camilli. 2004. Cyclic diguanylate (c-di-GMP) regulates Vibrio cholerae biofilm formation. Mol. Microbiol. 53857-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waters, C. M., W. Lu, J. D. Rabinowitz, and B. L. Bassler. 2008. Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT. J. Bacteriol. 1902527-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yildiz, F. H., N. A. Dolganov, and G. K. Schoolnik. 2001. VpsR, a member of the response regulators of the two-component regulatory systems, is required for expression of vps biosynthesis genes and EPSETr-associated phenotypes in Vibrio cholerae O1 El Tor. J. Bacteriol. 1831716-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yildiz, F. H., and G. K. Schoolnik. 1999. Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc. Natl. Acad. Sci. USA 964028-4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoon, S. S., and J. J. Mekalanos. 2006. 2,3-Butanediol synthesis and the emergence of the Vibrio cholerae El Tor biotype. Infect. Immun. 746547-6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu, J., and J. J. Mekalanos. 2003. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev. Cell 5647-656. [DOI] [PubMed] [Google Scholar]
- 56.Zhu, J., M. B. Miller, R. E. Vance, M. Dziejman, B. L. Bassler, and J. J. Mekalanos. 2002. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 993129-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.