

Abstract
Coronary arterioles from hypercholesterolemic swine display attenuated adenosine-mediated vasodilatation that is attributable to the elimination of voltage-dependent K+ (Kv) channel stimulation. For the present study, we tested the hypotheses that exercise training would correct impaired adenosine-induced dilatation in coronary arterioles from hypercholesterolemic pigs through restoration of adenosine activation of Kv channels and that vasodilatation to the receptor-independent adenylyl cyclase activator, forskolin, would also be attenuated in arterioles from hypercholesterolemic pigs. Pigs were randomly assigned to a control (NC) or high-fat, high-cholesterol (HC) diet for 20 wk. Four weeks after the diet was initiated, pigs from both groups were assigned to exercise training (Ex; 5 days/wk for 16 wk) or sedentary (Sed) protocols, resulting in four groups of pigs: NC-Sed, NC-Ex, HC-Sed, and HC-Ex. Arterioles (∼150 μm) from both HC-Sed and HC-Ex pigs displayed impaired adenosine-mediated dilatation that was attributable to the elimination of 4-aminopyridine (4-AP; 1 mM)-sensitive Kv channel activation compared with NC counterparts. Arteriolar smooth muscle whole cell Kv currents were significantly reduced in HC-Sed compared with NC-Sed, although HC-Ex and NC-Ex did not differ. Forskolin-mediated dilatation was attenuated by 4-AP (1 mM) and in a concentration-dependent manner by tetraethylammonium (TEA; 0.1–1 mM) in NC-Sed but not HC-Sed. Further, TEA-sensitive Kv currents were diminished in cells of HC-Sed compared with NC-Sed pigs. Quantitative RT-PCR revealed similar expression levels of Kv3.1 and 3.3 in arterioles of NC-Sed and HC-Sed swine with undetectable expression of Kv1.1, 3.2, and 3.4. Taken together, these results suggest that hypercholesterolemia-mediated attenuation of adenosine-induced vasodilatation in coronary arterioles is not corrected by exercise training and is likely attributable to an impairment in the pathway coupling adenylyl cyclase with a highly TEA-sensitive Kv channel isoform(s).
Keywords: electrophysiology, forskolin, vascular smooth muscle
hypercholesterolemic, asymptomatic subjects display reduced coronary flow reserve in response to intravenous infusion of adenosine or dipyridamole (12, 13, 33, 53). Interestingly, coronary flow reserve displays a significant negative correlation with increasing concentrations of both serum total and LDL cholesterol levels (13, 53). In vitro studies (22, 24, 39, 42) have also provided evidence of altered vascular reactivity in the coronary microcirculation in hypercholesterolemia. Specifically, studies (22) from our laboratory have demonstrated that adenosine-mediated vasodilatation is significantly attenuated in coronary arterioles isolated from hypercholesterolemic compared with control swine and is attributable to the elimination of adenosine stimulation of voltage-dependent K+ (Kv) channels in hypercholesterolemic animals. Based on previous investigations (24, 45, 48, 49) that have shown beneficial effects of exercise training on vascular reactivity, one purpose of the current study was to determine the effect of exercise training on Kv channel contribution to adenosine-mediated vasodilatation in coronary arterioles of hypercholesterolemic pigs. We hypothesized that exercise training would correct impaired adenosine-induced dilatation in coronary arterioles through restoration of adenosine activation of Kv channels.
Additionally, adenosine has been reported to mediate dilation through both endothelium-dependent nitric oxide production (23) and the endothelium-independent adenylyl cyclase pathway in smooth muscle (43). While our previous studies (20) demonstrated a role for nitric oxide in adenosine-mediated vasodilatation, nitric oxide synthase inhibition did not alter the dependence of adenosine on Kv channels for vasodilatation in arterioles from control swine, indicating a pathway independent of nitric oxide mediated Kv channel contribution to adenosine-induced dilatation. Thus another purpose of the current study was to examine the dependence of Kv-mediated vasodilatation on adenylyl cyclase activation using the membrane-permeant, receptor-independent adenylyl cyclase activator, forskolin. We also sought to determine the effect of hypercholesterolemia on forskolin-mediated dilatation in coronary arterioles. Therefore, we further hypothesized that forskolin-mediated dilatation would be dependent on Kv channel activation and that this mechanism of vasodilatation would be impaired in arterioles from hypercholesterolemic pigs.
Data from our previous study (20, 22) also revealed that the Kv channel isoforms stimulated by adenosine were sensitive to blockade by both the classic Kv channel blocker 4-aminopyridine (4-AP; 1 mM) and the nonselective K+ channel blocker tetraethylammonium (TEA; 1 mM) but not the selective large-conductance, Ca2+-dependent K+ channel (BKCa) blocker iberiotoxin (100 nM) or the selective KATP channel inhibitor glibenclamide (10 μM; Ref. 20). These data are consistent with a significant contribution of TEA-sensitive Kv channel isoforms to adenosine-mediated relaxation in the coronary microcirculation and with the notion that the attenuated dilatation in hypercholesterolemia appears to be attributable to impaired activation of TEA-sensitive Kv channels. Review of the literature suggests that at a concentration of 1 mM TEA produces marked inhibition of only a small number of K+ channels, including BKCa, KCNQ2, Kv1.1, and the Kv3 subfamily, including Kv3.1, 3.2, 3.3, and 3.4 (for review, see Refs. 8, 9). Since KCNQ2 is insensitive to 4-AP and the selective BKCa channel blocker iberiotoxin did not alter adenosine-mediated vasodilatation, we hypothesized that the 4-AP-sensitive and highly TEA-sensitive Kv channel isoforms, Kv1.1, and/or the Kv3 subfamily contribute to adenosine-mediated dilatation in the coronary microcirculation and initiated studies to examine the contribution of candidate isoforms to adenosine-mediated vasodilatation.
MATERIALS AND METHODS
Experimental animals.
Male Yucatan miniature swine were obtained from the breeder (Sinclair Farms, Auxvasse, MO) and housed in animal facilities at Texas A&M University Veterinary Medicine Park. Animal protocols were approved by the Texas A&M University Institutional Animal Care and Use Committee and in accordance with the “Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training.” Animals were randomly assigned to either a control or high-fat, high-cholesterol (HC) diet for 20 wk. Pigs on the control diet were fed Laboratory Mini-Pig Breeder Chow (PMI Feeds) with calories provided by 23% protein, 8% fat, and 69% carbohydrate. The HC diet consisted of Mini-Pig Chow supplemented (by weight) with 2.0% cholesterol, 17.1% coconut oil, 2.4% corn oil, and 0.7% sodium cholate with calories provided by 13% protein, 46% fat, and 41% carbohydrate. Pigs were fed to maintain a matched body mass throughout the study. Animals were fed 15–20 g/kg once daily and water was provided ad libitum.
Blood (∼20 ml) was obtained via venapuncture from the anterior vena cava at baseline (before initiation of dietary protocol), at 30 days after initiation of diet, and at termination. Pigs were anesthetized for blood collection with glycopyrrolate (0.004 mg/kg im), followed 10 min later by xylazine (2 mg/g im), followed 15 min later by a mixture of ketamine HCl (5 mg/g) and butorphanol tartrate (0.22 mg/g) administered intramuscularly. After blood collection was complete, yohimbine HCl (0.05 mg/g iv) was administered as an antagonist of the above anesthetic mixture to expedite recovery from the anesthetic. Blood sampling occurred after a 12- to 18-h fast. Serum cholesterol analyses were performed by the Comparative Pathology Laboratory at the University of California at Davis.
Exercise training.
Animals were randomly assigned to either sedentary or exercise training groups. Exercise-trained pigs underwent a progressive treadmill exercise training program, 5 days/wk for 16 wk, as described previously (24). Sedentary animals remained pen confined throughout the 16-wk duration. Effectiveness of the exercise training program was determined by comparing heart-to-body weight ratio and skeletal muscle citrate synthase activity as previously described (24).
Isolation of coronary arterioles.
After completion of the dietary and exercise training protocols, animals were anesthetized using ketamine (35 mg/kg im), rompun (2.25 mg/kg im), and pentothal sodium (20 mg/kg iv), followed by administration of heparin (1,000 U/kg iv). Animals were euthanized by removal of hearts, which were immediately placed in cold (0–4°C) Krebs bicarbonate buffer. The left ventricular free wall, near the apical region of the heart, was isolated and placed in a chilled (0–4°C) dissection chamber containing the following physiological saline solution (PSS; in mM): 138 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 20 HEPES, pH 7.4. Arterioles (∼150 μm in luminal diameter; ∼1.5 mm in length) were dissected free of surrounding myocardium with the aid of a dissection microscope and cannulated and pressurized or enzymatically dissociated for measurement of smooth muscle whole cell Kv currents.
Microvessel cannulation and videodimensional instrumentation.
For vascular reactivity studies, arterioles were transferred to a Lucite vessel chamber containing PSS and cannulated on one end with a glass micropipette filled with PSS-albumin. The arteriole was tied securely to the pipette using 11-0 ophthalmic suture and gently flushed, and the other end was cannulated with a second micropipette and tied. PSS-albumin contained the following (in mM): 145 NaCl, 4.7 KCl, 2 CaCl2, 1.17 MgSO4, 1.2 NaH2PO4, 5 glucose, 2 pyruvate, 0.02 EDTA, 3 MOPS pH 7.4, and 1 g/100 ml BSA. The cannulated arteriole was transferred to the stage of an inverted microscope (Olympus CKX41) equipped with a ×10 objective (numerical aperature of 0.25) and coupled with a video camera (Hitachi KP-M3AN), video monitor (Sony PVM97), and video micrometer (Colorado Video 307). Both micropipettes were connected to a single reservoir system adjusted to set the intraluminal pressure of the arteriole at 40 mmHg without allowing flow through the vessel lumen. Leaks were detected by verifying that intraluminal diameter of the pressurized arteriole remained constant when the valve to the reservoir system was closed. Only arterioles that were free of leaks were studied. The vessel chamber bath (PSS-albumin) was gradually warmed and maintained at 37 °C for the duration of the experiment. Luminal diameter was monitored continuously throughout the experiment.
Experimental protocol.
Arterioles underwent a 1-h equilibration period at 40 mmHg during which time the vessels established a stable basal tone. Arterioles were further preconstricted with endothelin-1 until a preconstriction level of ∼40–60% maximal diameter was attained. For experiments in which a K+ channel blocker was present, vessels were preconstricted to the same level (∼40–60%) using the inhibitor plus endothelin-1 as described previously (22). Adenosine and forskolin concentration-response relationships were determined by cumulative additions of concentrated stock solutions directly to the tissue bath. Adenosine or forskolin concentration was increased when the response to the previous concentration had stabilized. The order of the concentration-response curves (in the absence and presence of K+ channel blockade) was randomized to control for potential changes in vessel responsiveness over time. At the completion of the experimental protocol, maximal (passive) intraluminal diameters (Dp) of pressurized coronary arterioles were measured in Ca2+-free PSS containing 1 mM EGTA plus the Ca2+ channel blocker nifedipine (2 μM). All drugs applications were made to the tissue bath.
Smooth muscle cell dissociation.
All electrophysiology experiments were performed using freshly dispersed arteriolar smooth muscle cells. Coronary arterioles were placed in low-Ca2+ (0.1 mM) physiological buffer containing 294 U/ml collagenase, 5 U/ml elastase, 2 mg/ml BSA, 1 mg/ml soybean trypsin inhibitor, and 0.4 mg/ml DNase I. Cells were enzymatically dissociated by incubation in a 37°C water bath for 45 min. The enzyme solution was then replaced with enzyme-free low-Ca2+ solution, and the arterioles were dispersed with gentle trituration by micropipette for isolation of single smooth muscle cells. Smooth muscle cells were morphologically distinguishable from other cell types in the dispersion, such as endothelial cells and fibroblasts. Isolated cells were maintained in low-Ca2+ solution at 4°C until use (0–6 h).
Whole cell voltage clamp.
Whole cell K+ currents were obtained from single cells using standard whole cell voltage-clamp techniques as used routinely (20, 22). Experiments were conducted under physiological K+ concentrations. Because membrane depolarization activates both Kv and BKCa channels, we used low extracellular Ca2+ (0.1 mM) and 10 mM EGTA in the pipette to chelate intracellular Ca2+ and thereby minimized the contribution of BKCa current to outward K+ current (46). We also limited the depolarizing command pulses to +20 mV to minimize activation of BKCa channels (31). The contribution of KATP channels to whole cell K+ current was minimized by inclusion of 2 mM ATP in the pipette solution. These conditions allowed us to isolate Kv currents (20, 31, 46). Cells were initially superfused with PSS containing the following (in mM): 138 NaCl, 5 KCl, 0.1 CaCl2, 1 MgCl2, 10 glucose, and 20 HEPES, pH 7.4. Heat-polished glass pipettes (2–5 MΩ) were filled with a solution containing the following (in mM): 120 KCl, 10 NaCl, 1 MgCl2, 10 EGTA, 10 HEPES, 2 Na2ATP, and 0.5 Tris-GTP, pH 7.1 with KOH. Ionic currents were amplified with an Axopatch 200B patch-clamp amplifier (Axon Instruments). Currents were low-pass filtered with a cutoff frequency of 1,000 Hz, digitized at 2.5 kHz, and stored on a computer. Data acquisition and analysis were accomplished using pClamp 9.0 software (Axon Instruments). Leak subtraction was not performed. Cells were continuously perfused under gravity flow at room temperature (22–25°C).
Quantitative RT-PCR.
Primer sets used to amplify candidate Kv channel isoforms from porcine coronary arterioles were designed based primarily on published human sequences from the National Center for Biotechnology Information GenBank database (Table 1). Sus scrofa sequences were used when available. Expression of GAPDH was used as an internal reference. Conventional RT-PCR was performed in preliminary experiments to optimize MgCl2 concentration and annealing temperature for each primer pair. Further, PCR-amplified products were electrophoresed on a 1.5% agarose gel and visualized with ethidium bromide staining. PCR products of Kv channel isoforms were identified by size and nucleotide sequenced to confirm amplification of predicted Kv channel isoforms. Yucatan brain cDNA was used as positive control given that the candidate Kv isoform genes are expressed in brain (8).
Table 1.
Oligonucleotide sequence of primers used for RT-PCR experiments
| Isoform | Expected Fragment Size, bp | Sense and Antisense | Location |
|---|---|---|---|
| Kv1.1 | 389 | TTC TCC TTC GAG CTT GTG GT | 694–713 |
| (NM_000217) | TCG GGG ATA CTG GAA AAG TG | 1,063–1,082 | |
| Kv3.1 | 122 | TGT AAA TCT GTC GTA AAC TCT CC | 1,480–1,502 |
| (NM_004976) | CAG ATC GAC ATG CCT CTA AG | 1,581–1,601 | |
| Kv3.2 | 319 | ACT ACT CCT TGG CAA TGG C | 1,457–1,475 |
| (NM_139136) | CAC GTG TAA TCA CCT GTC G | 1,757–1,775 | |
| Kv3.3 | 197 | GCT CAG GAG GAG GTG ATT GA | 2,240–2,259 |
| (NM_004977) | GCA TAG TCG GTG AGG AGG AA | 2,417–2,436 | |
| Kv3.4 | 268 | GCC TTT GCC TCT CTC TTC TT | 841–860 |
| (NM_004978) | CGA TGA TGT TGA GCA GGT TC | 1,089–1,108 | |
| GAPDH | 91 | CCC TAC TGC CAA CGT GTC G | 1,040–1,058 |
| (AF017079) | CGC CTG CTT CAC CAC CTT C | 1,112–1,130 |
GenBank accession number is shown in parentheses.
Total RNA was extracted from coronary arterioles (8–10 arterioles per animal) using the RNeasy micro kit (Qiagen), including RNase-free DNase treatment. RNA yield and purity were determined by either spectrophotometer or Agilent 2100 bioanalyzer. First-strand cDNA synthesis was performed using RT and oligo(dT)20 to prime the reaction (Superscript III First Strand Synthesis System; Invitrogen). Quantitative real-time RT-PCR was performed using iQ SYBR Green Supermix (Bio-Rad) and both sense and anti-sense primers (500 nM each) and 10 μl cDNA (30 ng reverse transcribed total RNA) in 25 μl of total reaction volume. The PCR reaction was performed in an iQ5 real-time PCR detection system (Bio-Rad) with an initial denaturation at 95°C, followed by 35 cycles at 95°C (30 s), 60°C (30 s), and 72°C (30 s). At the end of the amplification reaction, melt curves were generated to confirm amplification of a single product. The 2−ΔΔCt method was used to compare expression of Kv channel isoforms in arterioles from control and hypercholesterolemic animals, using the following equation:
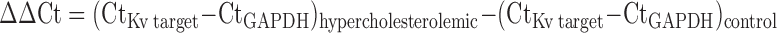 |
The mean threshold cycle (Ct) values for duplicate samples of the candidate Kv channel isoform and for the reference gene (GAPDH) were determined for each animal.
Drugs and solutions.
Stock solution of adenosine, dendrotoxin-K, blood depressing substance (BDS)-I, iberiotoxin, and TEA were prepared in distilled H2O. Stock solution of 4-AP was prepared with distilled H2O and 1 M HCl to a final pH of 7.4. Forskolin stock was prepared in DMSO. Endothelin-1 stock was prepared in PSS. Nifedipine stock solution was prepared in ethanol. Vehicle concentrations did not exceed 0.1%. Chemicals were obtained from Sigma Chemical unless otherwise noted. Smooth muscle cell dispersion chemicals were obtained from Worthington Chemicals, and endothelin-1 was from Peninsula Laboratories.
Data analysis.
For endothelin-1 preconstriction, data are presented as percent possible constriction, [(DP − DSS)/DP] × 100, where DP is the passive internal diameter and DSS is the steady-state internal diameter in the presence of endothelin-1. One-way ANOVA was used to evaluate differences between group means where a single parameter was evaluated. Dilatation responses to adenosine and forskolin are presented as the percent increase in internal diameter relative to the maximal possible relaxation [(DSS − DB)/(DP − DB)] × 100, where DB is the endothelin-1-preconstricted baseline diameter to normalize for differences in initial and passive diameters between vessels. Concentration-response curves and current-voltage (I>V) relationships were analyzed using two-way ANOVA with repeated measures. Voltage-clamp data was analyzed on a per cell basis to account for potential cell-to-cell heterogeneity in smooth muscle channel function. Mean differences were ascertained using Bonferroni multiple comparison tests when either the main interaction or drug effect was significant. For all analyses, a P value <0.05 was considered significant. Data are presented as means ± SE, and n values in parentheses reflect the number of animals for concentration-response curves and the number of animals, cells for voltage-clamp studies.
RESULTS
Effectiveness of the HC diet.
Animal body weight at the time of termination was not significantly different between groups with the exception of HC-sedentary (Sed) compared with NC-exercised-trained (Ex) animals (Table 2). Total serum cholesterol and LDL cholesterol levels were both significantly elevated by 4 wk after initiation of the HC diet compared with levels in the animals maintained on a normal cholesterol diet and remained at greater levels throughout the study (Table 2) as described previously (22).
Table 2.
Animal characteristics
| NC-Sed | NC-Ex | HC-Sed | HC-Ex | |
|---|---|---|---|---|
| Body weight, kg | 42.9±1.1 (40) | 39.9±1.1 (20) | 45.5±1.1† (37) | 41.8±1.2 (16) |
| Heart wt/body wt, g/kg | 4.3±0.1 | 5.6±0.1‡ | 3.9±0.1 | 5.6±0.2‡ |
| Total cholesterol, mg/dl | ||||
| Baseline | 64±2 (40) | 56±4 (20) | 61±3 (36) | 58±3 (15) |
| 4 wk | 58±2 | 51±4 | 366±21* | 353±45* |
| Termination | 58±2 | 58±2 | 416±25* | 433±40* |
| LDL cholesterol, mg/dl | ||||
| Baseline | 23±2 (24) | 21±5 (5) | 23±2 (22) | 24±1 (8) |
| 4 wk | 19±2 | 15±3 | 167±17* | 196±33* |
| Termination | 19±2 | 22±2 | 190±18* | 218±22* |
| Citrate synthase, μmol·min−1·g−1 Deltoid | 15.8±1.2 (22) | 20.7±1.1‡ (20) | 14.7±0.8 (21) | 22.4±1.3‡ (16) |
| Triceps brachii | ||||
| Long head | 12.4±0.9 | 18.9±1.9‡ | 11.5±0.8 | 16.1±1.3‡ |
| Lateral head | 9.7±0.8 | 14.9±1.1‡ | 11.1±0.9 | 17.4±1.1‡ |
| Medial head | 13.5±0.9 | 18.8±1.5‡ | 15.2±1.0 | 22.4±0.8‡ |
Values are mean ± SE. Numbers in parentheses are the number of animals studied for each data subset. NC, normal cholesterol; HC, hypercholesterolemic; Sed, sedentary; Ex, exercise trained.
P ≤ 0.05 HC vs. NC;
P ≤ 0.05 HC-Sed vs. NC-Ex;
P ≤ 0.05 Ex vs. Sed.
Efficacy of the exercise training program.
Effectiveness of the 16-wk exercise training regimen was demonstrated by significant increases in skeletal muscle oxidative enzyme capacity and heart-to-body weight ratios in exercise-trained compared with sedentary pigs in both NC and HC groups (Table 2).
Characteristics of arterioles.
Maximal (passive) intraluminal diameters (DP) of cannulated coronary arterioles measured at 40 mmHg of intraluminal pressure in Ca2+-free PSS plus nifedipine were not significantly different between the four animal groups (Table 3). The level of preconstriction was similar across groups with the exception of arterioles from the NC-Ex compared with the NC-Sed and HC-Sed as indicated in Table 3. Importantly, data from our laboratory (n = 33) suggest that varying the level of preconstriction does not alter adenosine concentration-response curves (data not shown). The concentration of endothelin-1 required to attain this level of preconstriction was not significantly different between arterioles from the four animal groups (Table 3).
Table 3.
Dimensional characteristics of coronary arterioles
| NC-Sed (n = 67) | NC-Ex (n = 15) | HC-Sed (n = 33) | HC-Ex (n = 14) | |
|---|---|---|---|---|
| Passive diameter, μm | 146±5 | 173±10 | 147±6 | 164±11 |
| %Preconstriction | 58±2 | 45±8* | 64±2 | 49±6 |
| ET-1, [nM] | 0.5±0.1 | 0.5±0.1 | 0.5±0.1 | 0.5±0.1 |
Values are mean ± SE; n indicates number of arterioles studied. NC, normal cholesterol; HC, hypercholesterolemic; Sed, sedentary; Ex, exercise trained; %preconstriction, %maximal diameter to which arterioles were preconstricted; ET-1, concentration of endothelin-1 required to obtain %preconstriction.
Significantly lower level of preconstriction than NC-Sed and HC-Sed.
Adenosine-mediated vasodilatation.
The effects of hypercholesterolemia and exercise training on adenosine-mediated vasodilatation in coronary arterioles are presented in Fig. 1. Concentration-dependent dilatation in response to adenosine was significantly attenuated in arterioles from HC-Sed compared with NC-Sed pigs (Fig. 1A), as we have reported previously (22). Similarly, adenosine-induced vasodilatation was significantly impaired in arterioles from HC-Ex compared with NC-Ex animals (Fig. 1B). Comparison of Fig. 1A and Fig. 1B demonstrates that exercise training does not alter the response of coronary arterioles to adenosine in NC or HC pigs.
Fig. 1.

Effect of hypercholesterolemic diet and exercise training on adenosine-mediated concentration-response curves in porcine coronary arterioles. Adenosine-mediated vasodilatation was significantly attenuated in arterioles from hypercholesterolemic (HC) compared with control (NC) animals in both sedentary (Sed; A) and exercise-trained (Ex; B) groups. Arterioles were preconstricted with endothelin-1. Values are means ± SE of the number of animals in parentheses. *P ≤ 0.05.
Kv channel contribution to basal tone and adenosine-mediated vasodilatation.
We also explored the contribution of Kv channels to basal vascular tone and adenosine-mediated dilatation in coronary arterioles from all animal groups (Fig. 2). The reduction in basal diameter in response to the classic Kv channel blocker 4-AP (1 mM) was statistically similar in coronary arterioles from all groups [NC-Sed: 34.9 ± 4.5% (n = 7); NC-Ex: 33.4 ± 8.9% (n = 5); HC-Sed: 31.2 ± 4.0% (n = 12); and HC-Ex: 31.6 ± 7.1% (n = 10)]. Pretreatment with 4-AP significantly attenuated adenosine-induced dilatation in arterioles from NC-Sed (Fig. 2A) and NC-Ex (Fig. 2B) pigs. In contrast, 4-AP did not alter dilatation responses in coronary arterioles from HC-Sed (Fig. 2C) and HC-Ex (Fig. 2D) pigs, suggesting that adenosine activation of Kv channels is eliminated in hypercholesterolemia and not restored with concomitant exercise training.
Fig. 2.

Effect of Kv channel blockade on adenosine-mediated concentration-response curves in porcine coronary arterioles. Adenosine-mediated vasodilatation was significantly attenuated by the classic Kv channel inhibitor 4-aminopyridine (4-AP) in arterioles from both NC-Sed (A) and NC-Ex (B) animals. In contrast, Kv channel blockade did not significantly alter adenosine-induced dilatation in coronary arterioles from hypercholesterolemic pigs of sedentary (HC-Sed; C) and exercise-trained (HC-Ex; D) groups. A subset of data in 2C has been published previously (19). Arterioles were preconstricted with endothelin-1. Values are means ± SE of the number of animals in parentheses. *P ≤ 0.05.
Whole cell Kv channel current.
To examine potential mechanisms behind the loss of Kv channel contribution to adenosine-mediated vasodilatation, we determined the effects of hypercholesterolemia and exercise training on coronary arteriolar smooth muscle Kv channel currents. Previous studies (40, 41, 52) have reported that endothelin-1 alters Kv channel currents in pulmonary and coronary arterial smooth muscle cells. Thus we anticipated a potential need for inclusion of endothelin-1 in our voltage-clamp experiments to maintain consistent experimental conditions with our isolated arteriole experiments. However, we found in preliminary studies that endothelin-1, at concentrations of 0.1 and 1 nM, did not alter Kv channel currents from control levels in arteriolar smooth muscle cells from our pigs (data not shown). Thus endothelin-1 was not included in our voltage-clamp experiments. As presented in Fig. 3, currents were elicited by 500-ms step depolarizations to potentials ranging from −60 to +20 mV from a holding potential of −80 mV (Fig. 3A). Current is plotted as the mean value of the outward current for the last 50 ms of each test potential and is normalized to cell membrane capacitance (pA/pF). Cell capacitance was significantly (P < 0.05) greater in smooth muscle cells isolated from HC-Ex (13.7 ± 0.8 pF; n = 49) compared with NC-Ex (11.5 ± 0.4 pF; n = 89) pigs, whereas all other comparisons were not statistically different [NC-Sed: 12.0 ± 0.4 pF (n = 126) and HC-Sed: 13.1 ± 0.4 pF (n = 114)]. Comparison of the I–V relationships indicated significantly reduced whole cell Kv currents in cells from HC-Sed compared with NC-Sed animals (Fig. 3B). In contrast, I–V relationships were not significantly different between cells from HC-Ex and NC-Ex pigs (Fig. 3C). These data indicate that a reduction in whole cell Kv channel availability may be partially responsible for the loss of adenosine activation of Kv channels in arterioles from HC-Sed but not in arterioles from HC-Ex animals.
Fig. 3.

Effect of hypercholesterolemia on whole cell Kv current in coronary arteriolar smooth muscle cells. A: representative current traces for whole cell Kv current of cells from NC-Sed, HC-Sed, NC-Ex, and HC-Ex animals. Currents were elicited by 500-ms step depolarizations (tp) to potentials ranging from −60 to +20 (in 10-mV increments) from a holding potential (hp) of −80 mV. B and C: comparison of current-voltage (I-V) relationships obtained by plotting mean current at the end of the steps as a function of the indicated test potential. Whole cell Kv current was significantly diminished in myocytes from HC-Sed compared with NC-Sed animals (B). In contrast, Kv currents in cells from NC-Ex and HC-Ex did not differ (C). A subset of data in B has been published previously (19). Numbers in parentheses indicate number of pigs, cells. Values are means ± SE of the number of cells in parentheses. *P ≤ 0.05.
Effect of TEA on whole cell Kv currents.
We previously reported that the nonselective K+ channel blocker TEA (1 mM) significantly attenuated adenosine-mediated vasodilatation in arterioles from NC-Sed but not HC-Sed, whereas selective blockers of BKCa channels (iberiotoxin; 100 nM) and KATP channels (glibenclamide; 10 μM) did not alter adenosine-mediated dilatation. One of our goals for the present study was to thoroughly compare TEA-sensitive Kv currents in arteriolar cells from NC-Sed and HC-Sed pigs. As detailed in materials and methods, we designed whole cell voltage-clamp conditions to optimize measurement of Kv currents with minimal contribution of BKCa currents. These conditions allowed us to evaluate K+ channel blocker effects primarily on Kv currents (20, 46). We demonstrate that under these conditions a contribution of BKCa channels to total outward current was not evident at test potentials negative or equal to +20 mV (Fig. 4, A and B), as evidenced by the lack of inhibition of outward K+ current by iberiotoxin in the superfusate and thus the use of iberiotoxin in subsequent experiments was unnecessary. In contrast to our findings with iberiotoxin, we demonstrate that the nonselective K+ channel blocker TEA decreased Kv currents in a concentration-dependent manner, supporting our assertion that TEA effectively inhibits a subset of Kv channel isoforms (Fig. 4A). It is also noteworthy that >40% of the whole cell Kv current was inhibited by 1 mM TEA, supporting our previous observations in porcine arteriolar smooth muscle cells (20).
Fig. 4.

Concentration-dependent effect of the K+ channel blocker tetraethylammonium (TEA) on whole cell K+ currents in coronary arteriolar smooth muscle cells. A: representative current traces from a cell of a control pig demonstrating that outward K+ current was not affected by inclusion of iberiotoxin (IbTx; 100 nM) in superfusate at step depolarizations to +20 mV (hP = −80 mV) indicating no contamination of the selective large-conductance, Ca2+-dependent K+ channel (BKCa) current in whole cell measures [control current in presence (a) and absence (b) of IbTx, respectively]. In contrast, in the presence of IbTx, TEA effectively inhibited K+ current in a concentration-dependent manner [0.1 (c), 0.3 (d), 0.5 (e), and 1 (f) mM TEA, respectively]. B: mean current data (n = 13 cells) illustrating lack of effect of IbTx on whole cell Kv currents at Vm = +20 mV. C. Mean data representing TEA-sensitive currents in cells from NC-Sed and HC-Sed pigs. Data are average of 12–26 cells from 4–6 animals. Values are means ± SE. *P ≤ 0.05 HC-Sed vs. NC-Sed.
TEA-sensitive currents were obtained by subtraction of currents in the presence of TEA from control currents (difference currents) in cells from both NC-Sed and HC-Sed animals. Currents were elicited by 500-ms step depolarizations to +20 mV from a holding potential of −80 mV as shown in Fig. 4A. Comparison of the TEA-sensitive current indicated that cells from HC-Sed pigs tended to display reduced currents when compared with NC-Sed pigs at each TEA concentration, although statistical significance was attained only at 500 μM TEA (Fig. 4C). Additionally, treatment with 500 μM TEA abolished the difference between whole cell Kv currents in cells from NC-Sed and HC-Sed pigs. These data suggest that smooth muscle cells from HC-Sed animals exhibit reduced expression and/or function of a highly TEA-sensitive Kv channel that may potentially be activated by adenosine.
Forskolin-mediated vasodilatation.
We previously reported that adenosine activation of Kv channels is independent of nitric oxide (20) suggesting that adenosine may activate Kv channels through the adenylyl cyclase/cAMP/PKA pathway. We therefore sought to examine the effect of hypercholesterolemia on the vasodilatory response to the receptor-independent activator of adenlyl cyclase, forskolin. Concentration-dependent vasodilatation in response to forskolin in arterioles from NC-Sed and HC-Sed animals are compared in Fig. 5. These data demonstrate that forskolin-mediated vasodilatation was significantly attenuated in arterioles from HC-Sed compared with NC-Sed pigs (Fig. 5A). Furthermore, administration of 4-AP (1 mM) or TEA (1 mM) significantly attenuated forskolin-mediated vasodilatation in arterioles from NC-Sed (Fig. 5B) but not HC-Sed (Fig. 5C) pigs. Indeed, after 4-AP or TEA pretreatment, forskolin-mediated vasodilatation in arterioles from NC-Sed animals was virtually superimposable on forskolin-mediated vasodilatation in arterioles from HC-Sed pigs. Evaluation of the concentration-dependent effect of TEA on forskolin-induced dilatation revealed that 100 μM TEA tended to attenuate the vasodilatation response to forskolin (P = 0.08), whereas both 300 and 500 μM TEA significantly impaired forskolin-mediated dilatation in arterioles from NC-Sed animals (Fig. 6). Taken together, these data provide evidence that forskolin and adenosine are coupled with a highly TEA-sensitive Kv channel in arterioles from control pigs and the loss of this coupling in hypercholesterolemia is independent of the G protein-coupled adenosine receptor.
Fig. 5.

Effect of hypercholesterolemic diet on forskolin-mediated concentration-response curves in porcine coronary arterioles. A: forskolin-mediated vasodilatation was significantly attenuated in arterioles from HC-Sed compared with NC-Sed. Forskolin-mediated vasodilatation was significantly attenuated by 4-AP and TEA in arterioles from NC-Sed (B) but not HC-Sed (C) animals. Arterioles were preconstricted with endothelin-1. Values are means ± SE of the number of animals in parentheses. *P ≤ 0.05.
Fig. 6.

Concentration-dependent effect of the K+ channel blocker, TEA, on forskolin-mediated vasodilatation in coronary arterioles from NC-Sed pigs. TEA attenuated forskolin-mediated dilatation in a concentration-dependent manner in arterioles from NC-Sed pigs. Values are means ± SE of the number of animals in parentheses. *P ≤ 0.05, 300 μM TEA vs. CTL; †P ≤ 0.05, 500 μM TEA vs. CTL.
mRNA detection of candidate Kv channel isoforms.
RT-PCR experiments were performed to determine the expression of the candidate highly TEA-sensitive Kv channel isoforms in coronary arterioles. These experiments established that transcripts of Kv3.1 and Kv3.3 were expressed in coronary arterioles, whereas Kv1.1, Kv3.2, and Kv3.4 mRNA were not detected (Fig. 7A). Quantitative real-time RT-PCR analysis revealed that expression levels of Kv3.1 and Kv3.3 were not altered by the hypercholesterolemic diet (Fig. 7B). As a positive control, all candidate Kv channel isoforms (Fig. 7C) and GAPDH (Fig. 7D) were amplified using cDNA prepared from Yucatan brain. PCR products were identified by size and nucleotide sequenced to confirm amplification of genes of interest. Furthermore, completion of additional functional experiments using pharmacological inhibitors selective for Kv1.1, dendrotoxin-K (10 nM; Refs. 3, 19), and Kv3.4, BDS-I (100 nM; Ref. 15), demonstrated no effect of these inhibitors on vasodilatory responses to adenosine or forskolin in NC-Sed or HC-Sed animals, as well as no effect of these blockers on whole cell Kv currents, providing confirmation that these isoforms are absent from porcine coronary arteriolar smooth muscle (data not shown).
Fig. 7.

RT-PCR analysis of candidate Kv channel isoform expression in coronary arterioles. A: RT-PCR experiments established that transcripts of Kv3.1 and Kv3.3 were expressed in coronary arterioles, whereas Kv1.1, Kv3.2, and Kv3.4 mRNA were not detected. B: quantitative real-time PCR analysis revealed that expression levels of Kv3.1 and Kv3.3 were not altered by the hypercholesterolemic diet as determined using the 2−ΔΔCT method. Expression levels are reported relative to GAPDH in the same samples. C: RT-PCR amplification of all candidate isoforms with cDNA prepared from porcine brain mRNA as a positive control. PCR-amplified products were electrophoresed on a 1.5% agarose gel and visualized with ethidium bromide staining. Marker is 123-bp DNA ladder. D: amplification of GAPDH from Yucatan brain in the presence (+RT) and absence (−RT; negative control) of reverse transcriptase in the reaction mixture. A no-template control sample is also included using GAPDH primers. Marker is 50-bp DNA ladder. Values in B are means ± SE of the number of animals in parentheses. P ≤ 0.05.
DISCUSSION
These studies are the first to document that exercise training does not correct attenuated adenosine-mediated vasodilatation in coronary arterioles of hypercholesterolemic animals and that the attenuated dilatation is attributable to impaired adenosine activation of Kv channels in arterioles of both HC-Sed and HC-Ex pigs. These studies also document the novel finding that forskolin-mediated vasodilatation is impaired in coronary arterioles of hypercholesterolemic animals and that this attenuated dilation is attributable to the reduced contribution of a highly TEA-sensitive Kv channel isoform(s). These data advance our previous findings (22) that adenosine-induced dilatation is significantly attenuated in arterioles of sedentary hypercholesterolemic pigs and provide indirect evidence that the altered response is downstream of the G protein-coupled adenosine receptor.
Hypercholesterolemia has been reported to be associated with altered vascular reactivity and ion channel function of the coronary microcirculation (22, 24, 29, 30, 39, 42). Both hypercholesterolemic patients and animal models display impaired vasodilatory responses of the coronary microcirculation that are generally attributed to endothelium dysfunction (16, 39, 54). However, numerous studies (12, 13, 33, 53) have documented reduced coronary flow reserve in hypercholesterolemic patients in response to intravenous infusion of adenosine or dipyridamole, which also act directly on smooth muscle. These reports indicate that changes in vascular reactivity of the coronary microcirculation in response to hypercholesterolemia may also be attributable to alterations in smooth muscle function, independent of endothelial function. Furthermore, in vitro studies have revealed impaired relaxation responses to endothelium-independent vasodilators (22, 36) and enhanced contractile reactivity (11, 17, 45) in vascular segments, providing evidence of altered smooth muscle function in hypercholesterolemia.
Numerous investigations have demonstrated reduced K+ channel activity in coronary arteries (18, 22, 30, 32, 34) and other vascular beds (11, 25) under hypercholesterolemic conditions, contributing to blunted vasodilatory responses of affected arteries (11, 22, 30, 34). It is noteworthy in our study that basal Kv channel activity appeared unaltered by hypercholesterolemia; an adaptation that may be novel to the microcirculation (4). Furthermore, our laboratories have previously demonstrated that exercise training increases the contribution of Kv channels to basal active tone in epicardial coronary arteries from both control (6) and chronically occluded (21) pigs. On the contrary, the contribution of Kv channels to basal tone was not altered by exercise training in the present study, suggesting heterogeneous adaptations in Kv channels in the macrocirculation compared with the microcirculation. Vascular K+ channels are key contributors to vasomotor responsiveness. Increased smooth muscle K+ channel opening and subsequent K+ efflux produce membrane hyperpolarization, closing voltage-gated calcium channels and decreasing calcium entry, leading to vasodilatation. Thus modifications in K+ channel availability or function ultimately alters the vasodilatory response of affected arteries.
Few studies (14, 28, 37) have examined the molecular identity of Kv channel isoforms in the coronary circulation. We specifically set out to determine the expression of candidate Kv channel isoforms that may contribute to adenosine- and forskolin-induced vasodilatation in the coronary microcirculation. Candidate Kv channel isoforms that are sensitive to 4-AP and highly sensitive to TEA include Kv1.1 and the Kv3 subfamily of isoforms (8, 9). Examination of mRNA expression of these isoforms revealed that only Kv3.1 and Kv3.3 were amplified from coronary arteriolar mRNA. Transcripts of Kv1.1, Kv3.2, and Kv3.4 were not detected in our preparation. Undetectable levels of Kv1.1 and Kv3.4 mRNA are supported by our findings that selective inhibition of these isoforms using dendrotoxin-K and BDS-I, respectively, did not alter whole cell K+ current in coronary arteriolar smooth muscle cells. Previous studies (47, 50, 51) have reported expression of Kv3 subfamily isoforms in various vascular tissues; however, the role of these isoforms in vasculature has not been explored attributable to the lack of selective pharmacological blockers for most of the Kv3 family. Furthermore, other investigators (14) recently demonstrated a significant role for Kv channels in blood flow responses to intracoronary adenosine infusion, as well as in the regulation of resting coronary blood flow and reactive hyperemia in dogs. These studies also demonstrated a significant role for the Kv1 subfamily of isoforms in adenosine-mediated vasodilatation in isolated coronary arterioles. While our data reveal that the Kv1.1 isoform does not contribute to adenosine-mediated dilatation in porcine coronary arterioles, the role of other isoforms from the Kv1 subfamily was not evaluated. Data from our study suggest that adenosine and forskolin are coupled with a highly TEA-sensitive Kv channel isoform in arterioles from NC, but not HC, pigs. Although the literature suggests that the Kv1 channel subfamily, with the exception of Kv1.1, is fairly insensitive to TEA, future examination of the Kv1 subfamily in our studies may be of interest.
Although we observed reduced whole cell Kv current in arteriolar smooth muscle cells isolated from HC-Sed compared with NC-Sed pigs, Kv currents were not significantly altered in NC-Ex compared with HC-Ex animals. Taken together, these data suggest that altered Kv channel availability does not fully account for the impaired adenosine-mediated dilatation in arterioles from HC-Ex pigs. However, we pursued measurement of TEA-sensitive current in cells from NC-Sed and HC-Sed animals to determine if a potential reduction in TEA-sensitive channel activity might contribute to impaired adenosine- and forskolin-mediated vasodilatation in coronary arterioles from sedentary pigs. TEA-sensitive currents were reduced in cells from HC-Sed compared with NC-Sed, suggesting that a reduction in the availability of TEA-sensitive Kv channels may contribute to the attenuated adenosine-mediated vasodilatation in arterioles from HC-Sed pigs. However, the lack of a reduction in whole cell Kv currents in HC-Ex compared with NC-Ex suggests another mechanism is likely the primary mediator behind the attenuation of Kv channel activation by adenosine in hypercholesterolemia. Thus we also initiated studies to further pursue a potential role for the adenlyl cyclase/cAMP/PKA pathway in the coupling of adenosine with Kv channels. Studies using the receptor-independent, membrane-permeant activator of adenlyl cyclase forskolin provided similar results to those found with adenosine indicating that an impairment in the adenlyl cyclase/cAMP/PKA cascade that is downstream of the G-protein coupled adenosine receptor as the key mechanism by which adenosine activation of Kv channels is lost with hypercholesterolemia.
Recent studies (52) have examined whole cell K+ channel currents in smooth muscle cells from large coronary arteries of pigs similar to the animals used in our studies. These studies demonstrated that K+ currents were not significantly altered by hypercholesterolemia or exercise training compared with sedentary normal cholesterol-fed animals. In contrast, hypercholesterolemia significantly reduced K+ currents in exercise-trained animals. While the effect of hypercholesterolemia on K+ currents of exercise-trained pigs in cells of the large arteries is substantially different from our findings in arteriolar cells, methodological considerations may have contributed. For example, the previous investigation (52) utilized a greater range of depolarizing commands (−70 to +80 mV) than the present study. Limiting depolarizing commands to +20 mV to isolate Kv channel currents, as in the present study, may have produced a different statistical outcome. As the depolarizing commands become more positive, the contribution of BKCa currents to whole cell K+ currents increases markedly as demonstrated by the iberiotoxin-sensitive current in the previous study (52). Lastly, the previous study (52) utilized smooth muscle cells isolated from large epicardial coronary arteries, whereas the present study uses cells isolated from coronary arterioles. Our group (4, 5) has previously demonstrated heterogenous adaptations in ion channel currents between smooth muscle cells of coronary conduit arteries compared with coronary arterioles. Thus unique adaptations between arteries of different caliber are not unfounded.
An increasing body of evidence generated from our laboratory (20, 22) and others (1, 2, 10, 26) indicates that cAMP-dependent PKA and substances known to enhance cellular PKA levels stimulate Kv channels in coronary and other smooth muscle cell types. Stimulation of β-adrenoceptors (isoproterenol) or adenylyl cyclase (forskolin) both produce a reversible increase in a 4-AP-sensitive, delayed rectifier current that is inhibited by a specific peptide inhibitor of PKA (PKI) in myocytes from both rabbit coronary artery and portal vein (2, 10). Additional studies (1) by these investigators indicate PKA-mediated phosphorylation of an ∼15-pS delayed rectifier channel in portal vein. Other studies (26) have reported that the catalytic subunit of PKA increases the open probability of an ∼20-pS delayed rectifier channel in smooth muscle cells from the colonic circular layer. The conductances of these channels are similar to that observed for Kv1.1 and the Kv3 subfamily when expressed in Xenopus oocyte expression systems (10–27 pS; Ref. 9); however, the specific Kv channel isoform(s) activated by PKA in native tissue has not been determined. While our pursuit of the contribution of these candidate isoforms was limited by the lack of selective pharmacological inhibitors, we were able to definitely eliminate Kv1.1 and Kv3.4 as candidate isoforms for activation by both forskolin and adenosine. It is important to note that the family of Kv channels is quite extensive and the TEA sensitivity of all Kv channel isoforms has not been fully realized; thus determination of the specific Kv channel isoform(s) activated by adenosine may prove difficult.
Limitations
Data from the present study demonstrate a significant contribution of Kv channels to both adenosine- and forskolin-mediated vasodilatation in coronary arterioles from control swine that is abolished in arterioles from hypercholesterolemic animals. However, our studies did not examine the effects of hypercholesterolemia on adenosine or forskolin activation of Kv currents under voltage-clamp conditions. Studies (7) by other investigators using a rodent model of diabetes mellitus demonstrated that forskolin increased whole cell Kv currents in coronary smooth muscle cells of control but not diabetic rats, suggesting that the coupling of adenylyl cyclase with Kv channels is also impaired in diabetes. Similarly, we would anticipate adenosine activation of Kv channel currents in arteriolar smooth muscle cells of control pigs that is abolished by hypercholesterolemia.
Clinical Implications
In addition to our previous observation that coronary arterioles from hypercholesterolemic pigs display diminished responsiveness to adenosine, our current findings indicate a much broader effect of hypercholesterolemia on arteriolar vasodilatation. Like adenosine, forskolin mediates its effects via activation of the adenylyl cyclase pathway. However, forskolin is a membrane-permeant, receptor-independent activator of adenylyl cyclase (38), providing evidence that the impaired coupling of adenylyl cyclase with plasmalemmal Kv channels is independent of receptor activation. This novel finding suggests that other agonists that mediate vasodilatory effects via the coupling of the adenylyl cyclase pathway with Kv channels may be impaired in pathological conditions such as hypercholesterolemia. Studies (27) by other investigators have indicated that pathophysiological conditions associated with oxidative stress impair PKA-mediated vasodilatation of coronary arterioles, attributed in part, to reduced activity of Kv channels. The deleterious effects of hypercholesterolemia on vascular reactivity of coronary arterioles have also been associated with increased oxidative stress (35), suggesting increased oxidative stress associated with hypercholesterolemia as a potential mechanism for the impaired coupling of the adenylyl cyclase pathway with Kv channels observed in the present study. Identification of the mechanisms by which agonist-mediated activation of Kv channels is impaired in early stages of coronary artery disease, such as hypercholesterolemia, may provide viable K+ channel targets for regulation of coronary microvascular tone and improvements in coronary flow reserve.
It is also important to note that the effects of hypercholesterolemia on coronary flow reserve in our porcine model have not been determined; however, other investigators (44) also utilizing a porcine model of hypercholesterolemia have demonstrated significantly reduced endomyocardial coronary flow reserve in hypercholesterolemic compared with control pigs, whereas epicardial flow reserve was not significantly impaired. Arterioles for the present study were isolated primarily from the subepicardium, and although our data demonstrate a tendency for reduced maximal adenosine-mediated vasodilatation in arterioles from hypercholesterolemic compared with control animals (Fig. 1), these differences are not statistically significant. However, taken together, these data lead us to speculate that our observation of impaired responsiveness to adenosine may be exacerbated in the endocardium and thus may contribute to reductions in coronary flow reserve.
Conclusion
Results from the current study indicate that exercise training does not correct the impaired adenosine-induced vasodilatation observed in hypercholesterolemia. Furthermore, attenuated forskolin-mediated vasodilatation in coronary arterioles from hypercholesterolemic swine is attributable to impaired activation of TEA-sensitive Kv channels. We propose several potential hypotheses regarding the reduced contribution of these channels to impaired forskolin-mediated vasodilatation. First, the signaling cascade coupling forskolin and adenosine to highly TEA-sensitive Kv channels may be impaired in arterioles from hypercholesterolemic pigs, suggesting altered function of the adenylyl cyclase second messenger pathway. Second, the candidate Kv channels may be modified in hypercholesterolemic conditions in such a way that reduces the activation (e.g., phosphorylation) of these channels. Third, a complete loss of function of a candidate TEA-sensitive isoform could account fully for the reduced TEA-sensitive current in arterioles from HC-Sed pigs, but this does not resolve the lack of a reduction in whole cell Kv current in HC-Ex animals, despite attenuation of Kv channel contribution to adenosine-mediated dilatation. Further studies will be required to determine the precise mechanism by which hypercholesterolemia abolishes agonist-mediated activation of voltage-dependent K+ channels.
GRANTS
These studies were supported by research funds from the American Heart Association Grant 0330252N (to C. L. Heaps), National Heart, Lung, and Blood Institute Grants HL-70500 (to C. L. Heaps) and HL-52490 (to D. K. Bowles and E. M. Price), and Centers for Disease Control and Prevention Grant 623086 (to G. A. Laine).
Acknowledgments
We thank Rebecca Shaw, Pam Thorne, David Harrah, Erin Ashmore, and Mildred Mattox for technical contributions.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Aiello EA, Malcolm AT, Walsh MP, Cole WC. β-adrenoceptor activation and PKA regulate delayed rectifier K+ channels of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 275: H448–H459, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Aiello EA, Walsh MP, Cole WC. Phosphorylation by protein kinase A enhances delayed rectifier K+ current in rabbit vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 268: H926–H934, 1995. [DOI] [PubMed] [Google Scholar]
- 3.Akhtar S, Shamotienko O, Papakosta M, Ali F, Dolly JO. Characteristics of brain Kv1 channels tailored to mimic native counterparts by tandem linkage of αsubunits. J Biol Chem 277: 16376–16382, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Bowles DK, Heaps CL, Turk JR, Maddali KK, Price EM. Hypercholesterolemia decreases L-type Ca2+ current in coronary macro-, not microcirculation. J Appl Physiol 96: 2240–2248, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Bowles DK, Hu Q, Laughlin MH, Sturek M. Heterogeneity of L-type calcium current density in coronary smooth muscle. Am J Physiol Heart Circ Physiol 273: H2083–H2089, 1997. [DOI] [PubMed] [Google Scholar]
- 6.Bowles DK, Laughlin MH, Sturek M. Exercise training increases K+ channel contribution to regulation of coronary arterial tone. J Appl Physiol 84: 1225–1233, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Bubolz AH, Li H, Wu Q, Liu Y. Enhanced oxidative stress impairs cAMP-mediated dilation by reducing Kv channel function in small coronary arteries of diabetic rats. Am J Physiol Heart Circ Physiol 289: H1873–H1880, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Chandy KG, Gutman GA. Voltage-gated potassium channel genes. In: Handbook of Receptors and Channels: Ligand- and Voltage-Gated Ion Channels, edited by North RA. Boca Raton, FL: CRC, 1995, p. 1–71.
- 9.Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci 868: 233–285, 1999. [DOI] [PubMed] [Google Scholar]
- 10.Cole WC, Clement-Chomienne O, Aiello EA. Regulation of 4-aminopyridine-sensitive delayed rectifier K+ channels in vascular smooth muscle by phosphorylation. Biochem Cell Biol 74: 439–447, 1996. [DOI] [PubMed] [Google Scholar]
- 11.Cox RH, Tulenko TN. Altered contractile and ion channel function in rabbit portal vein with dietary atherosclerosis. Am J Physiol Heart Circ Physiol 268: H2522–H2530, 1995. [DOI] [PubMed] [Google Scholar]
- 12.Czernin J, Barnard RJ, Sun KT, Krivokapich J, Nitzsche E, Dorsey D, Phelps ME, Schelbert HR. Effect of short-term cardiovascular conditioning and low-fat diet on myocardial blood flow and flow reserve. Circulation 92: 197–204, 1995. [DOI] [PubMed] [Google Scholar]
- 13.Dayanikli F, Grambow D, Muzik O, Mosca L, Rubenfire M, Schwaiger M. Early detection of abnormal coronary flow reserve in asymptomatic men at high risk for coronary artery disease using positron emission tomography. Circulation 90: 808–817, 1994. [DOI] [PubMed] [Google Scholar]
- 14.Dick GM, Bratz IN, Borbouse L, Payne GA, Dincer UD, Knudson JD, Rogers PA, Tune JD. Voltage-dependent K+ channels regulate the duration of reactive hyperemia in the canine coronary circulation. Am J Physiol Heart Circ Physiol 294: H2371–H2381, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Diochot S, Schweitz H, Beress L, Lazdunski M. Sea anemone peptide with a specific blocking activity against the fast inactivating potassium channel Kv3.4. J Biol Chem 273: 6744–6749, 1998. [DOI] [PubMed] [Google Scholar]
- 16.Egashira K, Inou T, Hirooka Y, Yamada A, Maruoka Y, Kai H, Sugimachi M, Suzuki S, Takeshita A. Impaired coronary blood flow response to acetylcholine in patients with coronary risk factors and proximal atherosclerotic lesions. J Clin Invest 91: 29–37, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galle J, Busse R, Bassenge E. Hypercholesterolemia and atherosclerosis change vascular reactivity in rabbits by different mechanisms. Arterioscler Thromb 11: 1712–1718, 1991. [DOI] [PubMed] [Google Scholar]
- 18.Genda S, Miura T, Miki T, Ichikawa Y, Shimamoto K. KATP channel opening is an endogenous mechanism of protection against the no-reflow phenomenon but its function is compromised by hypercholesterolemia. J Am Coll Cardiol 40: 1339–1346, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Hatton WJ, Mason HS, Carl A, Doherty P, Latten MJ, Kenyon JL, Sanders KM, Horowitz B. Functional and molecular expression of a voltage-dependent K+ channel (Kv1.1) in interstitial cells of cajal. J Physiol 533: 315–327, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heaps CL, Bowles DK. Gender-specific K+-channel contribution to adenosine-induced relaxation in coronary arterioles. J Appl Physiol 92: 550–558, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Heaps CL, Mattox ML, Kelly KA, Meininger CJ, Parker JL. Exercise training increases basal tone in arterioles distal to chronic coronary occlusion. Am J Physiol Heart Circ Physiol 290: H1128–H1135, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heaps CL, Tharp DL, Bowles DK. Hypercholesterolemia abolishes voltage-dependent K+ channel contribution to adenosine-mediated relaxation in coronary arterioles. Am J Physiol Heart Circ Physiol 288: H568–H578, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Hein TW, Kuo L. cAMP-independent dilation of coronary arterioles to adenosine: role of nitric oxide, G proteins, and KATP channels. Circ Res 85: 634–642, 1999. [DOI] [PubMed] [Google Scholar]
- 24.Henderson KK, Turk JR, Rush JWE, Laughlin MH. Endothelial function in coronary arterioles from pigs with early-stage coronary disease induced by high-fat, high-cholesterol diet: effect of exercise. J Appl Physiol 97: 1159–1168, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Jeremy RW, McCarron H. Effect of hypercholesterolemia on Ca2+-dependent K+ channel-mediated vasodilatation in vivo. Am J Physiol Heart Circ Physiol 279: H1600–H1608, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Koh SD, Sanders KM, Carl A. Regulation of smooth muscle delayed rectifier K+ channels by protein kinase A. Pflügers Arch 432: 401–412, 1996. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Chai Q, Gutterman DD, Liu Y. Elevated glucose impairs cAMP-mediated dilation by reducing Kv channel activity in rat small coronary smooth muscle cells. Am J Physiol Heart Circ Physiol 285: H1213–H1219, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Gutterman DD, Rusch NJ, Bubolz A, Liu Y. Nitration and functional loss of voltage-gated K+ channels in rat coronary microvessels exposed to high glucose. Diabetes 53: 2436–2442, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Mathew V, Cannan CR, Miller VM, Barber DA, Hasdai D, Schwartz RS, Holmes DR, Lerman A. Enhanced endothelin-mediated coronary vasoconstriction and attenuated basal nitric oxide activity in experimental hypercholesterolemia. Circulation 96: 1930–1936, 1997. [DOI] [PubMed] [Google Scholar]
- 30.Mathew V, Lerman A. Altered effects of potassium channel modulation in the coronary circulation in experimental hypercholesterolemia. Atherosclerosis 154: 329–335, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Miller AL, Morales E, Leblanc NR, Cole WC. Metabolic inhibition enhances Ca2+-activated K+ current in smooth muscle cells of rabbit portal vein. Am J Physiol Heart Circ Physiol 265: H2184–H2195, 1993. [DOI] [PubMed] [Google Scholar]
- 32.Mokelke EA, Hu Q, Song M, Toro L, Reddy HK, Sturek M. Altered functional coupling of coronary K+ channels in diabetic dyslipidemic pigs is prevented by exercise. J Appl Physiol 95: 1179–1193, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Pitkanen OP, Raitakari O, Niinikoski H, Nuutila P, Iida H, Voipio-Pulkki LM. Coronary flow reserve is impaired in young men with familial hypercholesterolemia. J Am Coll Cardiol 28: 1705–1711, 1996. [DOI] [PubMed] [Google Scholar]
- 34.Pongo E, Balla Z, Mubagwa K, Flameng W, Edes I, Szilvassy Z, Ferdinandy P. Deterioration of the protein kinase C-KATP channel pathway in regulation of coronary flow in hypercholesterolaemic rabbits. Eur J Pharmacol 418: 217–223, 2001. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Porcel M, Lerman LO, Herrmann J, Sawamura T, Napoli C, Lerman A. Hypercholesterolemia and hypertension have synergistic deleterious effects on coronary endothelial function. Arterioscler Thromb Vasc Biol 23: 885–891, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Rozsa Z, Pataricza J, Nemeth J, Papp JG. Differential efficacy of vasodilators in hypercholesterolaemic rabbits. J Pharm Pharmacol 50: 1035–1044, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Schmalz F, Kinsella JL, Koh SD, Vogalis F, Schneider A, Flynn ERM, Kenyon JL, Horowitz B. Molecular identification of a component of delayed rectifier current in gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol 274: G901–G911, 1998. [DOI] [PubMed] [Google Scholar]
- 38.Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci USA 78: 3363–3367, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sellke FW, Armstrong ML, Harrison DG. Endothelium-dependent vascular relaxation is abnormal in the coronary microcirculation of atherosclerotic primates. Circulation 81: 1586–1593, 1990. [DOI] [PubMed] [Google Scholar]
- 40.Shimoda LA, Sylvester JT, Booth GM, Shimoda TH, Meeker S, Undem BJ, Sham JS. Inhibition of voltage-gated K+ currents by endothelin-1 in human pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 281: L1115–L1122, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Shimoda LA, Sylvester JT, Sham JS. Inhibition of voltage-gated K+ current in rat intrapulmonary arterial myocytes by endothelin-1. Am J Physiol Lung Cell Mol Physiol 274: L842–L853, 1998. [DOI] [PubMed] [Google Scholar]
- 42.Shioiri H, Komaru T, Sato K, Takahashi K, Takeda S, Kanatsuka H, Watanabe J, Shirato K. Impact of hypercholesterolemia on acidosis-induced coronary microvascular dilation. Basic Res Cardiol 98: 76–83, 2003. [DOI] [PubMed] [Google Scholar]
- 43.Silver PJ, Walus K, DiSalvo J. Adenosine-mediated relaxation and activation of cyclic AMP-dependent protein kinase in coronary arterial smooth muscle. J Pharmacol Exper Ther 228: 342–347, 1984. [PubMed] [Google Scholar]
- 44.Theilmeier G, Verhamme P, Dymarkowski S, Beck H, Bernar H, Lox M, Janssens S, Herregods MC, Verbeken E, Collen D, Plate K, Flameng W, Holvoet P. Hypercholesterolemia in minipigs impairs left ventricular response to stress: association with decreased coronary flow reserve and reduced capillary density. Circulation 106: 1140–1146, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Thompson MA, Henderson KK, Woodman CR, Turk JR, Rush JWE, Price EM, Laughlin MH. Exercise preserves endothelium-dependent relaxation in coronary arteries of hypercholesterolemic male pigs. J Appl Physiol 96: 1114–1126, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Volk KA, Matsuda JJ, Shibata EF. A voltage-dependent potassium current in rabbit coronary artery smooth muscle cells. J Physiol 439: 751–768, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Weigand L, Wang W, Sylvester JT, Shimoda LA. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am J Physiol Lung Cell Mol Physiol 288: L1049–L1058, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Woodman CR, Turk JR, Rush JWE, Laughlin MH. Exercise attenuates the effects of hypercholesterolemia on endothelium-dependent relaxation in coronary arteries from adult female pigs. J Appl Physiol 96: 1105–1113, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Woodman CR, Turk JR, Williams DP, Laughlin MH. Exercise training preserves endothelium-dependent relaxation in brachial arteries from hyperlipidemic pigs. J Appl Physiol 94: 2017–2026, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Xu C, Lu Y, Tang G, Wang R. Expression of voltage-dependent K+ channel genes in mesenteric artery smooth muscle cells. Am J Physiol Gastrointest Liver Physiol 277: G1055–G1063, 1999. [DOI] [PubMed] [Google Scholar]
- 51.Xu C, Tang G, Lu Y, Wang R. Molecular basis of voltage-dependent delayed rectifier K+ channels in smooth muscle cells from rat tail artery. Life Sci 66: 2023–2033, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Yang Y, Jones AW, Thomas TR, Rubin LJ. Influence of sex, high-fat diet, and exercise training on potassium currents of swine coronary smooth muscle. Am J Physiol Heart Circ Physiol 293: H1553–H1563, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Yokoyama I, Ohtake T, Momomura S, Nishikawa J, Sasaki Y, Omata M. Reduced coronary flow reserve in hypercholesterolemic patients without overt coronary stenosis. Circulation 94: 3232–3238, 1996. [DOI] [PubMed] [Google Scholar]
- 54.Zeiher AM, Drexler H, Saurbier B, Just H. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest 92: 652–662, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]