

Abstract
The E2f3 locus encodes two Rb-binding gene products, E2F3a and E2F3b, which are differentially regulated during the cell cycle and are thought to be critical for cell cycle progression. We targeted the individual inactivation of E2f3a or E2f3b in mice and examined their contributions to cell proliferation and development. Chromatin immunoprecipitation and gene expression experiments using mouse embryo fibroblasts deficient in each isoform showed that E2F3a and E2F3b contribute to G1/S-specific gene expression and cell proliferation. Expression of E2f3a or E2f3b was sufficient to support E2F target gene expression and cell proliferation in the absence of other E2F activators, E2f1 and E2f2, suggesting that these isoforms have redundant functions. Consistent with this notion, E2f3a−/− and E2f3b−/− embryos developed normally, whereas embryos lacking both isoforms (E2f3−/−) died in utero. We also find that E2f3a and E2f3b have redundant and nonredundant roles in the context of Rb mutation. Analysis of double-knockout embryos suggests that the ectopic proliferation and apoptosis in Rb−/− embryos is mainly mediated by E2f3a in the placenta and nervous system and by both E2f3a and E2f3b in lens fiber cells. Together, we conclude that the contributions of E2F3a and E2F3b in cell proliferation and development are context dependent.
The retinoblastoma gene (RB) was the first tumor suppressor identified. Inheritance of a mutant RB allele leads to early childhood retinoblastoma and predisposes patients to a variety of other malignancies (29). In addition, somatic mutation in RB is a frequent event observed in small-cell lung carcinoma and breast cancer (9, 30). Mice with mutation in a single Rb allele, however, do not develop retinoblastoma but instead develop pituitary and thyroid tumors (3, 14, 16). The underlying basis for this difference in tumor spectrum between humans and mice remains unclear. Consistent with having tumor suppressor function, loss of both copies of Rb in mice leads to defects in proliferation, apoptosis, and differentiation (3, 14, 16). Rb−/− embryos have widespread ectopic proliferation and apoptosis in the central nervous system (CNS), peripheral nervous system (PNS), placenta, and a number of other tissues. In addition, cells of neuronal, erythroid, and skeletal muscle lineage fail to terminally differentiate. Consequently embryos suffer from anemia, systemic edema, and placental dysfunction and die at embryonic day 13.5 (E13.5) or soon after.
The Rb tumor suppressor pathway is believed to play a critical role in the control of cellular proliferation by regulating E2F transcriptional activities. To date, eight different E2F family members have been identified (4, 7, 18, 21). Among these, E2F3 has emerged as a critical Rb-binding factor important for mediating many of Rb's functions during embryonic development and tumorigenesis (24, 35, 36). For instance, loss of E2f3 profoundly suppresses ectopic proliferation and inappropriate apoptosis in the CNS, PNS, and lens fiber cells of Rb−/− embryos. Moreover, development of pituitary tumors in Rb+/− mice lacking one or two copies of E2f3 is decreased, but surprisingly the incidence and invasiveness of medullary thyroid carcinomas are increased (35).
The molecular basis for why E2F3 is uniquely important for mediating Rb function may be related to the complexity of the E2f3 genomic organization (17). The E2f3 locus drives the expression of two related gene products, E2F3a and E2F3b, through the use of two distinct promoters (1). As a result, E2f3a and E2f3b have unique first exons encoding 122 and 6 N-terminal amino acids, respectively. Other than at their N termini, these two related proteins share the same coding sequence, including regions important for specific DNA binding, transactivation, and association with the Retinoblastoma-encoded tumor suppressor protein and related pocket proteins. The control of their expression is complex and involves both transcriptional and posttranscriptional mechanisms (1). E2F3b protein is expressed in quiescent cells and remains constant throughout the cell cycle, whereas E2F3a protein accumulates maximally at the G1/S transition (17). The interesting dichotomy of the E2f3 locus in relation to the control of cell proliferation remains to be clearly understood. To rigorously evaluate the individual contributions of the two isoforms in the control of cell proliferation and development, we specifically targeted the inactivation of E2f3a or E2f3b in mice. Here, we provide evidence suggesting that both E2F3a and E2F3b contribute to the activation of E2F target gene expression and cell proliferation but that E2F3a is the main isoform that contributes to the unscheduled proliferation, apoptosis, and early lethality observed in Rb−/− embryos.
MATERIALS AND METHODS
MEF proliferation and ChIP analysis.
For analysis of cell proliferation, passage 2 mouse embryo fibroblasts (MEFs) with the indicated genotypes were plated and counted every day for 6 days. To bring cells to quiescence, cultures at 40 to 50% confluence were incubated in Dulbecco modified Eagle medium containing 0.2% serum for 60 h. Synchronized cells were then stimulated to proliferate by the addition of Dulbecco modified Eagle medium supplemented with 15% fetal bovine serum (FBS) and 10 μM bromodeoxyuridine (BrdU). At the desired time points, cells were then fixed with methanol and acetone (1:1), processed for immunofluorescence using anti-BrdU antibody (Ab-3; Oncogene), and counterstained with 4,6-diamidino-2-phenylindole (DAPI). A minimum of 500 DAPI-positive nuclei were scored for each time point. For chromatin immunoprecipitation (ChIP) experiments, approximately 10 × 106 cells were harvested for each antibody reaction. Formaldehyde was added directly to the culture medium at a final concentration of 1%. Cross-linking was allowed to proceed for 10 min at room temperature and was then stopped by the addition of glycine to a final concentration of 0.125 M. Cross-linked cells were washed twice with phosphate-buffered saline and scraped off the plate, and lysates were sonicated in order to shear the genomic DNA into fragments of 200 to 1,000 bp. Four micrograms of anti-E2F3 antibody (sc-876x) was added to each sample and incubated overnight at 4°C. Antibody-protein-DNA complexes were recovered by adding 60 μl of salmon sperm DNA-protein G agarose slurry and incubated for 1 h at 4°C. Following extensive washing, complexes were eluted and cross-linking was reversed by heating the samples at 65°C for 4 h. The eluted material was purified and resuspended in 30 μl of water. Quantitative real-time PCR (RT-PCR) was performed with 1 μl of DNA and 100 nM primers diluted to a final volume of 25 μl in Sybr green reaction mix (Bio-Rad). Accumulation of fluorescent products was monitored by RT-PCR using the Bio-Rad iCycler PCR machine. Reactions were performed in triplicate, and values were normalized using the threshold cycle number from the total input sample.
RT-PCR.
Total RNA from E13.5 Rb−/− or Rb+/+ placentas and MEFs was isolated by using Trizol (Invitrogen) according to the manufacturer's instruction. cDNA was synthesized from 2 μg of total RNA using Superscript III reverse transcriptase (Invitrogen) at 50°C for 60 min. After the completion of the reverse transcription, the final volume was brought to 100 μl by adding 80 μl of water. RT-PCR was performed in the Bio-Rad iCycler PCR machine. Each PCR mixture contained 0.5 μl of cDNA template and 100 nM of gene-specific primers in a final volume of 25 μl of Sybr green reaction mix (Bio-Rad). Each quantitative PCR yielded only the expected amplicon as determined by the melting-temperature profiles of the final products and by gel electrophoresis. Standard curves were generated using cDNA to determine the linear range of amplification for each primer pair. Reactions were performed in triplicate, and relative amounts of cDNAs were normalized to the Gapdh gene. Primers used for RT-PCR have been described previously (21).
BrdU staining and terminal deoxynucleotidyltransferase-mediated dUTP- biotin nick end labeling (TUNEL) analysis.
Pregnant females (13.5 days postcoitum) were injected intraperitoneally with BrdU (100 μg/gram of body weight) 2 h prior to harvesting of embryos. Desired tissues from placentas and embryos were fixed in 10% neutral buffered formalin upon harvesting, and paraffin-embedded sections (5 μm) were used for immunohistochemistry analysis after deparaffinization. Anti-BrdU antibody (Dako Co.; MO-0744) and Vectastain Elite ABC reagent (Vector Labs) were used to detect BrdU incorporation according to the manufacturer's instructions, followed by hematoxylin counterstaining. For lens fiber cells, we used a secondary antibody conjugated with Alexa 594 and DAPI for nuclear staining.
Apoptotic cells were determined using TUNEL assays according to the manufacturer's protocol (Chemican S7101), except that the terminal deoxynucleotidyltransferase enzyme was incubated at room temperature for 1.5 h and the antidigoxigenin conjugate was incubated at room temperature for 1 h. All slides were counterstained with hematoxylin. Quantification of proliferation and apoptosis was achieved by calculating the percentage of cells with positive signals in the desired tissue sections.
Giemsa staining.
Peripheral blood samples (4 μl) were spread on slides. Giemsa staining (Sigma; diluted 1:20) was performed according to the manufacturer's protocol to determine whether red blood cells were nucleated. The same embryos were used both for hematoxylin and eosin (H&E) staining and for immunohistochemical analysis.
RESULTS
E2F3 isoforms contribute to cell proliferation in MEFs.
The E2f3 locus plays a critical role in the control of cell proliferation. Ablation of both E2f3a and E2f3b (E2f3−/−) in MEFs profoundly decreases E2F target gene expression and cell proliferation (11). To evaluate the individual contributions of each isoform in the control of cell proliferation, we investigated the molecular consequences of inactivating E2f3a and E2f3b in MEFs. The targeted inactivation of E2f3a and E2f3b in mice was described previously (Fig. 1A) (26). MEFs were derived from E2f3a+/− or E2f3b+/− intercrosses, and the specific ablation of E2F3a or E2F3b proteins was confirmed by Western blot assays using total E2F3-specific antibodies (Fig. 1B). Standard proliferation assays revealed a measurable defect in the proliferation rate of E2f3a−/− and E2f3b−/− MEFs relative to E2f3+/+ littermate control cells (Fig. 1C and D). We also analyzed the ability of quiescent E2f3a−/− and E2f3b−/− MEFs to enter the cell cycle in response to mitogenic signals. S-phase entry was monitored by the incorporation of BrdU into genomic DNA. These assays also revealed a slight delay in the entry of E2f3a−/− and E2f3b−/− MEFs into the cell cycle, relative to E2f3+/+ MEFs (Fig. 1C and D).
FIG. 1.

E2F3a and E2F3b contribute to transcriptional activation. (A) Illustration of the E2f3 locus and the E2f3a and E2f3b targeted alleles. (B) Western analysis of E2F3a and E2F3b in cell extracts derived from E2f3+/+, E2f3a−/−, and E2f3b−/− MEFs. (C and D) (Left panels) Exponential growth curves of control (E2f3a+/+ or E2f3b+/+; gray), E2f3a−/− (red), and E2f3b−/− (blue) MEFs. (Right panels) BrdU incorporation of control (E2f3a+/+ or E2f3b+/+; gray), E2f3a−/− (red), and E2f3b−/− (blue) MEFs. Quiescent MEFs with the indicated genotypes were restimulated with medium containing 15% FBS and assessed for BrdU incorporation at the indicated time points. Three independent experiments for proliferation assays, BrdU incorporation, and gene expression were performed in triplicate. The P value (*) comes from all three independent experiments. (E and F) RT-PCR analysis of E2F target gene expression. Total RNA from MEFs harvested as described for panels C and D was used to measure the levels of expression of the indicated E2F target genes. The y axis represents the average induction in gene expression, where the expression level at the 0-h time point is equal to 1. Statistical analyses for proliferation assays, BrdU incorporation, and gene expression were performed by using generalized linear models. The interaction “group by time” was included in both models. The P values shown in the plots correspond to the total “group” effect, which indicates the interactions of “group by time.”
Given the established roles for E2F family members in the control of cell cycle-dependent gene expression, we measured the levels of known E2F target genes in isoform-specific mutant MEFs that were stimulated to enter the cell cycle. Consistent with previous reports, expression of E2F-responsive genes such as cdc6, cyclin E, mcm3, polα, dhfr, and pcna in wild-type MEFs peaked at the G1/S transition (Fig. 1E and F, E2f3a+/+ and E2f3b+/+; see also Fig. S1 in the supplemental material). In contrast, this serum-dependent induction of E2F target gene expression was significantly delayed in E2f3a−/− and E2f3b−/− MEFs and peak levels never reached those observed in littermate control cells. Together, these observations and recent work by Danielian and colleagues (5) suggest that E2F3a and E2F3b proteins individually contribute to the growth factor-dependent activation of E2F target genes during the G1/S transition and to the proliferative capacity of MEFs.
Proliferation of MEFs with a single E2F isoform.
Considerable functional redundancy has been observed among members of the E2F activator subclass (26). Perhaps the most striking example of redundancy was demonstrated by previous studies comparing the proliferative capacities of MEFs deficient in E2f1, E2f2, and E2f3, singly or in combination. These studies showed that loss of E2f3 (E2f3a and E2f3b) significantly compromised the proliferation of MEFs but the combined loss of E2f1, E2f2, and E2f3 completely abrogated proliferation (33). To examine the potential redundancy among these E2Fs, we generated fibroblasts from E13.5 E2f1−/− E2f2−/− E2f3a−/− and E2f1−/− E2f2−/− E2f3b−/− embryos and once again examined E2F target gene expression and cell proliferation under the same conditions as described above. While entry into S phase was delayed and proliferation was compromised in E2f1−/− E2f2−/− E2f3a−/− and E2f1−/− E2f2−/− E2f3b−/− (TKO) MEFs relative to littermate-derived control cells (Fig. 2A and B), these TKO cells did not proliferate appreciably slower than cells lacking only in E2f3a or E2f3b. These results suggest that one of the two E2f3 isoforms is sufficient to support cell proliferation (Fig. 2A and B). While some variation was observed among different MEF preparations of the same genotype, we could reproducibly measure a delay and attenuation in the expression of many E2F target genes as TKO cells were stimulated to enter the cell cycle (Fig. 2C and D; see also Fig. S2 in the supplemental material). From these experiments we conclude that E2f3a and E2f3b have redundant functions and that expression of either one is sufficient to support E2F target gene expression and cell proliferation.
FIG. 2.

E2f3a and E2f3b are sufficient to support cellular proliferation. (A and B) (Left panels) Growth curves of control (gray), E2f1−/− E2f2−/− E2f3a−/− (red), and E2f1−/− E2f2−/− E2f3b−/− (blue) MEFs. Passage 2 MEFs with the indicated genotypes were plated in duplicate and counted every day for 6 days. (Right panels) BrdU incorporation in control (gray), E2f1−/− E2f2−/− E2f3a−/− (red), and E2f1−/− E2f2−/− E2f3b−/− (blue) MEFs. (C and D) RT-PCR analysis of E2F target gene expression from MEFs harvested as described for panels A and B. The statistical analyses for proliferation assays, BrdU incorporation, and gene expression were performed as described for Fig. 1. Three independent experiments were performed for proliferation assays, BrdU incorporation, and gene expression. The P value comes from all three independent experiments.
E2F3a and E2F3b act as transcriptional activators.
The E2f3a and E2f3b genes are differentially expressed during the cell cycle. E2f3a mRNA levels are low in quiescent cells, increase in late G1, and peak at G1/S, whereas E2f3b mRNA levels are constitutively expressed throughout the cell cycle. Consistent with their expression patterns, previous biochemical studies described E2F3a as a transcription activator that functions at G1/S and E2F3b as a repressor that functions during G0 (17). To determine the extent to which E2F3a and E2F3b proteins might contribute to the control of E2F target gene expression during the cell cycle, we performed ChIP assays on wild-type, E2f3a−/−, and E2f3b−/− MEFs with antibodies that recognize both isoforms. The specificity of these antibodies was confirmed by comparing the recruitment of E2F3 proteins to two target promoters, cdc6 and E2f1, in E2f3+/+ and E2f3−/− cells (Fig. 3A). Using these antibodies, we could detect E2F3-specific proteins on target promoters during G0 in E2f3+/+ cells (Fig. 3B, 0-h samples). Consistent with the selective expression of E2f3b in quiescent cells, the G0-specific recruitment of total E2F3 was markedly reduced in E2f3b−/− cells but was unaltered in E2f3a−/− MEFs. The recruitment of E2F3 proteins to target promoters increased two- to threefold when E2f3+/+ cells were stimulated to enter G1/S (Fig. 3B, 18-h samples). This increase was abolished in E2f3a−/− cells, indicating that E2F3a was the major isoform recruited at G1/S. But clearly, E2F3b continued to occupy E2F binding sites on target promoters at G1/S since an equal amount of E2F3 protein could be detected in E2f3a−/− cells at G0 and G1/S. From these assays we conclude that E2F3b is the principal isoform bound to target promoters in quiescent cells and that both E2F3a and E2F3b contribute to the loading of target promoters during G1/S. Exogenous expression of E2F3a or E2F3b activated an E2F-responsive luciferase reporter to an equal extent (Fig. 3C). Together, these findings are consistent with their role in transcription activation at G1/S and provide a molecular basis for their redundant roles in cell proliferation.
FIG. 3.

E2F3a and E2F3b proteins can bind and activate E2F-responsive promoters. (A) Specificity of anti-E2F3 antibodies used in ChIP assays. Control ChIP assays using anti-total E2F3 antibodies in proliferating E2f3+/+ (gray) or E2f3−/− (white) MEFs. (B) ChIP assays of cell lysates from synchronized E2f3a+/+ or E2f3b+/+ (gray), E2f3a−/− (red), and E2f3b−/− (blue) MEFs using antibodies against total E2F3. Immunoprecipitated DNA was measured by RT-PCR using primers flanking the E2F binding sites on the E2f1 and cdc6 promoters. RT-PCR was performed in triplicate, and cycle numbers were normalized to 1% of the input DNA. Results are shown as induction relative to the quiescent (0-h) E2f3a+/+ sample; 18 h, quiescent MEFs stimulated with 15% FBS for 18 h. Statistical analyses for ChIP analysis were performed for two independent experiments using the Student t test method, and P values are indicated within the graph. (C) E2F3a and E2F3b proteins can activate E2F-responsive promoters. Cells were cotransfected with the thymidine kinase Renilla luciferase reporter plasmids (as an internal control), with either an E2F-responsive reporter (E2F-reporter) or a control reporter construct (control reporter), and with 0.1 μg or 0.3 μg of a vector expressing E2F3a (3a), E2F3b (3b), or an empty control (con). Relative luciferase reporter activity was internally normalized to the Renilla luciferase activity. (D) E2F4 recruitment to E2F target promoters during quiescence. ChIP assays of cell lysates from synchronized wild-type (E2f3a+/+ and E2f3b+/+; gray), E2f3a−/− (red), and E2f3b−/− (blue) MEFs using antibodies against E2F4 (sc-1082).
Previous work suggested that E2F3b functions as a transcriptional repressor (17). The fact that loss of E2f3b did not result in widespread derepression of classic E2F target genes, especially during G0, is inconsistent with a general model for E2F3b in transcriptional repression. One possibility for the normal levels of E2F target gene expression in quiescent E2f3b−/− MEFs is the potential for E2F4, which is known to function as a repressor during G0, to compensate for the absence of E2F3b. ChIP assays using E2F4-specific antibodies in E2f3b−/− MEFs revealed a 1.5- to 2-fold increase in the loading of E2F4 to at least some E2F target promoters (Fig. 3D). Given the relatively high abundance of E2F4 in MEFs, as determined by gel shift assays (13, 17), this increase in E2F4 loading could represent a compensatory mechanism in quiescent cells to repress E2F target genes in the absence of E2F3b.
Loss of E2f3a extends the life span of Rb−/− embryos.
One physiological circumstance where E2Fs have been shown to contribute to the control of proliferation in vivo is when there is a limiting amount of the Rb tumor suppressor protein. Loss of E2f1 or E2f3 (E2f3a and E2f3b) can suppress the unscheduled proliferation and apoptosis observed in Rb−/− embryos and extend their viability from approximately E13.5 to E17.5 (24, 25, 36). To determine which E2F3 isoform contributes to the phenotypes caused by the loss of Rb, we set up intercrosses between Rb+/−; E2f3a+/− mice and either Rb+/−; E2f3a+/− or Rb+/−; E2f3a−/− mice (Table 1), as well as intercrosses between Rb+/−; E2f3b−/− and Rb+/−; E2f3b−/− mice (Table 2). Genotypic analysis of the resulting embryos at various stages of gestation is summarized in Tables 1 and 2. In contrast to Rb−/− and Rb−/−; E2f3b−/− embryos, which died by E13.5, Rb−/−; E2f3a−/− embryos were found alive as late as E17.5, suggesting that loss of E2f3a suppresses the lethality of Rb mutant embryos to the same extent as does loss of E2f3 (36).
TABLE 1.
Genotypic analysis of embryos derived from intercrosses between Rb+/−; E2f3a+/− mice and either Rb+/−; E2f3a+/− or Rb+/−; E2f3a−/− micea
| Genotype | No. of embryos at day:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| E13.5
|
E15.5
|
E16.5
|
E17.5
|
|||||
| Found | Expected | Found | Expected | Found | Expected | Found | Expected | |
| Rb+/+ | ||||||||
| 3a+/+ | 6 | 9 | 12 | 9 | − | − | 2 | 1 |
| 3a+/− | 20 | 15 | 15 | 13 | 6 | 3 | 3 | 3 |
| 3a−/− | 7 | 9 | 9A | 9 | 7 | 5 | 4 | 6 |
| Rb+/− | ||||||||
| 3a+/+ | 11 | 13 | 16 (1) | 19 | − | − | 5 | 3 |
| 3a+/− | 33 (1) | 31 | 31 (1) | 26 | 5 | 7 | 4 | 6 |
| 3a−/− | 16 (1) | 22 | 11 | 18 | 8 | 8 | 12 | 13 |
| Rb−/− | ||||||||
| 3a+/+ | 4 (2) | 7 | 0 (9)B | 9 | − | − | 0 (1) | 1 |
| 3a+/− | 14 (5) | 15 | 2 (9) | 13 | 0 | 3 | 0 (2) | 3 |
| 3a−/− | 6 (2) | 9 | 5 (3)C | 9 | 2 (4) | 5 | 1 (7) | 6 |
| Total | 128 | 124 | 32 | 41 | ||||
All embryos, including dead embryos, were genotyped. The numbers of genotyped dead fetuses are indicated in parentheses. −, no embryos with this genotype were found. Chi-square analysis was used for association (superscript capital letters): A, P > 0.1 compared to Rb+/+ mice; B, P < 0.005 compared to Rb+/+ mice; C, P < 0.005 compared to Rb−/− mice.
TABLE 2.
Genotypic analysis of embryos derived from Rb+/−; E2f3b−/− intercrossesa
| Genotype | No. of embryos at day:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| E13.5
|
E15.5
|
E16.5
|
E17.5
|
|||||
| Found | Expected | Found | Expected | Found | Expected | Found | Expected | |
| Rb+/+3b−− | 13 | 14 | − | − | 17 | 14 | − | − |
| Rb+/−3b−− | 30 (2) | 28 | − | − | 24 | 28 | − | − |
| Rb−/−3b−− | 9 (5)A | 14 | − | − | 0B | 14 | − | − |
| Total | 59 | 41 | ||||||
All embryos, including dead embryos, were genotyped. The numbers of genotyped dead fetuses are indicated in parentheses. −, no embryos with this genotype were found. Chi-square analysis was used for association (superscript capital letters): A, P > 0.1 compared to Rb+/+ mice; B, P < 0.001 compared to Rb+/+ mice.
Loss of E2f3a suppresses placental abnormalities in Rb−/− embryos.
Previous work demonstrated that the extraembryonic function of Rb is critical for embryonic viability (6, 34). Indeed, the specific deletion of Rb in extraembryonic trophoblast cells was sufficient to cause embryonic lethality (31). Conversely, Rb−/− fetuses supplied with a wild-type placenta could be carried to term. Therefore, we reasoned that the significant extension of Rb−/− embryonic viability by loss of E2f3a could be due to the suppression of Rb mutant placental phenotypes. To investigate this possibility, we first analyzed the expression of E2f3a, E2f3b, and a number of known E2F target genes in Rb−/− placentas. Loss of Rb resulted in a specific increase of E2f3a expression, without affecting E2f3b expression, and a concomitant increase in E2F target genes (Fig. 4A and B). These observations raised the possibility that E2f3 isoforms may differentially contribute to Rb mutant placental phenotypes.
FIG. 4.

(A) Analysis of E2f3a and E2f3b expression in Rb+/+ and Rb−/− placentas by RT-PCR. (B) Gene expression analysis of known E2F target genes in Rb+/+ and Rb−/− placentas by RT-PCR.
We thus compared placental development in Rb−/−, Rb−/−; E2f3a−/−, and Rb−/−; E2f3b−/− embryos. Consistent with previous observations, histological examination of Rb−/− placentas by H&E staining revealed a severe disruption of labyrinth architecture, with extensive accumulation of densely packed trophoblast cells at the expense of maternal and fetal blood spaces (Fig. 5A). This increase in cellularity was, at least in part, due to an increase in proliferation of trophoblast cells (Fig. 5A to C; see also Fig. S3 in the supplemental material). Remarkably, Rb−/−; E2f3a−/− placentas appeared relatively normal, with few clustered trophoblast and blood cells populating the ample and well-distributed supply of blood spaces present within the labyrinth cell layer. In contrast, the inactivation of E2f3b did not ameliorate the placental abnormalities of Rb mutant embryos (Fig. 5A; see also Fig. S3 in the supplemental material). Quantification of BrdU-positive trophoblast cells showed that loss of E2f3a, but not E2f3b, suppressed much of the ectopic proliferation evident in Rb−/− placentas (Fig. 5B and C). Although we cannot completely rule out other potential functions, such as differentiation (2), these data suggest that elevated and unharnessed E2F3a activity contributes to the ectopic trophoblast proliferation and severe disruption of placental architecture in Rb mutant embryos.
FIG. 5.

Loss of E2f3a, but not E2f3b, rescues placental defects in Rb−/− embryos. (A) Placenta sections (5 μm) from E13.5 Rb+/+; E2f3a+/+, Rb−/−; E2f3a+/+, Rb−/−; E2f3a−/−, Rb−/−; E2f3b−/−, Rb+/+; E2f3a−/−, and Rb+/+; E2f3b−/− embryos were stained with H&E, processed for immunohistochemical analysis using anti-BrdU antibodies, or processed for TUNEL analysis (not shown). (B) Quantification of BrdU-positive trophoblast cells from embryos with the indicated genotypes. 3a, E2f3a; 3b, E2f3b. *, t test, P = 0.0076. (C) Quantification of TUNEL-positive trophoblast cells in embryos with the indicated genotypes. At least two different sections were evaluated per placenta.
E2f3a is the major isoform contributing to Rb mutant phenotypes in the CNS and PNS.
During embryonic development, Rb controls proliferation, apoptosis, and cell differentiation through cell-autonomous and nonautonomous mechanisms (6, 19, 22, 32, 34). To determine the extent to which E2F3 isoforms contribute to these processes in developing fetuses, we analyzed Rb−/−; E2f3a−/− and Rb−/−; E2f3b−/− fetal tissues of the nervous system that are typically affected by the simple loss of Rb, including neurons of the CNS and PNS.
In the hindbrain of the CNS, proliferation of neuronal progenitors is restricted to a region lining the ventricles that is known as the “ventricular zone.” As progenitors are induced to differentiate, they migrate from the ventricular zone into the intermediate and marginal zones, exit the cell cycle, and exhibit a terminal differentiation program of gene expression. The functional integrity of postmitotic neurons requires the timely exit of the cell cycle and maintenance of a G0 state. The Rb tumor suppressor protein is critical for establishing and maintaining this G0 state. Indeed, Rb inactivation results in increased proliferation and apoptosis of neurons that should normally be postmitotic (3, 14, 16). Along with this unscheduled proliferation and apoptosis, loss of Rb function results in elevated levels of free E2Fs and increased expression of E2F target genes known to be involved in the control of cell proliferation (20). To determine whether E2F3a and/or E2F3b contributes to the unscheduled proliferation observed in Rb−/− neuronal tissues, we analyzed postmitotic neurons in the intermediate zone adjacent to the fourth ventricle of the hindbrain of doubly null Rb/E2F embryos for their ability to replicate DNA. As reported previously (20), loss of Rb resulted in increased neuronal proliferation, which was accompanied by a wave of apoptosis (Fig. 6A to F). The additional inactivation of E2f3a, but not E2f3b, significantly suppressed the unscheduled proliferation of neurons in the fourth ventricle of the CNS (Fig. 6A and C). Similarly, deletion of E2f3a significantly suppressed the massive apoptosis arising in the CNS of Rb-deficient embryos (Fig. 6A and E). These results suggest that E2f3a is a particularly important mediator of Rb mutant phenotypes manifested in the CNS, but these findings do not show a direct role of either Rb or E2f3a in these tissues.
FIG. 6.

Analysis of proliferation and apoptosis in the CNS and PNS of Rb+/+; E2f3a+/+, Rb−/−; E2f3a+/+, Rb−/−; E2f3a−/−, Rb−/−; E2f3b−/−, Rb+/+; E2f3a−/−, and Rb+/+; E2f3b−/− embryos. (A) BrdU and TUNEL staining in the fourth ventricle of the hindbrain of the CNS derived from E13.5 embryos with the indicated genotypes. (B) BrdU staining and TUNEL assays in dorsal root ganglia of the PNS in embryos with the indicated genotypes. (C) Loss of E2f3a suppresses proliferation in the CNS of Rb−/− embryos. Percentage of BrdU-positive cells in hindbrain sections of embryos with the indicated genotypes. 3a, E2f3a; 3b, E2f3b. *, t test, P = 0.03; **, t test, P = 0.16. (D) Loss of E2f3a suppresses proliferation in the PNS of Rb−/− embryos. Percentage of BrdU-positive cells in dorsal root ganglion sections of embryos with the indicated genotypes; *, t test, P = 0.03. (E) Loss of E2f3a suppresses apoptosis in the CNS of Rb−/− embryos. Quantification of TUNEL-positive cells in the fourth ventricle of the hindbrains of embryos with the indicated genotypes; *, t test, P = 0.0017; **, t test, P = 0.18. (F) Loss of E2f3a does not suppress apoptosis in the PNS of Rb−/− embryos. Quantification of TUNEL-positive cells in dorsal root ganglia of embryos with the indicated genotypes. At least two different sections were evaluated per embryo.
We next analyzed proliferation and apoptosis in the PNS. While proliferation of neurons in the dorsal root ganglia was significantly reduced in Rb−/−; E2f3a−/− embryos relative to Rb−/− embryos, apoptosis was not affected (Fig. 6B, D, and F). In contrast, loss of E2f3b did not suppress either the ectopic proliferation or apoptosis in these Rb−/− neurons of dorsal root ganglia. Together, these results demonstrate that E2f3a is the main isoform responsible for mediating the tissue-specific ectopic proliferation and apoptosis observed in Rb-deficient embryos.
Limited role of individual E2f3 isoforms in Rb−/− lens fiber cells and erythrocytes.
We also analyzed lens development and erythropoiesis, two additional processes severely affected by the loss of Rb. By E10.5 the developing eye has already formed into a semihollow sphere of cells that have anterior and posterior orientation, with posterior cells lining the interior of the eye. Proliferative epithelial cells at the anterior side continue to divide laterally until they reach the lens equator, at which point they exit the cell cycle and begin to migrate toward the inside of the lens. E13.5 postmitotic lens cells then express copious amounts of crystallin proteins and elongate vertically to form lens fiber cells that eventually fill the cavity of the lens vesicle (15). Rb is an important negative regulator of proliferation that is necessary for cell cycle exit at the lens equator (10, 23). The inactivation of Rb not only leads to ectopic proliferation of lens fiber cells within the cavity of the lens vesicle but also results in apoptosis and failure of nuclei to properly migrate to the center of the vesicle, leading to a disorganized lens cavity filled with nuclei (Fig. 7A to C). In contrast to the loss of both E2F3 isoforms, which was previously shown to restore normal proliferation and lens architecture in Rb−/− embryos, the individual inactivation of E2f3a or E2f3b did not significantly suppress ectopic proliferation and apoptosis, nor did it restore lens architecture (Fig. 7A to C). From these results we conclude that E2f3a and E2f3b both contribute to the disruption of lens development in Rb−/− embryos.
FIG. 7.

Analysis of proliferation and apoptosis in lenses from Rb+/+; E2f3a+/+, Rb−/−; E2f3a+/+, Rb−/−; E2f3a−/−, Rb−/−; E2f3b−/−, Rb+/+; E2f3a−/−, and Rb+/+; E2f3b−/− embryos. (A) Sections of paraffin-embedded lenses from embryos with the indicated genotypes were used for H&E, anti-BrdU immunofluorescence, and DAPI staining as well as for TUNEL analysis. Arrows indicate apoptosis-positive cells. At least two different sections were evaluated per lens. (B) Quantification of BrdU-positive lens fiber cells from embryos with the indicated genotypes. 3a, E2f3a; 3b, E2f3b. *, t test, P = 0.065. (C) Quantification of TUNEL-positive lens fiber cells in embryos with the indicated genotypes.
Impaired red blood cell differentiation leading to anemia is another hallmark phenotype of Rb-knockout mice. This role of Rb is mediated through both cell-autonomous and nonautonomous functions in liver-specific macrophages (12, 27, 28). Consistent with previous observations, there were few enucleated red blood cells in E13.5 Rb−/− embryos (Fig. 8A and B), and this defect was not corrected by the loss of either E2f3a or E2f3b (Fig. 8A and B). Our observation that loss of E2f3a extended the life span of Rb mutant embryos without alleviating the anemia is consistent with the notion that E2F2 is the key downstream target of Rb in erythroblasts and that lethality of Rb−/− embryos is not linked to anemia (8, 34).
FIG. 8.
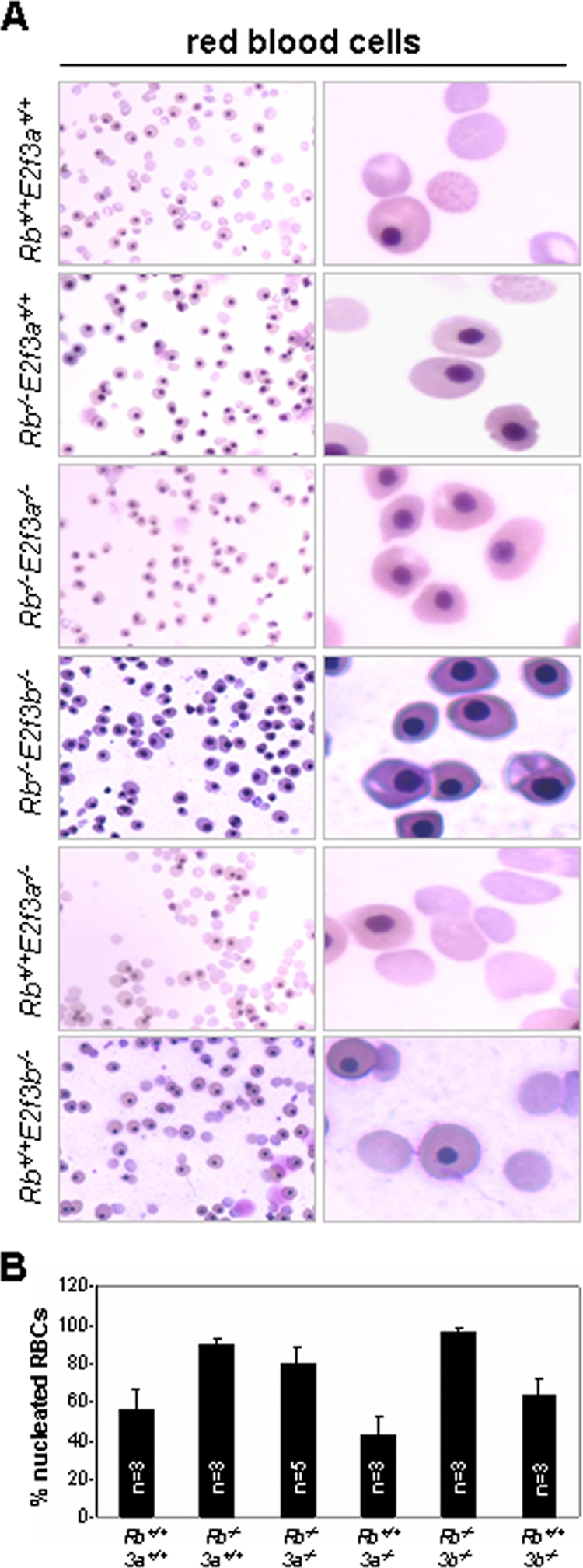
Analysis of defective enucleation of peripheral red blood cells from Rb+/+; E2f3a+/+, Rb−/−; E2f3a+/+, Rb−/−; E2f3a−/−, Rb−/−; E2f3b−/−, Rb+/+; E2f3a−/−, and Rb+/+; E2f3b−/− embryos. (A) Peripheral blood smears from E13.5 embryos with the indicated genotypes were analyzed by Giemsa staining. (B) Percentage of enucleated red blood cells (RBCs) in embryos with the indicated genotypes. 3a, E2f3a; 3b, E2f3b.
DISCUSSION
The E2f3 locus encodes two Rb-binding proteins, E2F3a and E2F3b, thought to be important for mouse development and cell proliferation. E2f3a expression is regulated during the cell cycle with maximum expression in G1/S, while E2f3b expression is constitutive throughout the cell cycle. Previous studies analyzed the inactivation of both E2f3 isoforms (11, 24, 33, 36), but the extent to which their individual functions contribute to mouse development and cell proliferation remained unclear. To investigate their functions, we targeted the specific inactivation of E2f3a and E2f3b in mice. In contrast to the ablation of both E2f3a and E2f3b, which resulted in embryonic lethality, inactivation of either isoform alone had no measurable effect on mouse embryonic development. On the other hand, loss of E2f3a or E2f3b significantly rescued selected phenotypes observed in Rb mutant embryos. The data would suggest that E2F3a and E2F3b proteins contribute to the control of proliferation in Rb mutant embryos in a tissue-specific manner, with E2F3a playing a major role in the placenta and nervous system and with E2F3a and E2F3b having critical roles in the lens. The suppression of placental phenotypes in Rb-E2f3a compound-knockout embryos likely accounts, at least in part, for the rescue of fetal phenotypes. This notion is consistent with previous findings showing that Rb mutant fetuses supplied with a wild-type placenta could be carried to term (34).
Currently, it is thought that E2F3a acts as a transcription activator. Given that E2F3b complexes specifically with Rb in quiescent cells, it has been assumed that E2F3b functions as a repressor (17). Consistent with functional redundancy among repressors, ChIP experiments revealed that in quiescent cells, an increase in E2F4 recruitment to target promoters may compensate for loss of E2F3b. However, it is difficult to envision how E2F3b would contribute, in collaboration with E2F3a, to the proliferation of Rb−/− lens fiber cells by repressing gene expression. Given that loss of both E2f3a and E2f3b, but not either alone, can suppress proliferation in these cells, it is more likely that E2F3b, like E2F3a, contributes by activating gene expression in this cellular context. In this view, either E2F3a or E2F3b activity would be sufficient to promote unscheduled proliferation in Rb mutant lenses. Several additional lines of in vivo evidence now suggest that E2F3a and E2F3b proteins can function in the same capacity in other cell types. Analysis of MEFs deficient in various E2F family members showed that either E2F3a or E2F3b can rescue the proliferation defect of cells lacking all known activators (E2f1, E2f2, and E2f3a/b) (33). Moreover, recent analysis of knock-in mice showed that expression of E2f3b from the E2f3a locus (E2f3a3bki) suppressed all the postnatal phenotypes associated with the inactivation of E2f1 and E2f3a (26). Finally, results from this study show that expression of either E2F3 isoform (E2f1−/− E2f2−/− E2f3a−/− and E2f1−/− E2f2−/− E2f3b−/−) is sufficient to support cell proliferation. We therefore suggest that E2F3b is important for normal cell cycle progression and, like E2F3a, functions as a transcription activator of E2F-responsive genes.
Why then is E2F3b protein loaded onto target promoters in quiescent cells? The association of E2F3b with target promoters during G0 could be viewed as a mechanism for cells to quickly respond to growth-stimulatory signals by having E2F3b loaded and poised to activate gene expression upon mitogenic induction. In G0 cells, E2F3b exists as a complex with Rb and hence is blocked from activating gene expression. Phosphorylation of Rb as cells are stimulated to enter the cell cycle would release Rb from E2F3b and allow it to activate transcription. The accumulation of E2F3a protein later in G1 would then ensure that the amount of total E2F3 activity, composed of E2F3a and E2F3b proteins, is available to maximally activate gene expression at the G1/S transition.
We also find that E2f3a and E2f3b have redundant and nonredundant roles in the context of Rb mutation, with E2f3a playing a more dominant role in most Rb−/− tissues affected. It is notable that loss of E2f3a, E2f3b, or both did not decrease cell proliferation in Rb mutant tissues below levels normally found in wild-type tissues. In other words, basal levels of proliferation were not affected in Rb-E2f3a or Rb-E2f3b compound-knockout embryos. Because the inactivation of E2f3a or E2f3b in an otherwise normal embryo failed to affect basal levels of proliferation and apoptosis, we conclude that the in vivo roles for these two isoforms are manifested mainly in the context of inactivation of Rb. The contribution of E2F3a and E2F3b to Rb mutant phenotypes in developing embryos may thus represent a special circumstance that could be relevant to the cancer phenotype. We suggest that E2F3a, and to a lesser extent E2F3b, is a critical component of the Rb tumor suppressor axis.
Supplementary Material
Acknowledgments
We thank J. Moffitt and J. Opavska for technical assistance and H.-Z. Chen for critical comments on the manuscript. We also thank A. de Bruin and S. Naidu for assistance in analyzing histological slides.
This work was funded by NIH grants to G.L. (R01CA85619, R01CA82259, R01HD04470, and P01CA097189). J.-L.C. is the recipient of a DoD award (BC061730).
Footnotes
Published ahead of print on 17 November 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Adams, M. R., R. Sears, F. Nuckolls, G. Leone, and J. R. Nevins. 2000. Complex transcriptional regulatory mechanisms control expression of the E2f3 locus. Mol. Cell. Biol. 203633-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen, D., R. Opavsky, M. Pacal, N. Tanimoto, P. Wenzel, M. W. Seeliger, G. Leone, and R. Bremner. 2007. Rb-mediated neuronal differentiation through cell-cycle-independent regulation of E2f3a. PLoS Biol. 51504-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke, A. R., E. R. Maandag, M. van Roon, N. M. van der Lugt, M. van der Valk, M. L. Hooper, A. Berns, and H. te Riele. 1992. Requirement for a functional Rb-1 gene in murine development. Nature 359328-330. [DOI] [PubMed] [Google Scholar]
- 4.Dagnino, L., C. J. Fry, S. M. Bartley, P. Farnham, B. L. Gallie, and R. A. Phillips. 1997. Expression patterns of the E2F family of transcription factors during murine epithelial development. Cell Growth Differ. 8553-563. [PubMed] [Google Scholar]
- 5.Danielian, P. S., L. B. Friesenhahn, A. M. Faust, J. C. West, A. M. Caron, R. T. Bronson, and J. A. Lees. 2008. E2f3a and E2f3b make overlapping but different contributions to total E2f3 activity. Oncogene 276561-6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Bruin, A., L. Wu, H. I. Saavedra, P. Wilson, Y. Yang, T. J. Rosol, M. Weinstein, M. L. Robinson, and G. Leone. 2003. Rb function in extraembryonic lineages suppresses apoptosis in the CNS of Rb-deficient mice. Proc. Natl. Acad. Sci. USA 1006546-6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimova, D. K., and N. J. Dyson. 2005. The E2F transcriptional network: old acquaintances with new faces. Oncogene 242810-2826. [DOI] [PubMed] [Google Scholar]
- 8.Dirlam, A., B. T. Spike, and K. F. Macleod. 2007. Deregulated E2f-2 underlies cell cycle and maturation defects in retinoblastoma null erythroblasts. Mol. Cell. Biol. 248713-8728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fearon, E. R. 1997. Human cancer syndromes: clues to the origin and nature of cancer. Science 2781043-1050. [DOI] [PubMed] [Google Scholar]
- 10.Fromm, L., W. Shawlot, K. Gunning, J. S. Butel, and P. A. Overbeek. 1994. The retinoblastoma protein-binding region of simian virus 40 large T antigen alters cell cycle regulation in lenses of transgenic mice. Mol. Cell. Biol. 146743-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humbert, P. O., R. Verona, J. M. Trimarchi, C. Rogers, S. Dandapani, and J. A. Lees. 2000. E2f3 is critical for normal cellular proliferation. Genes Dev. 14690-703. [PMC free article] [PubMed] [Google Scholar]
- 12.Iavarone, A., E. R. King, X. M. Dai, G. Leone, E. R. Stanley, and A. Lasorella. 2004. Retinoblastoma promotes definitive erythropoiesis by repressing Id2 in fetal liver macrophages. Nature 4321040-1045. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda, M. A., L. Jakoi, and J. Nevins. 1996. A unique role for the Rb protein in controlling E2F. Proc. Natl. Acad. Sci. USA 933215-3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacks, T., A. Fazeli, E. M. Schmitt, R. T. Bronson, M. A. Goodell, and R. A. Weinberg. 1992. Effects of an Rb mutation in the mouse. Nature 359295-300. [DOI] [PubMed] [Google Scholar]
- 15.Kondoh, H. 1999. Transcription factors for lens development assessed in vivo. Curr. Opin. Genet. Dev. 9301-308. [DOI] [PubMed] [Google Scholar]
- 16.Lee, E. Y., C. Y. Chang, N. Hu, Y. C. Wang, C. C. Lai, K. Herrup, W. H. Lee, and A. Bradley. 1992. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359288-294. [DOI] [PubMed] [Google Scholar]
- 17.Leone, G., F. Nuckolls, S. Ishida, M. Adams, R. Sears, L. Jakoi, A. Miron, and J. R. Nevins. 2000. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol. Cell. Biol. 203626-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Logan, N., A. Graham, X. Zhao, R. Fisher, B. Maiti, G. Leone, and N. B. La Thangue. 2005. E2F-8: an E2F family member with a similar organization of DNA-binding domains to E2F-7. Oncogene 245000-5004. [DOI] [PubMed] [Google Scholar]
- 19.Maandag, E. C., M. van der Valk, M. Vlaar, C. Feltkamp, J. O'Brien, M. van Roon, N. van der Lught, A. Berns, and H. te Riele. 1994. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 134260-4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macleod, K. F., Y. Hu, and T. Jacks. 1996. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 156178-6188. [PMC free article] [PubMed] [Google Scholar]
- 21.Maiti, B., J. Li, A. de Bruin, F. Gordon, C. Timmers, R. Opavsky, K. Patil, J. Tuttle, W. Cleghorn, and G. Leone. 2005. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J. Biol. Chem. 28018211-18220. [DOI] [PubMed] [Google Scholar]
- 22.Myers, T. R., and I. Greenwald. 2005. lin-35 Rb acts in the major hypodermis to oppose ras-mediated vulval induction in C. elegans. Dev. Cell 8117-123. [DOI] [PubMed] [Google Scholar]
- 23.Pan, H., and A. E. Griep. 1994. Altered cell cycle regulation in the lens of HPV-16 E6 or E7 transgenic mice: implications for tumor suppressor gene function in development. Genes Dev. 81285-1299. [DOI] [PubMed] [Google Scholar]
- 24.Saavedra, H. I., L. Wu, A. de Bruin, C. Timmers, T. J. Rosol, M. Weinstein, M. L. Robinson, and G. Leone. 2002. Specificity of E2F1, E2F2, and E2F3 in mediating phenotypes induced by loss of Rb. Cell Growth Differ. 13215-225. [PubMed] [Google Scholar]
- 25.Tsai, K. Y., Y. Hu, K. F. Macleod, D. Crowley, L. Yamasaki, and T. Jacks. 1998. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell 2293-304. [DOI] [PubMed] [Google Scholar]
- 26.Tsai, S. Y., R. Opavsky, N. Sharma, L. Wu, S. Naidu, E. Nolan, E. Feria-Arias, C. Timmers, J. Opavska, A. de Bruin, J.-C. Chong, P. Trikha, P. Stromberg, R. J. Rosol, and G. Leone. 2008. Mouse development with a single E2F activator. Nature 4541137-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walkley, C. R., and S. H. Orkin. 2006. Rb is dispensable for self-renewal and multilineage differentiation of adult hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1039057-9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walkley, C. R., J. M. Shea, N. A. Sims, L. E. Purton, and S. H. Orkin. 2007. Rb regulates interactions between hematopoietic stem cell and their bone marrow microenvironment. Cell 1291081-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinberg, R. A. 1992. The retinoblastoma gene and gene product. Cancer Surv. 1243-57. [PubMed] [Google Scholar]
- 30.Weinberg, R. A. 1995. The retinoblastoma protein and cell cycle control. Cell 81323-330. [DOI] [PubMed] [Google Scholar]
- 31.Wenzel, P. L., L. Wu, A. de Bruin, J. L Chong, W. Y. Chen, G. Dureska, E. Sites, T. Pan, A. Sharma, K. Huang, R. Ridgway, K. Mosaliganti, R. Sharp, R. Machiraju, J. Saltz, H. Yamaoto, J. C. Cross, M. L. Robinson, and G. Leone. 2007. Rb is critical in a mammalian tissue stem cell population. Genes Dev. 2185-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams, B. O., E. M. Schmitt, L. Remington, R. T. Bronson, D. M. Albert, R. A. Weinberg, and T. Jacks. 1994. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 134251-4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu, L., C. Timmer, B. Maiti, H. I. Saavedra, L. Sang, G. T. Chong, F. Nuckolls, P. Giangrande, F. A. Wright, S. J. Field, M. E. Greenberg, S. Orkin, J. R. Nevins, M. L. Robinson, and G. Leone. 2001. The E2F1-3 transcription factors are essential for cellular proliferation. Nature 414457-462. [DOI] [PubMed] [Google Scholar]
- 34.Wu, L., A. de Bruin, H. I. Saavedra, M. Starovic, A. Trimboli, Y. Yang, J. Opavska, P. Wilson, J. C. Thompson, M. C. Ostrowski, T. J. Rosol, L. A. Woollett, M. Weinstein, J. C. Cross, M. I. Robison, and G. Leone. 2003. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 421942-947. [DOI] [PubMed] [Google Scholar]
- 35.Ziebold, U., E. Y. Lee, R. T. Bronson, and J. A. Lees. 2003. E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol. Cell. Biol. 236542-6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziebold, U., T. Reza, A. Caron, and J. A. Lees. 2001. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 15386-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.