

Abstract
Cell death related nuclease 4 (CRN-4) is one of the apoptotic nucleases involved in DNA degradation in Caenorhabditis elegans. To understand how CRN-4 is involved in apoptotic DNA fragmentation, we analyzed CRN-4's biochemical properties, in vivo cell functions, and the crystal structures of CRN-4 in apo-form, Mn2+-bound active form, and Er3+-bound inactive form. CRN-4 is a dimeric nuclease with the optimal enzyme activity in cleaving double-stranded DNA in apoptotic salt conditions. Both mutational studies and the structures of the Mn2+-bound CRN-4 revealed the geometry of the functional nuclease active site in the N-terminal DEDDh domain. The C-terminal domain, termed the Zn-domain, contains basic surface residues ideal for nucleic acid recognition and is involved in DNA binding, as confirmed by deletion assays. Cell death analysis in C. elegans further demonstrated that both the nuclease active site and the Zn-domain are required for crn-4's function in apoptosis. Combining all of the data, we suggest a structural model where chromosomal DNA is bound at the Zn-domain and cleaved at the DEDDh nuclease domain in CRN-4 when the cell is undergoing apoptosis.
Apoptosis is a fundamental process essential for the development and maintenance of tissue homeostasis, the elimination of damaged cells, and antiviral defense (1, 22, 35). One of the major biochemical features of apoptosis is chromosome DNA fragmentation implemented by apoptotic nucleases. Inactivation of these apoptotic nucleases produces undigested DNA and is linked to a number of autoimmune disorders (38). Two major apoptotic DNases in eukaryotes, CAD (15, 31) and Endo G (26, 37), have been shown by biochemical experiments and genetic screens to be activated in apoptosis. Both proteins are endonucleases that attack chromatin to yield 5′-phosphate groups first at the level of 50- to 300-kb cleavage products and next at the level of internucleosomal DNA fragmentation.
Nevertheless, CAD was not identified in Caenorhabditis elegans, the model organism for the study of apoptosis. An Endo G homologue, mitochondrial CPS-6, and a type II DNase, NUC-1, have been implicated in mediating apoptotic DNA fragmentation in C. elegans (37, 47). In addition to CPS-6 and NUC-1, seven additional cell death-related nucleases (CRNs) have been identified by the RNA interference (RNAi)-based functional genomic screen (39). A number of CRNs interact with CPS-6 to form a degradeosome, a protein complex assembled during apoptosis and composed of CPS-6, CYP-13, WAH-1, CRN-1, CRN-4, and CRN-5. Some of the CRNs, such as CRN-2, CRN-3, and CRN-6, appear not associated with degradeosome (39). It has been suggested that apoptotic DNA degradation is a complicated process, requiring nucleases with different substrate specificities, such as DNA nicking, gap-dependent endonuclease, and exonuclease digestion activities, so that these nucleases work cooperatively to promote fragmentation of chromatin at various stages in a sequential way (38). The newly identified CRNs are all predicted by sequence analysis to have some homology to known nuclease domains. For instance, CRN-1 is homologous to human FEN1, containing two XPG nuclease domains (19, 30), and CRN-5 is homologous to the exosome component protein Rrp46, containing an RNase PH domain (39).
Similar to other CRNs, CRN-4 also contains a “classical” nuclease domain, i.e., a 3′-5′ exoribonuclease DEDDh domain, and additionally a C-terminal domain with unknown function (see Fig. 1). RNAi knockdown of the crn-4 gene results in accumulation of TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling)-positive nuclei and delays the appearance of embryonic cell corpses during development, indicating its involvement in apoptotic DNA fragmentation (39). The DEDDh superfamily (classified as DnaQ-like 3′-5′ exonuclease family in SCOP) consists of both DNases and RNases that are involved in various aspects of RNA metabolism and degradation and also DNA proofreading and repair. Several typical DEDDh family proteins that share high sequence identities (∼30% at the DEDDh domain) to CRN-4 are shown in Fig. 1, including C. elegans ERI-1a involved in the regulation of RNAi (14, 23), human 3′hExo involved in histone mRNA degradation (12, 48), human TREX2 involved in DNA ends processing during DNA metabolism and cellular proliferation (5, 32, 40), bacterial RNase T essential for 5S and 23S rRNA maturation (27, 28) and tRNA turnover (11), and the epsilon subunit of DNA polymerase III (ɛ186) involved in a proofreading step during DNA replication (13, 18). However, the biological function of CRN-4, as well as its substrates, either RNA or DNA, in nonapoptotic cells is yet to be identified.
FIG. 1.

Domain structures and the sequence alignment of CRN-4 with other DEDDh family of nucleases. The upper panel shows a comparison of the domain structure of CRN-4 to ERI-1a (14, 23), 3′hExo (12, 48), TREX2 (5, 32, 40), ɛ subunit of proofreading DNA polymerase III (ɛ186) (13, 18), and RNase T (27, 28). CRN-4 has a classical 3′-5′exonuclease DEDDh domain and an additional C-terminal function-unknown domain. ERI-1a and 3′hExo have an additional N-terminal SAP domain. The lower panel shows the sequence alignment at the DEDDh active site regions. The five highly conserved residues in CRN-4—Asp15, Glu17, Asp115, Asp184, and His179—are displayed in red and green.
The additional C-terminal domain in CRN-4 does not display significant sequence homology to other proteins currently found in available databases. To determine the function of each domain of CRN-4 and to reveal the molecular basis of DNA fragmentation in apoptosis, we characterized the biochemical properties and determined the crystal structure of CRN-4. The biochemical, structural, and functional assays consistently suggest that the C-terminal novel-fold Zn-domain of CRN-4 is involved in DNA binding and that the N-terminal nuclease domain is responsible for DNA degradation. The present study therefore provides new insights into the DEDDh family of nucleases in DNA fragmentation in apoptosis.
MATERIALS AND METHODS
Protein expression and purification.
The plasmid of pGEX4T-2-CRN-4 (39) was transformed into Escherichia coli BL21(DE3)/pLysS strain cultured in LB medium supplemented with 50 μg of ampicillin/ml to an optical density at 600 nm of 0.5 to 0.6. Expression of glutathione S-transferase (GST)-fused CRN-4 was induced by 0.5 to 0.8 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 18°C for 20 h. The harvested cells were disrupted by microfluidizer in phosphate-buffered saline (1 mM KH2PO4, 10 mM Na2HPO4, 20 mM NaCl, 2.7 mM KCl [pH 7.4]). The crude cell extract was passed through a glutathione-Sepharose column (GE Healthcare Biosciences Co., Piscataway, NJ) in a standard protocol, and the collected protein fractions were thrombin digested to remove the GST tag at room temperature for 16 h. The protein sample was then applied to a HiTrap heparin column (GE Healthcare), followed by application to a gel filtration chromatography column (Superdex 200; GE Healthcare). Purified CRN-4 samples were concentrated to 10 to 15 mg/ml in 150 mM NaCl and 50 mM Tris-HCl (pH 7.5). All of the CRN-4 point mutants were generated by QuikChange site-directed mutagenesis kits (Stratagene, La Jolla, CA) and purified by the same procedures as for wild-type CRN-4.
The genes of the Zn-domain deletion mutant (residues 1 to 206) was amplified by PCR and subcloned into NdeI/XhoI sites of expression vectors pET-28a (Novagen, Madison, WI) to generate the N-terminal His-tagged construct. The construct was transformed into E. coli BL21-CodonPlus(DE3)-RIPL strain (Stratagene) and cultured in LB medium supplemented with 30 μg of kanamycin/ml to an optical density at 600 nm of 0.5 to 0.6. Expression of the Zn-domain deletion mutant was induced by 0.5 to 0.8 mM IPTG at 18°C for 20 h. The harvested cells were disrupted by a microfluidizer in buffer (50 mM Tris-HCl, 300 mM NaCl [pH 7.5]) and loaded into a Ni-nitrilotriacetic acid resin affinity column (Qiagen, Inc., Chatsworth, CA), followed by application to a Q column (GE Healthcare) and a gel filtration chromatography column (Superdex 200; GE Healthcare). The purified Zn-domain deletion mutant was concentrated to 10 to 15 mg/ml in 150 mM NaCl and 50 mM Tris-HCl (pH 7.5).
DNase activity assay.
DNase activity assays were performed using plasmid DNA, single-stranded DNA (ssDNA; forward strand. 5′-GACGCTGCCGAATTCTGGCGTTAGGAGATACCGATAAGCTTCGGCTTAA-3′) and double-stranded DNA (dsDNA; annealed forward and reverse strands, reverse strand, 5′-CTTAAGCCGAAGCTTATCGGTATCTCCTAACGCCAGAATTCGGCAGCGT-3′) as the substrates. The purified CRN-4 (0.1 to 50 μM) was incubated with either 300 ng of pQE30 plasmid DNA, 0.3 μM ssDNA, or 0.3 μM dsDNA in a solution of 100 mM NaCl and 20 mM Tris-HCl (pH 7.5) with a final volume of 10 μl at room temperature for 30 or 120 min. DNA digestion reactions were then stopped by the addition of 10 mM protease K for 30 min to remove CRN-4. The plasmid digestion patterns were resolved on 1% agarose gels, whereas the ssDNA and dsDNA digestion patterns were resolved on 20% Tris-borate-EDTA gels. All gels were stained with ethidium bromide.
For the exonuclease activity assays (see Fig. 2F), the 10-mer single-stranded DNA (5′-ACAAATACTC-3′) were labeled at the 5′ end with [γ-32P]ATP by T4 polynucleotide kinase and at the 3′ end with [α-32P]dCTP by terminal deoxynucleotidyltransferase. The 5′-labeled or 3′-labeled ssDNA (20 nM) was then incubated with purified CRN-4 at various concentrations (0 to 20 μM) in a solution of 100 mM NaCl and 20 mM Tris-HCl (pH 7.5) at room temperature for 2 h. The reaction was then stopped by the addition of DNA loading dye containing formamide at 95°C for 5 min.
FIG. 2.

In vitro nuclease activity assays of CRN-4. (A) The gel filtration profile of CRN-4 suggested a size of ∼70 kDa, which was about double its calculated molecular mass of 34,641.9 Da, suggesting that CRN-4 was a homodimer in solution in 150 mM NaCl and 50 mM Tris-HCl at pH 7.5. The markers used here include blue dextran (2,000 kDa), conalbumin (75 kDa), and ovalbumin (43 kDa). Native gel analysis of purified CRN-4 also shows that CRN-4 has a molecular mass of ∼70 kDa. (B) CRN-4 digested pQE30 plasmid DNAs in the presence of 1 mM magnesium ions in an enzyme concentration-dependent manner (lanes 2 to 4) but did not digest DNA in the presence of 5 mM EDTA (lane 5). The starting supercoiled substrate (s) was cleaved by CRN-4 to generate linear DNA (l) and open-circle DNA (o). (C) The DNase activity of 1-μM CRN-4 in plasmid digestion is close to that of 0.001 μM DNase I (bovine; Sigma). (D) RNA digestion assays show that a single-stranded 20-mer RNA was digested by CRN-4 in the presence of 1 mM magnesium ions in 100 mM NaCl and 20 mM Tris-HCl (pH 7.5) (lanes 2 to 4). The RNase activity was abolished in the presence of 5 mM EDTA (lane 5). (E) CRN-4 digested a double-stranded 49-mer DNA more efficiently than ssDNA. (F) The ssDNA labeled at either the 5′ or the 3′ end with 32P was incubated with CRN-4. Only the 5′-end-labeled DNA was digested into ladder bands, suggesting that CRN-4 has 3′-5′ exonuclease activity.
The DNA digestion patterns were resolved on 20% denaturing polyacrylamide gels and visualized by autoradiography (Fujifilm FLA-5000).
RNase activity assay.
The 20-mer single-stranded RNA (5′-ACUGGACAAAUACUCCGAGG-3′) was first labeled at the 5′ end with [γ-32P]ATP by T4 polynucleotide kinase and then purified on a Microspin G-25 column (GE Healthcare) to remove the unincorporated nucleotides. The 5′-labeled ssRNA (20 nM) was then incubated with purified CRN-4 at various concentrations (0.1 to 0.9 μM) in a solution of 100 mM NaCl and 20 mM Tris-HCl (pH 7.5) at room temperature for 20 min. The reaction was then stopped by the addition of RNA loading dye containing formamide at 95°C for 5 min. The RNA digestion patterns were resolved on 20% denaturing polyacrylamide gels and visualized by autoradiography (Fujifilm FLA-5000).
DNA-binding assays.
Electrophoresis mobility shift assays were performed using a 31-mer dsDNA as the substrate (forward strand, 5′-TTTGTCCGGGTAAAGAGGGCCGTTGGGTTCG-3′; reverse strand, 5′-CGAACCCAACGGCCCTCTTTACCCGGACAAA-3′). The DNA substrates were labeled first at their 5′ ends with [γ-32P]ATP by T4 polynucleotide kinase and then purified by using a Microspin G-25 column (GE Healthcare) to remove the unincorporated nucleotides. The 5′-labeled dsDNA substrate (20 nM) was then incubated with different concentrations of CRN-4 and Zn-domain deletion mutant (5 to 80 μM) in a buffer containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 5 mM EDTA at 23°C for 20 min. After incubation, the reaction mixture was applied to 20% Tris-borate-EDTA gels, which were exposed to the phosphorimaging plate (Fujifilm) for autoradiographic visualization.
Crystallization and X-ray diffraction data collection.
Native and selenomethionine-labeled crystals were grown by the hanging-drop vapor diffusion method at 4°C. The crystallization drop was made by mixing 1 μl of protein solution and 1 μl of reservoir solution. Native CRN-4 was crystallized in three different conditions with a reservoir containing: (i) 0.2 M sodium formate and 20% PEG 3350; (ii) 0.2 M ammonium acetate, 10 mM calcium chloride, 50 mM sodium cacodylate, and 10% PEG 4000; or (3) 5% iso-propanol, 0.1 M HEPES (pH 7.5), and 10% PEG 4000. Selenomethionine-labeled CRN-4 was crystallized in 5% isopropanol, 0.1 M HEPES (pH 7.5), and 10% PEG 4000 by the microseeding method, using crushed native crystals as the crystal seeds. The single-wavelength diffraction data of the native CRN-4 crystal and the multiwavelength anomalous diffraction data of the selenomethionine-labeled CRN-4 crystal were collected at 100 K at the 13C1 and 13B1 beamlines, respectively, at the National Synchrotron Radiation Research Center (NSRRC), Taiwan.
The Mn-bound CRN-4 crystals were obtained by soaking the native CRN-4 crystals in buffer solutions containing 5 mM MnCl2, 5 mM AMP, 5% iso-propanol, 0.1 M HEPES (pH 7.5), and 10% PEG 4000 for 12 h. The Er-bound CRN-4 crystals were grown from a well solution containing 5 mM ErCl3-6H2O, 5 mM AMP, 0.2 M sodium formate, and 20% PEG 3350. The crystals were then back-soaked in 0.2 M sodium formate-20% PEG 3350 for 1 h before data collection. The X-ray diffraction data of Mn-bound and Er-bound CRN-4 were collected at 100 K at the 13C1 beamline at the NSRRC. All of the diffraction data were processed by HKL2000 (36), and the diffraction statistics are listed in Table 1.
TABLE 1.
Crystallographic statistics of CRN-4
| Parametera | CRN-4 | Se-Met-labeled CRN-4
|
CRN-4/Mn | CRN-4/Er | ||
|---|---|---|---|---|---|---|
| Peak | High-remote | Infection | ||||
| Data collection and processing | ||||||
| Wavelength (Å) | 0.999 | 0.978993 | 0.963960 | 0.979185 | 0.999 | 0.999 |
| Space group | P21212 | P21212 | P21212 | P21212 | ||
| Cell dimensions (a/b/c) (Å) | 126.47/165.38/64.26 | 126.13/164.72/64.83 | 125.13/165.43/64.59 | 130.28/157.80/65.24 | ||
| Resolution range (Å) | 30.0-2.5 | 30.0-3.4 | 30.0-3.1 | 30.0-3.2 | 30.0-2.8 | 30.0-2.6 |
| No. of observed reflections | 194,089 | 193,113 | 251,889 | 228,853 | 122,074 | 212,008 |
| No. of unique reflections | 45,644 | 19,400 | 25,181 | 23,055 | 30,689 | 43,082 |
| Redundancy* | 4.3 (3.8) | 10.0 (10.3) | 10.0 (10.2) | 9.9 (9.4) | 4.1 (3.8) | 4.9 (5.1) |
| Completeness* (%) | 96.4 (91.5) | 99.8 (99.5) | 99.7 (99.2) | 99.6 (98.5) | 92.2 (88.1) | 99.8 (100) |
| Rsym* (%) | 5.2 (42.6) | 8.9 (51.0) | 7.6 (43.8) | 8.5 (47.8) | 6.2 (42.4) | 7.7 (43.6) |
| I/σ(I)* | 21.5 (3.0) | 22.9 (4.3) | 27.5 (4.6) | 24.3 (4.1) | 20.5 (3.4) | 19.6 (3.7) |
| Refinement statistics | ||||||
| Resolution range (Å) | 3.0-2.5 | 3.0-2.8 | 30.0-2.6 | |||
| No. of reflections (work/test) | 43,319/2,301 | 20,978/1,564 | 39,961/2,119 | |||
| R-factor/R-free (%) | 20.6/24.7 | 20.1/23.9 | 22.5/26.5 | |||
| No. of nonhydrogen atoms | ||||||
| Protein | 4,822 | 4,792 | 4,806 | |||
| Metal ion | 2 | 4 | 4 | |||
| No. of solvent molecules | 173 | 81 | 154 | |||
| Model quality | ||||||
| RMS deviations in: | ||||||
| Bond length (Å) | 0.009 | 0.013 | 0.012 | |||
| Bond angle (°) | 1.145 | 1.443 | 1.320 | |||
| Avg B-factor (Å2) | 39 | 42 | 40 | |||
| Ramachandran plot (%) | ||||||
| Most favored | 91.2 | 89.5 | 91.2 | |||
| Additionally allowed | 8.8 | 10.5 | 8.8 | |||
| Generously allowed | 0 | 0 | 0 | |||
| Disallowed | 0 | 0 | 0 | |||
Last shell values are indicated in parentheses.
Structure determination and refinement.
CRN-4 crystallized in the orthorhombic space group P21212 with two molecules per asymmetric unit. The reflection phases were determined by multiwavelength anomalous diffraction (MAD) using SHELX97 (42) at a 3.5-Å resolution. The MAD phases were extended to 2.5 Å by phase extension (DM in CCP4) using the high-resolution data from native CRN-4. The protein model was constructed by the program Coot with water molecules added by ARP/wARP and refined by REFMAC-5 (45). Two structural Zn ions were added into the model at the final stage of refinement. The Mn-bound and Er-bound CRN-4 structures were solved by molecular replacement and refined by REFMAC-5. The final structural models had good stereochemistry as indicated by PROCHECK. Structural coordinates and diffraction structure factors have been deposited in the RCSB Protein Data Bank (PDB) with PDB codes of 3CG7 for apo-form CRN-4, 3CM5 for Mn-bound CRN-4, and 3CM6 for Er-bound CRN-4.
In vivo cell death assay.
C. elegans strains were maintained by using standard procedures (3). The CRN-4 expression constructs (at 25 μg/ml) were injected into crn-4(tm1415) animals as previously described (33), using the pRF4 (at 25 μg/ml) as a coinjection marker. The numbers of cell corpses in living transgenic embryos were determined by using Nomarski optics as described previously (44).
RESULTS
CRN-4 is a dimeric enzyme with both DNase and RNase activities.
GST-fused CRN-4 was overexpressed in the E. coli BL21(DE3)/pLysS strain. The GST-tag was removed by thrombin digestion after elution of the protein samples through a glutathione-Sepharose column. The full-length CRN-4 (residues 1 to 298) was further purified on a heparin and a gel filtration column. The gel filtration profiles showed that CRN-4 was mostly folded into a dimer with a molecular weight of ∼70 kDa at pH 7.5, with a small portion of CRN-4 folded into a tetramer (Fig. 2A). Native-gel analysis and dynamic light scattering (data not shown) gave consistent results of a homodimeric CRN-4 at physiological conditions.
To determine the DNase and RNase activities of CRN-4, recombinant dimeric CRN-4 was incubated with DNA and RNA substrates in the presence or absence of magnesium ions. CRN-4 digested a plasmid DNA in the presence of Mg2+ in an enzyme concentration-dependent manner (Fig. 2B). In contrast, CRN-4 did not digest DNA in the presence of EDTA, which chelated the magnesium ions in solution. The plasmid digestion assays at the low enzyme concentration (0.5 μM) produced open-circular and linear DNA products with only a single and two nicks; therefore, it remains to be determined whether CRN-4 has any sequence preferences in DNA digestion. Moreover, the DNase activity of CRN-4 in plasmid DNA digestion was ∼1,000-fold lower than that of DNase I, suggesting that CRN-4 was a weak enzyme (Fig. 2C). Similarly, CRN-4 was able to cleave a 20-mer single-stranded RNA in the presence of magnesium ions but did not cleave RNA substrates in the presence of EDTA (Fig. 2D). These results suggest that CRN-4 is a metal ion-dependent nuclease containing both DNase and RNase activities.
To analyze its substrate preference, CRN-4 was incubated with either a 49-mer ssDNA or a dsDNA. CRN-4 digested the dsDNA completely at concentrations greater than 5 μM, whereas CRN-4 did not completely digest the ssDNA at concentrations up to 50 μM (Fig. 2E). Similar results were observed using a 20-mer and a 31-mer ssDNA and dsDNA of different sequences (data not show). In summary, CRN-4 cleaves nucleic acid substrates with efficiencies in the following order: ssRNA > dsDNA > ssDNA. To further find out whether CRN-4 has an exonuclease activity, a ssDNA labeled at the 5′ end or at 3′ end with 32P was incubated with CRN-4. Only the 5′-end-labeled DNA, and not the 3′-end-labeled DNA, produced ladder bands after digestion, suggesting that CRN-4 has 3′→5′ exoribonuclease activity in digestion of ssDNA (Fig. 2F).
CRN-4 is a more active DNase in apoptotic conditions over normal cellular conditions.
The concentrations of Ca2+, K+, and Na+ ions, as well as pH values, are different in normal growing cells and in apoptotic cells (16, 25). Previous studies have shown that several enzymes and nucleases involved in apoptosis are activated by divalent metal ions. The increased cytosolic Ca2+ concentration may stimulate these apoptotic enzymes during apoptosis (41, 49). Moreover, when a cell is triggered for apoptosis, the cellular concentrations of K+ decrease from 150 to 50 mM, and Na+ concentrations increase from 15 to 30 mM (2). The optimal pH range of apoptotic enzymes is estimated from 7.5 to 9.5 (10).
To find out the optimal pH for CRN-4 DNase function, CRN-4 was incubated with plasmid DNAs over a pH range from 5.5 to 10.0. We found that CRN-4 cleaved plasmid DNA substrates (91 to 97% of supercoiled DNA) most efficiently at the pH range of 6.5 to 9.0, while CRN-4 had a reduced DNase activity (77 to 87%) in acidic conditions at pH 5.5 to 6.5 or in basic conditions at pH 9.5 to 10.0 (Fig. 3A). CRN-4 was then incubated with plasmid DNAs in the presence or absence of Mg2+ or Ca2+ ions at pH 7.5. As shown in Fig. 3B, Ca2+ was a weak cofactor, with only slight enzyme activation at the high concentration of 5 mM (lanes 3 and 6). Moreover, Ca2+ did not function as an inhibitor when Mg2+ was present (lane 5). This result suggests that CRN-4 is an Mg2+-dependent enzyme in vivo.
FIG. 3.

DNase activity assays of CRN-4 in different pH and salt conditions. (A) pQE30 plasmids were digested by CRN-4 in 1 mM Mg2+ and 100 mM NaCl buffered at different pH (20 mM MES at pH 5.5 and 6.0; 20 mM HEPES at pH 6.5, 7.0, and 7.5; 20 mM Tris-HCl at pH 8.0, 8.5, and 9.0; 20 mM CAPS at pH 9.5 and 10.0). The optimal pH for CRN-4 was 6.5 to 9.0. The starting supercoiled substrate (s) was cleaved by CRN-4 to generate linear DNA (l) and open-circle DNA (o). The intensity of the supercoiled substrate gel band was quantified, and the substrate cleavage percentage was estimated and is indicated at the bottom of each lane. (B) Assays of plasmid digestion by CRN-4 in the presence of Mg2+ and/or Ca2+ show that Ca2+ is a weaker cofactor of CRN-4 (lanes 3 and 6) than Mg2+ (lane 4), suggesting that CRN-4 is a Mg2+-dependent enzyme. (C) Plasmid digestion by CRN-4 in the presence of various concentrations of K+. The supercoiled substrate cleavage percentages are indicated below the gel. (D) The DNase activity of CRN-4 increased as the Na+ concentration increased from 15 to 90 mM.
On the other hand, the DNase activity of CRN-4 was sensitive to the concentrations of K+ and Na+. A higher DNase activity (80.4%) was observed in 50 mM KCl (Fig. 3C, lane 4), with lower activity (39.9%) in 150 mM KCl (lane 8). The DNase activity of CRN-4 increased from 57 to 85.0% as the Na+ concentration rose from 15 to 90 mM (Fig. 3D). CRN-4's DNase activity was better in 30 mM NaCl (82.2%) than in 15 mM NaCl (57.0%). These results suggest that CRN-4 has optimal DNase activity under apoptotic conditions, i.e., 50 mM KCl and 30 mM NaCl at pH 6.5 to 9.
Overall structure of CRN-4.
The full-length apo-form CRN-4 was crystallized by hanging-drop vapor diffusion at 4°C. The native apo-form crystals were crushed into microseeds to assist the crystallization of selenomethionine-labeled CRN-4. The Mn-bound CRN-4 crystals were obtained by soaking the native CRN-4 crystals in 5 mM MnCl2 for 12 h, whereas the Er-bound crystals were grown from a solution containing 5 mM ErCl3. Single-wavelength X-ray diffraction data of apo-form, Mn-bound and Er-bound CRN-4, as well as multiwavelength anomalous diffraction data of Se-labeled CRN-4, were collected at 100 K at beamlines 13C1 and 13B1 at the NSRRC. The crystal structure of CRN-4 was then solved by MAD, and the structural model was used as the search model to solve the structures of Mn-bound and Er-bound CRN-4 by molecular replacement. The crystal structures of apo-form, Mn-bound, and Er-bound CRN-4 were further refined to resolutions of 2.5, 2.8, and 2.6 Å, respectively, with good refinement and model geometry statistics. All of the diffraction and refinement details are listed in Table 1.
The two CRN-4 molecules in one asymmetric unit of the orthorhombic P21212 cell are folded into a dimeric structure (Fig. 4A). The N-terminal DEDDh domain adopts an α/β globular fold and shares similar structure to those of other members of DEDDh exonucleases. The C-terminal domain has a novel mixed α/β fold with a zinc ion bound to four cysteine residues, and we therefore refer to this hereafter as the Zn-domain. This Zn-domain is intimately attached to the DEDDh domain with a buried interface of 1,780 Å2 (62% polar residues and 38% nonpolar residues in the interface). The interfaces between the two monomeric subunits are formed by both the DEDDh domain and the Zn-domain with a buried surface of 1,780 Å2. An estimated 56.5% of nonpolar atoms and 43.5% of polar atoms are buried in the dimeric interface, within which nine hydrogen bonds and one salt bridge stabilize the dimeric structure.
FIG. 4.

Overall structure, structural comparison, and active site of CRN-4. (A) The stereo view of the crystal structure of CRN-4 reveals a homodimeric architecture, with each subunit containing a DEDDh nuclease domain (in green) and a C-terminal Zn-domain (in salmon). The locations of the DEDDh active site and zinc-binding site in one of the subunits are marked by a pink and a blue box, respectively. (B) Superposition of the DEDDh nuclease domain of CRN-4 (green) on those of ɛ186 (yellow, PDB code 1J53), RNase T (blue, PDB code 2IS3), and 3′hExo (red, PDB code 1ZBU). CRN-4 contains an additional Zn-domain compared to ɛ186 and RNase T, while 3′hExo contains an extra SAP domain. (C) The nuclease active sites of Mn-bound and Er-bound CRN-4. The five conserved DEDDh residues in the catalytic domain are displayed in stick models. The blue balls indicate water molecules. The omit 2FoFc maps of Mn2+ and Er3+ are displayed in cyan and contoured at 4.0 σ. (D) CRN-4 was active in the presence of Mg2+ and Mn2+, but inactive with Er3+, as shown by plasmid digestion assays. (E) The CRN-4 mutants D15A/E17A, D115A, H179A, and D184A digested plasmid DNA less efficiently, with activities reduced by ∼5-fold, than did the wild-type CRN-4. The triple mutant D15A/E17A/D115A had almost no observable DNase activity. (F) The zinc ion was bound to four cysteine residues—Cys210, Cys260, Cys263, and Cys270—in the Zn-domain of CRN-4. The omit 2FoFc map of zinc ion is displayed in cyan and contoured at 7.0 σ.
Superposition of the DEDDh domain of CRN-4 over those of ɛ186, RNase T, and 3′hExo shows that the overall structures of the DEDDh domain in these nucleases are highly similar (Fig. 4B). The average root mean square deviations were 0.80 Å for 121 Cα atoms between CRN-4 and ɛ186, 0.91 Å for 97 Cα atoms between CRN-4 and RNase T, and 0.78 Å for 198 Cα atoms between CRN-4 and 3′hExo. The major difference of the overall fold between CRN-4 and these proteins is that CRN-4 has an additional C-terminal Zn-domain. 3′hExo also has an extra SAP domain; however, this SAP domain is located far away from the Zn-domain when the DEDDh domains of CRN-4 and 3′hExo are superimposed (Fig. 4B).
Nuclease active site of CRN-4.
The N-terminal DEDDh domain contains conserved catalytic residues Asp15, Glu17, Asp115, His179, and Asp184. The crystal structure of Mn-bound CRN-4 showed that Mn2+ was bound to Asp15, Glu17, and Asp184 and to a water molecule at the active site (Fig. 4C). Previous studies suggest that DEDDh family nucleases catalyze the hydrolysis of nucleic acids by a two-metal ion mechanism with two divalent metal ions bound at A and B sites, respectively (18, 43). In the structure of Mn-bound CRN-4, the Mn2+ was bound at the A site, and the B site was not occupied. The plasmid digestion assays further showed that Mn2+ was an active cofactor in CRN-4, whereas Er3+ was not an active cofactor (Fig. 4D). In the crystal structure of Er-bound CRN-4, the side chain of Asp15 was rotated so that the two carboxyl oxygen atoms were both bound to Er3+. The Asp15 was thus unable to bind to the second metal ion at site B. This change in metal coordination disrupted the functional structure of the active site and accounted for the inhibition of enzyme activity by Er3+.
The five conserved residues were then subjected to site-directed mutagenesis to produce a double mutant (D15A/E17A), three single-site mutants (D115A, H179A, and D184A), and a triple mutant (D15A/E17A/D115A). All of the single and double mutants had compromised enzyme activities, showing ∼5-fold reduction in nuclease activity, compared to that of the wild-type CRN-4 in plasmid digestion assays, whereas the triple mutant had almost no observable DNase activity (Fig. 4E). Taken together, all of these results suggest that the nuclease active site of CRN-4 is located at the Mn2+ binding site, formed by the conserved Asp15, Glu17, Asp115, His179, and Asp184 residues in the DEDDh domain.
The C-terminal Zn-domain is involved in DNA binding.
The crystal structure of CRN-4 revealed a novel folded C-terminal Zn-domain. The structural homology searches on DALI (20) and NCBI-VAST (17) did not identify any similar structural fold in the databases. A metal ion was found in this domain, bound to the four cysteine residues Cys210, Cys260, Cys263, and Cys270, suggesting that it was likely a zinc ion. Both X-ray fluorescence spectrometry and inductively coupled plasma-atomic emission spectrometry confirmed that the metal ion in the apo-CRN-4 was indeed zinc (data not shown). This Zn2+ ion was coordinated in a tetrahedral geometry with ideal Zn-S distances of 2.39 Å/2.26 Å to Cys260, 2.45 Å/2.29 Å to Cys263, 2.30 Å/2.36 Å to Cys210, and 2.24 Å/2.24 Å to Cys270 in the two subunits of homodimeric CRN-4 (Fig. 4F). Sequence searches further identified homologous Zn-domains only in C. elegans and C. briggsae proteins, including ERI-1a (23) and seven hypothetical proteins, that all bear similar domain organization to that of CRN-4 with a DEDDh domain, followed by a C-terminal Zn-domain.
The calculation of the electrostatic surface potential of CRN-4 revealed that the Zn-domain is highly basic, with five arginine residues exposed to the surface: Arg216, Arg248, Arg253, Arg261, and Arg264 (Fig. 5). This suggests that the Zn-domain is likely involved in nucleic acid binding. To test this hypothesis, we constructed a Zn-domain deletion mutant (residues 1 to 206) that contained only the N-terminal DEDDh nuclease domain. Without the presence of the Zn-domain for dimer formation, the Zn-domain deletion mutant was dissociated into a monomer as assayed by gel filtration and dynamic light scattering (data not shown). Compared to the wild-type CRN-4, the Zn-domain deletion mutant cleaved a double-stranded 49-mer DNA less efficiently than the wild-type CRN-4 (Fig. 5C). The Zn-domain deletion mutant also bound to a 31-mer dsDNA less efficiently, with an ∼8-fold reduction in binding affinity, as shown by gel shift experiments (Fig. 5D), where similar binding activities are observed for 5 μM wild-type enzyme (lane 2) and 40 μM mutant (lane 10). The loss of the DNA-binding activity of the deletion mutant could be due to the structural change from a dimer to a monomer. It is also likely that the C-terminal Zn-domain is directly involved in DNA binding and the reduced DNase activity of the Zn-domain deletion mutant is due to its reduced DNA-binding affinity.
FIG. 5.

Electrostatic surface potential and DNA-binding sites of CRN-4. (A) The front and top views of the electrostatic surface potential of CRN-4 show that the surface of Zn-domain is highly basic. The color scale was set from −5 kT/e (red) to 5 kT/e (blue), as calculated by Pymol (9). (B) The top view of the ribbon model of CRN-4 shows the five arginine residues—Arg216, Arg248, Arg253, Arg261, and Arg264—in the Zn-domain (in salmon) exposing on the surface. (C) CRN-4 digested a double-stranded 49-mer DNA more efficiently than the C-terminal Zn-domain deletion mutant. The DNA was digested at 23°C for 180 min because the deletion mutant was less stable. (D) The Zn-domain deletion mutant bound to double-stranded 31-mer DNA with an ∼8-fold reduced affinity compared to the wild-type CRN-4, as assayed by gel shift experiments. The three arrowheads mark the protein-DNA complexes.
Both the DEDDh active site and the C-terminal Zn-domain of CRN-4 are required for the function of crn-4 in vivo.
To test whether the nuclease active site and the C-terminal Zn-domain of CRN-4 are required for the activity of crn-4 in vivo, we performed the transformation rescue experiments in a crn-4(tm1415) deletion mutant by injecting a construct expressing the crn-4::gfp fusion under the control of the dpy-30 gene promoter (Pdpy-30crn-4::gfp), which directs ubiquitous gene expression in C. elegans (21). The crn-4(tm1415) mutant contains a 514-bp deletion that removes the forth and fifth exons of crn-4 and is a putative null allele (data not shown). Like crn-4(RNAi), crn-4(tm1415) causes a delay-of-cell-death defect during embryonic development (Fig. 6A) (39). Compared to wild-type embryos, fewer cell corpses are seen in comma and 1.5-fold stage crn-4(tm1415) embryos and more cell corpses are seen in later embryonic stages (2-, 2.5-, and 3-fold stages) (Fig. 6A). The wild-type Pdpy-30crn-4::gfp construct can fully rescue the delay-of-cell-death defect of the crn-4(tm1415) mutant (Fig. 6A). In contrast, the mutant Pdpy-30crn-4::gfp construct carrying either the D15A/E17A/D115A mutations (the active site mutant) or a carboxyl-terminal deletion without the Zn-domain [CRN-4(1-206)] failed to rescue the cell death defect of the crn-4(tm1415) mutant (Fig. 6B and C). These results suggest that both the nuclease active site and the C-terminal Zn-domain of CRN-4 are needed for the CRN-4 function in apoptotic DNA degradation.
FIG. 6.

Cell death assay in C. elegans. (A) Transgenic crn-4(tm1415) animals expressing wild-type CRN-4, (B) CRN-4(D15A/E17A/D115A), or (C) CRN-4(1-206) under the control of the dpy-30 promoter were generated. For each construct, data were collected from three independent transgenic lines (smEx). The stages of transgenic embryos examined were: comma, 1.5-fold, 2-fold, 2.5-fold, 3-fold, and 4-fold. The y axis represents the average number of cell corpses scored, and error bars indicate standard deviations. Fifteen embryos were counted for each developmental stage. Statistical analyses were carried out by using the Student t test. The data of significant difference are indicated with asterisks: *, P < 0.0001; and **, P < 0.0005. There are no statistical differences between the crn-4 mutant and transgenic animals.
DISCUSSION
CRN-4 binds DNA through its Zn-domain and cleaves DNA through its DEDDh domain.
Our structural and biochemical analyses demonstrate that CRN-4 contains a catalytic DEDDh nuclease domain and a DNA-binding Zn-domain. Several members of the DEDD nuclease family also contain additional domains next to the catalytic domain that function in substrate binding and/or reorganization. For example, 3′hExo, involved in histone mRNA degradation, contains an N-terminal SAP domain that specifically recognizes the stem-loop region of histone mRNA (7, 24). E. coli ExoI, a DNase involved in ssDNA processing in DNA recombination and repair, contains a SH3 domain and a C-terminal helical region next to its DEDD domain to help it to encircle ssDNA substrates (4).
CRN-4 is similar to these two nucleases in that it also contains an extra C-terminal Zn-domain for substrate binding. Our deletion and biochemical assays demonstrate that this Zn-domain is indeed involved in DNA binding. A close look at the electrostatic surface potential of CRN-4 reveals that the basic surface formed by the exposed arginine residues located at the Zn-domain is suitable for substrate binding. This basic surface has two cleft regions ideal for binding the two phosphate backbones of dsDNA (Fig. 7). We therefore constructed a structural model of a dsDNA bound at the Zn-domain of CRN-4 by using the docking program ZDOCK (6). Not only did the phosphate backbones fit nicely into the clefts, a protruding region of CRN-4 also fit snugly into the major groove of DNA. This bound DNA is further extended and can be cleaved at the active site in the DEDDh catalytic domain. We therefore suggest that these two domains in CRN-4, i.e., the Zn-domain responsible for DNA binding and the DEDDh domain responsible for DNA cleavage, work together in DNA degradation. Moreover, CRN-4 in this model seems ideal for making a ssDNA nick on a dsDNA or, alternatively, making two nicks through the two nuclease active sites located on the same side of the CRN-4 dimer. However, CRN-4's substrate specificity and DNA cleavage pattern remain to be further characterized.
FIG. 7.
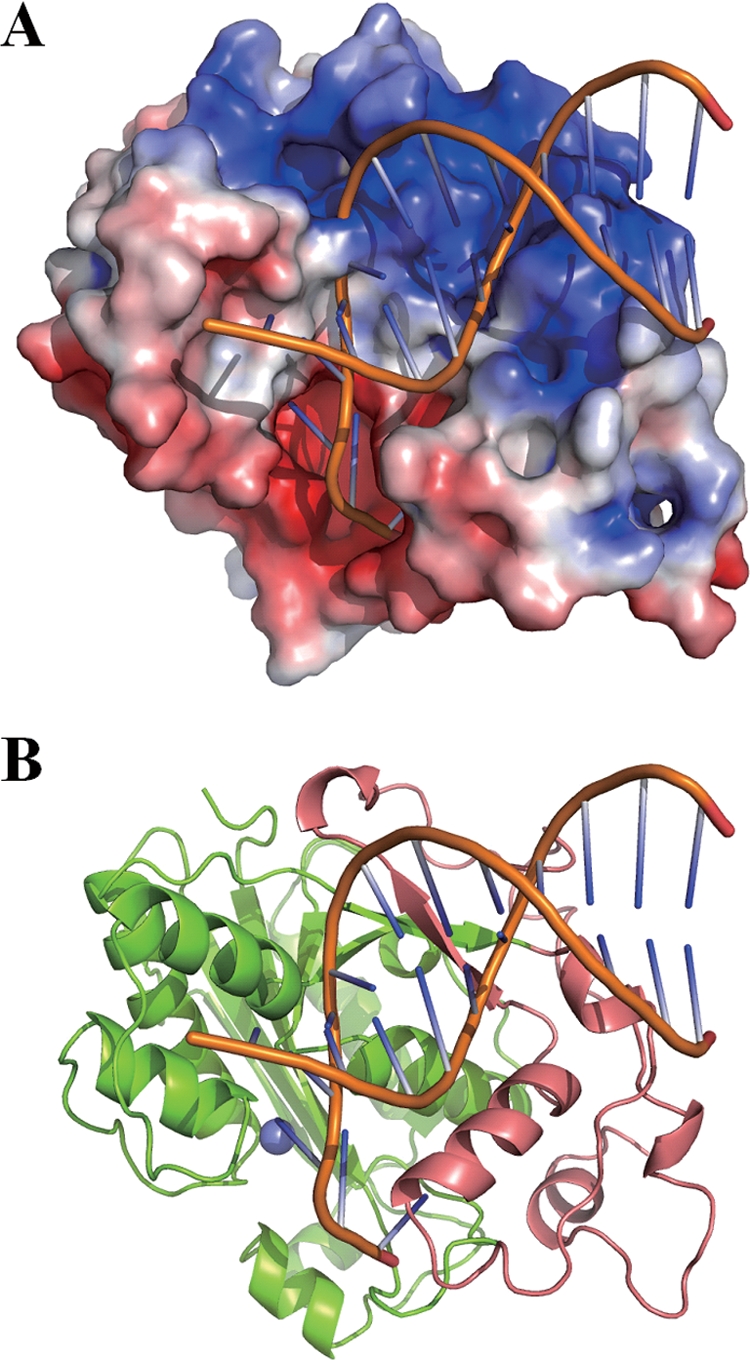
Structural model of CRN-4 bound with a DNA. (A) A model of a 12-mer dsDNA (PDB entry 1KLN) bound at the basic Zn-domain of CRN-4 was constructed by using Zdock (http://zdock.bu.edu/). The two phosphate backbones of DNA are bound snugly in the two cleft regions of the Zn-domain. For clarity, only one subunit of CRN-4 in the dimer is displayed. (B) The ribbon model of CRN-4 demonstrates that DNA is bound at the Zn-domain (in salmon) and further extended to the DEDDh domain (in green) for cleavage. The Mn2+ bound at the active site is displayed as a blue ball.
CRN-4 forms a dimer in a novel way.
Several DEDD proteins are homodimers, typical examples including bacterial RNase T, human PARN, and TREX2. RNase T is a stable dimer at various salt concentrations (29). It is involved in exonucleolytic trimming of tRNAs to yield a mature CCA end and generating the mature 3′ ends of 5S and 23S rRNAs. The crystal structure of the E. coli RNase T shows that the enzyme adopts an opposing dimeric arrangement, with the two catalytic DEDD active sites located at opposite sides of the dimer (Fig. 8) (51). Nevertheless, the DEDD active site in one of the monomers is located close to the conserved nucleic-acid binding sequence segments in the other monomer. It is therefore suggested that RNA substrates are bound in the nucleic-acid binding sequence regions in one subunit and cleaved at the DEDD active site in the other subunit in dimeric RNase T (50, 51). Oligoribonucleases, responsible for degrading small oligoribonucleotides to mononucleotides, also have a homodimeric arrangement similar to that of RNase T, indicating that this is one of the more common modes of dimerization in the DEDD family of nucleases (8).
FIG. 8.

Comparison of the different domain arrangement in dimeic DEDDh family proteins. Four DEDDh family nucleases, PARN, CRN-4, TREX2, and RNase T dimerize in different ways. One DEDDh domain, outlined by the dashed trapezoid, is displayed in the same orientation to show the different orientation of the second domain. A schematic diagram on the top panel represents different ways of dimerization identified in the DEDD family of nucleases. The arrows and ellipsoids mark the locations of twofold symmetry axes, yellow stars mark the active sites in the front side, and green stars mark the active sites in the opposite (back) side of the dimer. The PDB entry codes are 2A1R for PARN, 3CG7 for CRN-4, 1Y97 for TREX2, and 2IS3 for RNase T.
On the other hand, the human PARN and TREX2 dimerize in quite different orientations (Fig. 8). Dimeric PARN is involved in poly(A)-specific exoribonucleolytic digestion and dimer formation is required for its RNA-binding activity. The crystal structure of PARN reveals a quite different arrangement of the dimer, with two active sites facing opposite sides (46). TREX2 is a 3′-5′ exonuclease involved in the processing of DNA ends in DNA replication, repair, and recombination. The crystal structure of TREX2 reveals that the two active sites face the same side and are located at the outer edges, providing open access for DNA substrates (34). The DNA-binding site in TREX2 is juxtaposed to the active site in the same monomer, suggesting that each monomer works independently in DNA binding and cleavage. It is further suggested that TREX2 may simultaneously bind two DNA strands in DNA repair or recombination pathways and, therefore, the way it dimerizes may be linked to its role in processing of two 3′ ends of DNA from opposite directions (34).
In comparison to dimeric RNase T, PARN, and TREX2, CRN-4 dimerizes in a novel way. The two active sites are located on the same side of the dimer, but the relative orientation and distance are distinct from those observed in other DEDD family nucleases. This novel mode of dimerization could be related to CRN-4's substrate specificity or the regulation of its enzyme activity. In a normal growing cell, CRN-4 may have one specific function in RNA/DNA processing, whereas in an apoptotic cell, CRN-4 likely switches its role to participate in DNA degradation. Our in vitro nuclease activity assays demonstrated that CRN-4 is an efficient enzyme in cleaving dsDNA over ssDNA. A structural model of the CRN-4/DNA complex also suggests that CRN-4 is appropriate for binding and digestion of a dsDNA. Moreover, CRN-4 has higher activity in apoptotic salt conditions. All of these biochemical and structural data support that CRN-4 is a functional dimeric DNase for DNA degradation at apoptotic conditions. We thus suggest that CRN-4 is a dimer that prefers to digest dsDNA during apoptosis.
However, it is still largely unknown how CRN-4 and other nucleases are regulated and recruited into apoptotic machinery and how they switch functions in a normal growing cell versus an apoptotic cell. Different modes of oligomeric assembly provide a pointer to further structural and biochemical analysis of an enzyme's substrate specificity and activity. It is possible that CRN-4 forms different oligomeric assemblies or interacts with different proteins and thus targets different substrates when a cell is, or is not, committed to apoptosis. Our work on CRN-4 paves the way for future studies of the role of CRN-4 in the normal cell and of the determinant factors in the regulation of enzyme activity in different cell stages.
Acknowledgments
This study was supported by research grants from Academia Sinica and the National Science Council, Taiwan, Republic of China (H.S.Y.); NIH grant R01 GM79097 (D.X.); and a grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (S.M.). Portions of this research were carried out at the National Synchrotron Radiation Research Center (BL-13B1 and BL-13C1), a national user facility supported by the National Science Council of Taiwan, Republic of China. The Synchrotron Radiation Protein Crystallography Facility is supported by the National Research Program for Genomic Medicine.
We thank Harry Wilson of Academia Sinica for manuscript editing.
Footnotes
Published ahead of print on 3 November 2008.
REFERENCES
- 1.Barber, G. N. 2001. Host defense, viruses, and apoptosis. Cell Death Differ. 8113-126. [DOI] [PubMed] [Google Scholar]
- 2.Barbiero, G., F. Duranti, G. Bonelli, J. S. Amenta, and F. M. Baccino. 1995. Intracellular ionic variations in the apoptotic death of L cells by inhibitors of cell cycle progression. Exp. Cell Res. 217410-418. [DOI] [PubMed] [Google Scholar]
- 3.Brenner, S. 1974. The genetics of Caenorhabditis elegans. Genetics 7771-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breyer, W. A., and B. W. Matthews. 2000. Structure of Escherichia coli exonuclease I suggests how processivity is achieved. Nat. Struct. Biol. 71125-1128. [DOI] [PubMed] [Google Scholar]
- 5.Chen, M. J., S. M. Ma, L. C. Dumitrache, and P. Hasty. 2007. Biochemical and cellular characteristics of the 3′ → 5′ exonuclease TREX2. Nucleic Acids Res. 352682-2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, R., L. Li, and Z. Weng. 2003. ZDOCK: an initial-stage protein-docking algorithm. Proteins 5280-87. [DOI] [PubMed] [Google Scholar]
- 7.Cheng, Y., and D. J. Patel. 2004. Crystallographic structure of the nuclease domain of 3′hExo, a DEDDh family member, bound to rAMP. J. Mol. Biol. 343305-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin, K.-H., C.-Y. Yang, C.-C. Chou, A. H.-J. Wang, and S.-H. Chou. 2006. The crystal structure of XC847 from Xanthomonas campestris: a 3′-5′ oligoribonuclease of DnaQ fold family with a novel opposingly shifted helix. Proteins Struct. Funct. Bioinform. 651036-1040. [DOI] [PubMed] [Google Scholar]
- 9.Delano, W. L. 2002. The PyMOL molecular graphics system. Delano Scientific, Palo Alto, CA.
- 10.Deng, G., and E. R. Podack. 1995. Deoxyribonuclease induction in apoptotic cytotoxic T lymphocytes. FASEB J. 9665-669. [DOI] [PubMed] [Google Scholar]
- 11.Deutscher, M. P., C. W. Marlor, and R. Zaniewski. 1984. Ribonuclease T: new exoribonuclease possibly involved in end-turnover of tRNA. Proc. Natl. Acad. Sci. USA 814290-4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dominski, Z., X. C. Yang, H. Kaygun, M. Dadlez, and W. F. Marzluff. 2003. A 3′ exonuclease that specifically interacts with the 3′ end of histone mRNA. Mol. Cell 12295-305. [DOI] [PubMed] [Google Scholar]
- 13.Drake, J. W. 1991. Spontaneous mutation. Annu. Rev. Genet. 25125-146. [DOI] [PubMed] [Google Scholar]
- 14.Duchaine, T. F., J. A. Wohlschlegel, S. Kennedy, Y. Bei, D. Conte, Jr., K. Pang, D. R. Brownell, S. Harding, S. Mitani, G. Ruvkun, J. R. Yates III, and C. C. Mello. 2006. Functional proteomics reveals the biochemical niche of Caenorhabditis elegans DCR-1 in multiple small-RNA-mediated pathways. Cell 124:343-354. [DOI] [PubMed] [Google Scholar]
- 15.Enari, M., H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu, and S. Nagata. 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 39143-50. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Segura, E., F. J. Canizares, M. A. Cubero, A. Warley, and A. Campos. 1999. Changes in elemental content during apoptotic cell death studied by electron probe X-ray microanalysis. Exp. Cell Res. 253454-462. [DOI] [PubMed] [Google Scholar]
- 17.Gibrat, J. F., T. Madej, and S. H. Bryant. 1996. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 6377-385. [DOI] [PubMed] [Google Scholar]
- 18.Hamdan, S., P. D. Carr, S. E. Brown, D. L. Ollis, and N. E. Dixon. 2002. Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure 10535-546. [DOI] [PubMed] [Google Scholar]
- 19.Harrington, J. J., and M. R. Lieber. 1994. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J. 131235-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holm, L., and C. Sander. 1996. Mapping the protein universe. Science 273595-603. [DOI] [PubMed] [Google Scholar]
- 21.Hsu, D. R., and B. J. Meyer. 1994. The dpy-30 gene encodes an essential component of the Caenorhabditis elegans dosage compensation machinery. Genetics 137999-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson, M. D., M. Weil, and M. C. Raff. 1997. Programmed cell death in animal development. Cell 88347-354. [DOI] [PubMed] [Google Scholar]
- 23.Kennedy, S., D. Wang, and G. Ruvkun. 2004. A conserved siRNA-degrading RNase negatively regulates RNA interference in Caenorhabditis elegans. Nature 427645-649. [DOI] [PubMed] [Google Scholar]
- 24.Kipp, M., F. Gohring, T. Ostendorp, C. M. van Drunen, R. van Driel, M. Przybylski, and F. O. Fackelmayer. 2000. SAF-Box, a conserved protein domain that specifically recognizes scaffold attachment region DNA. Mol. Cell. Biol. 207480-7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang, F., S. M. Huber, I. Szabo, and E. Gulbins. 2007. Plasma membrane ion channels in suicidal cell death. Arch. Biochem. Biophys. 462189-194. [DOI] [PubMed] [Google Scholar]
- 26.Li, L. Y., X. Luo, and X. Wang. 2001. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 41295-99. [DOI] [PubMed] [Google Scholar]
- 27.Li, Z., and M. P. Deutscher. 1995. The tRNA processing enzyme RNase T is essential for maturation of 5S RNA. Proc. Natl. Acad. Sci. USA 926883-6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, Z., S. Pandit, and M. P. Deutscher. 1999. Maturation of 23S rRNA requires the exoribonuclease RNase T. RNA 5139-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, Z., L. Zhan, and M. P. Deutscher. 1996. Escherichia coli RNase T functions in vivo as a dimer dependent on cysteine 168. J. Biol. Chem. 2711133-1137. [DOI] [PubMed] [Google Scholar]
- 30.Lieber, M. R. 1997. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays 19233-240. [DOI] [PubMed] [Google Scholar]
- 31.Liu, X., H. Zou, C. Slaoghter, and X. Wang. 1997. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 89175-184. [DOI] [PubMed] [Google Scholar]
- 32.Mazur, D. J., and F. W. Perrino. 2001. Excision of 3′ termini by the Trex1 and TREX2 3′→5′ exonucleases. Characterization of the recombinant proteins. J. Biol. Chem. 27617022-17029. [DOI] [PubMed] [Google Scholar]
- 33.Mello, C. C., J. M. Krame, D. Stinchcomb, and V. Ambros. 1991. Efficient gene transfer in Caenorhabditis elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10:3959-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morita, M., G. Stamp, P. Robins, A. Dulic, I. Rosewell, G. Hrivnak, G. Daly, T. Lindahl, and D. E. Barnes. 2004. Gene-targeted mice lacking the Trex1 (DNase III) 3′→5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 246719-6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagata, S. 1997. Apoptosis by death factor. Cell 88355-365. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276307-326. [DOI] [PubMed] [Google Scholar]
- 37.Parrish, J., L. Li, K. Klotz, D. Ledwich, X. Wang, and D. Xue. 2001. Mitochondrial endonuclease G is important for apoptosis in Caenorhabditis elegans. Nature 41290-94. [DOI] [PubMed] [Google Scholar]
- 38.Parrish, J. Z., and D. Xue. 2006. Cuts can kill: the roles of apoptotic nucleases in cell death and animal development. Chromosoma 11589-97. [DOI] [PubMed] [Google Scholar]
- 39.Parrish, J. Z., and D. Xue. 2003. Functional genomic analysis of apoptotic DNA degradation in Caenorhabditis elegans. Mol. Cell 11987-996. [DOI] [PubMed] [Google Scholar]
- 40.Perrino, F. W., S. Harvey, S. McMillin, and T. Hollis. 2005. The human TREX2 3′→5′-exonuclease structure suggests a mechanism for efficient nonprocessive DNA catalysis. J. Biol. Chem. 28015212-15218. [DOI] [PubMed] [Google Scholar]
- 41.Rizzuto, R., P. Pinton, D. Ferrari, M. Chami, G. Szabadkai, P. J. Magalhaes, F. Di Virgilio, and T. Pozzan. 2003. Calcium and apoptosis: facts and hypotheses. Oncogene 228619-8627. [DOI] [PubMed] [Google Scholar]
- 42.Sheldrick, G., and T. 1997. Schneider. SHELXL: high-resolution refinement Methods Enzymol. 277319-343. [PubMed] [Google Scholar]
- 43.Steitz, T. A., and J. A. Steitz. 1993. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. USA 906498-6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang, X., C. Yang, J. Chai, Y. Shi, and D. Xue. 2002. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 2981587-1592. [DOI] [PubMed] [Google Scholar]
- 45.Winn, M. D., M. N. Isupov, and G. N. Murshudov. 2001. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst. D 57122-133. [DOI] [PubMed] [Google Scholar]
- 46.Wu, M., M. Reuter, H. Lilie, Y. Liu, E. Wahle, and H. Song. 2005. Structural insight into poly(A) binding and catalytic mechanism of human PARN. EMBO J. 244082-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu, Y. C., G. M. Stanfield, and H. R. Horvitz. 2000. NUC-1, a Caenorhabditis elegans DNase II homolog, functions in an intermediate step of DNA degradation during apoptosis. Genes Dev. 14536-548. [PMC free article] [PubMed] [Google Scholar]
- 48.Yang, X. C., M. Purdy, W. F. Marzluff, and Z. Dominski. 2006. Characterization of 3′hExo, a 3′ exonuclease specifically interacting with the 3′ end of histone mRNA. J. Biol. Chem. 28130447-30454. [DOI] [PubMed] [Google Scholar]
- 49.Zhivotovsky, B., B. Cedervall, S. Jiang, P. Nicotera, and S. Orrenius. 1994. Involvement of Ca2+ in the formation of high molecular weight DNA fragments in thymocyte apoptosis. Biochem. Biophys. Res. Commun. 202120-127. [DOI] [PubMed] [Google Scholar]
- 50.Zuo, Y., and M. P. Deutscher. 2002. Mechanism of action of RNase T. II. A structural and functional model of the enzyme. J. Biol. Chem. 27750160-50164. [DOI] [PubMed] [Google Scholar]
- 51.Zuo, Y., H. Zheng, Y. Wang, M. Chruszcz, M. Cymborowski, T. Skarina, A. Savchenko, A. Malhotra, and W. Minor. 2007. Crystal structure of RNase T, an exoribonuclease involved in tRNA maturation and end turnover. Structure 15417-428. [DOI] [PMC free article] [PubMed] [Google Scholar]