

Social integration is fundamental for good emotional and physical health. Indeed, socially isolated individuals with low perceived social support are at increased risk for many health conditions (reviewed in references Hawkley and Cacioppo1 and House et al.2), including cardiovascular disease.3–5 Development of animal models is required to understand the specific physiological mechanisms responsible for the effects of social isolation on health. Here, we provide evidence that social isolation in mice potentiates the pathophysiological responses to cardiac arrest (CA), which may explain, in part, why social isolation is as strong a predictor of 1-year mortality among acute myocardial infarction patients as some of the classic physiological risk factors, including high blood cholesterol concentrations and hypertension.5
To determine the impact of social environment on neuroinflammation, neuronal death and corticosteroid concentrations after global ischemia, we housed adult male C3H/e mice either alone (isolated, n = 7) or five per cage (social, n = 5) beginning 2 weeks prior to inducing 8 min of CA, followed by resuscitation by epinephrine injection and chest compressions (CA/ CPR (cardiopulmonary resuscitation)); complete description in Neigh et al.6). The control group consisted of mice that underwent the same cardiac arrest procedure, but ischemic influences on the brain were prevented through the use of hypothermia;6 hypothermic-isolated (n = 10) and hypothermic-social (n = 7) groups were collapsed into a single control group because they were not significantly different on any measure (P > 0.05 in all cases). Brain tissue and blood samples were collected either 24 h or 7 days after CA/CPR for gene expression or histological analysis, respectively. As expected, cardiac arrest induced neuronal degeneration throughout the hippocampus; however, the extent of damage was significantly increased among mice that were socially isolated prior to CA/CPR (Figure 1a, b and e). Specifically, CA/CPR evoked more neuronal degeneration (that is, Fluoro Jade-positive neurons (FJ+)) in the CA1 field (F2,24 = 20.15, P < 0.0001; Figure 1e), CA2 field (F2,24 = 21.14, P < 0.0001) and dentate gyrus (F2,24 = 16.06, P < 0.0001) of socially isolated versus socially housed and control mice. Social isolation also exacerbated the post-ischemic inflammatory responses throughout the hippocampus (Figure 1c, d, f and g). Microglial activation (that is, MAC-1 staining) following CA/CPR was higher in the CA1 field (F2,28 = 26.68, P < 0.0001; Figure 1f), CA2 field (F2,28 = 12.29, P < 0.0001), dentate gyrus (F2,28 = 20.56, P < 0.0001) and subiculum (F2,28 = 31.06, P < 0.0001) of socially isolated mice than socially housed mice and control (P < 0.05). Furthermore, mRNA expression of the proinflammatory cytokine tumor necrosis factor-α (TNF-α), measured 24 h after resuscitation, was significantly elevated in isolated mice, but not socially housed mice, relative to the control mice (F2,18 = 4.83, P < 0.0001; Figure 1f). Upregulation of microglia and the neurotoxic mediators they release, including TNF-α, in the developing ischemic lesion can contribute to secondary neuronal damage and infarct evolution.7 Thus, the near-complete attenuation of the inflammatory responses to cerebral ischemia in socially housed mice likely contributed to the observed reduction in neuronal damage (Figure 1a, b and e).
Figure 1.
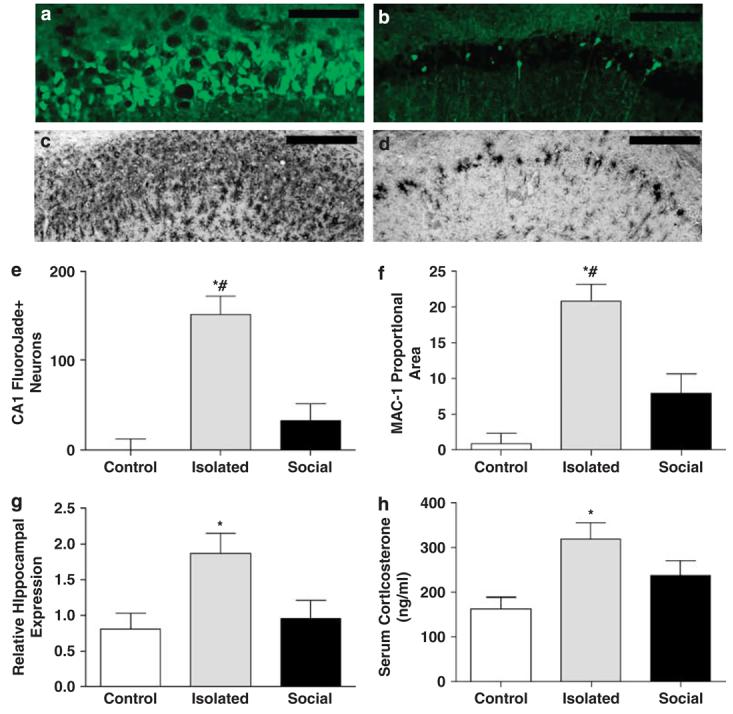
Social isolation potentiates ischemic damage. Fluoro Jade staining of hippocampal cell degeneration in socially isolated (a) and socially housed mice (b). Microglial activation in the socially isolated (c) and socially housed animals (d). Summary (mean±s.e.m.) of histological outcomes in the ischemia-vulnerable CA1 region; Fluoro Jade staining (e) and proportional microglial staining (f). mRNA expression of the proinflammatory cytokine tumor necrosis factor-α (TNF-α; mean±s.e.m.) 24 h after reperfusion (g). Circulating corticosterone (mean±s.e.m.) 24 h after reperfusion (h). An asterisk (*) indicates significantly different means from the control groups (P < 0.05), whereas a pound (#) indicates significantly different means from the social group (P < 0.05).
Social context also influenced early post-ischemic corticosterone concentrations. Isolated mice had significantly elevated serum corticosterone concentrations 24 h after ischemia relative to the control mice (F2,20 = 4.32, P < 0.0001; Figure 1h). Elevated post-ischemic corticosteroid concentrations have previously been linked to increased neuronal damage,8,9 and may have contributed to the elevated neuronal death among the isolated mice relative to the socially housed cohort.
Together, these data indicate that the pathophysiological responses of socially isolated and socially integrated mice to CA/CPR are quantitatively and qualitatively different; social isolation potentiates ischemia-induced neuronal damage, neuroinflammation and corticosteroid secretion, three important determinants of outcome following CA/CPR. The elevated inflammatory responses among socially isolated individuals also may indirectly compromise recovery by contributing to the development of post-ischemic affective disorders.10 Given the well-described influence of social relationships on health, and this new evidence that social isolation alters the physiological response to cardiac arrest, we emphasize the need for additional research aimed at characterizing and promoting the types of social relationships that promote health in humans, and the value of monitoring physiological measures in these studies.
References
- 1.Hawkley LC, Cacioppo JT. Brain Behav Immun. 2003;17(Suppl 1):S98–105. doi: 10.1016/s0889-1591(02)00073-9. [DOI] [PubMed] [Google Scholar]
- 2.House JS, Landis KR, Umberson D. Science. 1988;241:540–545. doi: 10.1126/science.3399889. [DOI] [PubMed] [Google Scholar]
- 3.Lett HS, Blumenthal JA, Babyak MA, Catellier DJ, Carney RM, Berkman LF. Health Psychol. 2007;26:418–427. doi: 10.1037/0278-6133.26.4.418. [DOI] [PubMed] [Google Scholar]
- 4.Brummett BH, Barefoot JC, Siegler IC, Clapp-Channing NE, Lytle BL, Bosworth HB. Psychosom Med. 2001;63:267–272. doi: 10.1097/00006842-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Mookadam F, Arthur HM. Arch Intern Med. 2004;164:1514–1518. doi: 10.1001/archinte.164.14.1514. [DOI] [PubMed] [Google Scholar]
- 6.Neigh GN, Kofler J, Meyers JL, Bergdall V, Perle KM, Traystman RJ. J Cereb Blood Flow Metab. 2004;24:372–382. doi: 10.1097/01.WCB.0000112323.75217.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoll G, Jander S, Schroeter M. Prog Neurobiol. 1998;56:149–171. doi: 10.1016/s0301-0082(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 8.DeVries AC, Joh HD, Bernard O, Hattori K, Hurn PD, Traystman RJ. Proc Natl Acad Sci USA. 2001;98:11824–11828. doi: 10.1073/pnas.201215298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sapolsky RM, Pulsinelli WA. Science. 1985;229:1397–1400. doi: 10.1126/science.4035356. [DOI] [PubMed] [Google Scholar]
- 10.Spalletta G, Bossu P, Ciaramella A, Bria P, Caltagirone C, Robinson RG. Mol Psychiatry. 2006;11:984–991. doi: 10.1038/sj.mp.4001879. [DOI] [PubMed] [Google Scholar]