

Abstract
Stress is a complex human experience having both positive and negative motivational properties. When chronic and uncontrollable, the adverse effects of stress on human health are considerable and yet poorly understood. Here, we report that the dysphoric properties of chronic stress are encoded by the endogenous opioid peptide dynorphin acting on specific stress-related neuronal circuits. Using different forms of stress presumed to evoke dysphoria in mice, we found that repeated forced swim and inescapable footshock both produced aversive behaviors that were blocked by a κ-opioid receptor (KOR) antagonist and absent in mice lacking dynorphin. Injection of corticotropin-releasing factor (CRF) or urocortin III, key mediators of the stress response, produced place aversion that was also blocked by dynorphin gene deletion or KOR antagonism. CRF-induced place aversion was blocked by the CRF2 receptor antagonist antisauvigine-30, but not by the CRF1 receptor antagonist antalarmin. In contrast, place aversion induced by the KOR agonist U50,488 was not blocked by antisauvigine-30. These results suggest that the aversive effects of stress were mediated by CRF2 receptor stimulation of dynorphin release and subsequent KOR activation. Using a phospho-selective antibody directed against the activated KOR to image sites of dynorphin action in the brain, we found that stress and CRF each caused dynorphin-dependent KOR activation in the basolateral amygdala, nucleus accumbens, dorsal raphe, and hippocampus. The convergence of stress-induced aversive inputs on the dynorphin system was unexpected, implicates dynorphin as a key mediator of dysphoria, and emphasizes κ-receptor antagonists as promising therapeutics.
Keywords: dynorphin, aversion, stress-induced aversion, drug addiction, κ-opioid receptor, corticotropin releasing factor
Introduction
Sustained stressful experience can induce despair and increase the risk of clinical depression and drug abuse (Gold and Chrousos, 2002; Volkow and Li, 2004; Fox et al., 2007; Koob and Kreek, 2007). Two key stress-related neuropeptides in the brain that may mediate these responses are corticotropin-releasing factor (CRF) and dynorphin (Bale et al., 2004; Nestler and Carlezon, 2006). Pharmacological activation of CRF produces conditioned place aversion (CPA) in rodents (Cador et al.,1992), and CRF receptor antagonists act as anti-depressants in humans (Zobel et al., 2000). Similarly, activation of the dynorphin receptor (κ-opioid receptor) causes place aversion in rodents and dysphoria in humans (Pfeiffer et al., 1986; Shippenberg and Herz, 1986). The degree of overlap in these behavioral effects suggests that CRF and dynorphin are critically linked in the stress response. However, a functional relationship between the CRF and dynorphin systems in the brain has not been defined.
CRF has been proposed to mediate stress-induced depression, stress-induced drug-seeking, and anxiety behaviors (Shaham et al., 1997; Erb et al., 1998; Bale and Vale, 2004; Keck et al., 2005; Wang B et al., 2005; Koob, 2006). The CRF receptor system is comprised of peptide ligands CRF and urocortin I, which act on the CRF1-receptor (CRF1-R) and the peptides urocortin II and urocortin III that are selective for CRF2-receptors (CRF2-R) (Bale and Vale, 2004). Activation of the CRF1-R is thought to be prodepressant and critical in mediating stress effects during drug withdrawal, whereas CRF2-R activation has been suggested to have compensatory, anti-depressive effects (Bale and Vale, 2004; Contarino and Papaleo, 2005). However, the activation of both receptors has been implicated in relapse to drug seeking and depression-like behaviors (Shaham et al., 1998; Hammack et al., 2003; Wang B et al., 2005, 2007; Wang J et al., 2005; Henry et al., 2006).
One possible mediator of the action of CRF on mood is the κ-opioid system comprised of the endogenous dynorphin neuropeptides and κ-opioid peptide receptor (KOR) (Chavkin et al., 1982). Dynorphins are released during stress exposure (McLaughlin et al., 2003a), and previous reports have identified the dynorphin/κ-opioid system as critical for stress-induced depression-like behaviors and reinstatement to drug seeking (Mague et al., 2003; McLaughlin et al., 2003a; Beardsley et al., 2005; Carlezon et al., 2006; Carey et al., 2007). We infer from this that stress-induced release of dynorphin affects mood to produce dysphoria, but this has never been examined directly. Thus, in this study, we tested the hypothesis that stress exposure activates dynorphin systems in the brain to cause the dysphoria responsible for subsequent aversive responses to stress, and we assessed the functional relationship between the CRF and dynorphin systems in brain. We demonstrate that the aversive property of two different stressors requires dynorphin signaling at KOR. We provide evidence that CRF mimics this aversive quality in the absence of stress, and that CRF requires activation of the KOR through dynorphin signaling. Finally, we use CRF receptor-selective compounds to show that CRF induces KOR-dependent aversion through activity at the CRF2-R. The results establish a previously unknown pathway leading from stress to dysphoria through the dynorphin/κ-system.
Materials and Methods
Animals.
Male C57Bl/6 mice (Charles River, Wilmington, MA) weighing 22–30 g were used. Prodynorphin (Pdyn) gene deletion on C57Bl/6 background were generated as described previously (McLaughlin et al., 2003a), and homozygous (−/−) and paired wild-type (+/+) littermate controls were generated by heterozygote crosses. Animal protocols were approved by the Institutional Animal Care and Use Committee.
Drug and chemicals.
Rat/human CRF, norbinaltorphimine (norBNI)-HCl, cyclo (31–34)[D-Phe12,Nle21,38,Glu31, Lys34]Ac-hCRF (4–41) (Stressin 1), and (±)U50,488 were obtained from Tocris Bioscience (Ellisville, MO). Additional U50,488 and norBNI were provided by the National Institute on Drug Abuse drug program. Antalarmin and (D-Phe11,His12)-Sauvigine, Fragment 11–40 trifluoroacetate salt (ASV-30, anti-sauvigine 30) were obtained from Sigma (St. Louis, MO). Mouse urocortin III was obtained from Phoenix Pharmaceuticals (Belmont, CA). Peptides were dissolved in saline/0.05% glacial acetic acid (added dropwise). For all experiments using peptides, the appropriate saline-vehicle controls were used. Antalarmin was dissolved in 5% Cremephor/5% DMSO (v/v), 90% saline as described previously (Griebel et al., 2002). U50,488 and norBNI were dissolved in saline. For systemic drug injections, 10 ml/kg (i.p.) was used. Volumes for intracerebroventricular injections are described for each experiment.
Cannulation and intracerebroventricular injections.
Isoflurane-anesthetized mice were mounted on a stereotaxic alignment system (David Kopf Instruments, Tujunga, CA), guide cannulas (26GA, 2.4 mm from pedestal) (Plastics One, Roanoke, VA) were placed in the lateral ventricle at 1.0 mm lateral, 0.4 mm posterior from bregma, and 3.0 mm below the skull (Paxinos and Franklin, 2004). Cannulas were anchored with screws using dental cement. After surgery, mice recovered in their home cage for 6–8 d. Mice were injected intracerebroventricularly by placing a 33 guage internal cannula (Plastics One) into the guide cannula and attached to a flared PE20 tube connected to a 10 μl Hamilton syringe. Injection volumes (1 or 5 μl) of saline or drug (CRF, urocortin III, or stressin I) were performed during a 2 min period. Internal cannulas were removed 1 min after injection, and mice were immediately placed in the testing chamber.
Conditioned place aversion.
Cannulated mice were trained in a balanced three-compartment conditioning apparatus as described previously (McLaughlin et al., 2003a). Mice were pretested by placing individual animals in the small central compartment and allowing them to explore the entire apparatus for 30 min. Mice that did not explore both side compartments within 2 min were guided to the unexplored compartment. Time spent in each compartment was recorded with a video camera (Canon ZR90) and analyzed using Ethovision software (Noldus, Wageningen, The Netherlands). Mice were randomly assigned to drug and saline training compartments and received saline (2 μl, i.c.v.) in the morning and either saline (5 μl, i.c.v.), CRF (1 μg/5 μl, i.c.v.), or urocortin III (0.5 μg/5 μl, i.c.v.) in the afternoon on days 2 and 3. Animals pretreated with norBNI (10 mg/kg, i.p.), antalarmin (10 mg/kg, i.p.), or anti-sauvigine 30 (0.5 μg/1 μl, i.c.v.) were injected at 1 h, 30 min, and 15 min before conditioning, respectively. CPA was assessed on day 4 by allowing the mice to roam freely in all three compartments and recording the time spent in each. Scores were calculated by subtracting the time spent in the drug compartment posttest minus the pretest.
Odorant-swim stress aversion.
Animals were pre-exposed to a Plexiglas T-Maze (73 cm long × 40 cm wide × 15 cm high) for 3 min before stress-aversion conditioning. Stress was induced using a modified Porsolt repeated forced-swim as described previously (Porsolt et al., 1977; McLaughlin et al., 2003a). Mice were exposed to one 15 min swim on day 1 and four 6 min swims on day 2, in 30°C water, without opportunity for escape. Two minutes before each swim, animals received four-quarter Nestletts (PharmaServ, Framingham, MA) with 20 μl of imitation almond extract (Kroger, Cincinnati, OH) in their home cage. When they were transferred to the forced swim test, each animal was exposed to a Nestlett with 20 μl of almond extract during the swim, 5–10 cm above the swim chamber. On day 3, animals were tested for odorant-aversion in a T-Maze with a Nestlett with imitation almond odorant cue (20 μl) randomly placed in one arm and an untreated Nestlett at the end of the other short arm. Animals were placed in the long arm and given 14 min to explore the maze. Time spent in each of the two distal ends of the short arms was recorded and analyzed using Ethovision. Aversion was scored as the time near the odorant subtracted from the time in the opposite end.
KOR agonist-induced odorant aversion.
On day 1, animals were injected with U50,488 at either 0, 2.5, 5, or 10 mg/kg (i.p.). This was immediately followed by introduction of four-quarter Nestletts, each containing 20 μl of almond extract, into the home cage. The Nestletts remained in the cage for 30 min and were then removed. This was repeated on day 2. On day 3, animals were tested in the T-maze as described above.
Odorant-conditioned place preference.
During the pretest, animals were placed in the neutral center chamber and allowed to explore the two side chambers that had equivalent visual and tactile cues (both chambers had the same black and white stripes and the same bedding) but varied in the odorant cue that was placed in the chamber (either lemon or vanilla; Kroger). The time spent in each chamber was recorded and analyzed using a video camera (Canon ZR-90) and Ethovision, respectively. We used a biased design for the place preference conditioning, meaning animals were paired with cocaine in the chamber with the odorant cue they preferred less. Thus, during the 2 d conditioning phase, animals received saline in the morning and were restricted to the chamber that was more preferred for 30 min and received cocaine (15 mg/kg, s.c.) in the afternoon and were restricted to the side less preferred for 30 min. Animals received four total injections: two saline and two cocaine. The posttest phase was identical to the pretest phase in that animals had free access to the three compartments for 30 min. Time in each chamber was again recorded, and the difference between the time spent in the cocaine-paired side during the posttest was subtracted from the time in the cocaine-paired during the pretest to give a preference measure.
Footshock stress-paired conditioned place aversion.
A shock box with two chambers connected by a guillotine door was used (35 cm wide × 18 cm deep × 28 cm high; Coulbourn Instruments, Allentown, PA). The shock chamber had vertical stripes (alternating black and white); the nonshock chamber had horizontal stripes. In the morning of day 1, mice were confined to the nonshock chamber for 15 min. Approximately 4 h later, animals were confined to the shock chamber for 15 min and received either 10 shocks (0.5 s duration, 0.6 mA) at semirandom intervals or no shock. On day 2, animals were allowed to freely explore both chambers of the shock box for 15 min, with the time spent in the shock chamber recorded.
Immunoblotting.
Cannulated mice were injected with drugs as described above, decapitated, and striata were dissected and homogenized with a dounce tissue homogenizer in lysis buffer (50 mm Tris, 300 mm NaCl, 1 mm EDTA, 1 mm Na3VO4, 1 mm NaF, 1× protease and phosphatase inhibitor mixture; Calbiochem, La Jolla, CA) as described previously (Bruchas et al., 2007). Samples were centrifuged at 30,000 × g for 25 min at 4°C. Supernatants were discarded, and the membrane pellet was resuspended in lysis buffer and centrifuged again. Membrane pellets were then resolubilized on ice for 1 h in buffer containing the following: 1%-N-docecyl-β-d-maltoside, 50 mm Tris, 150 mm NaCl, 1 mm CaCl2, 1 mm MgCl2, 10% glycerol. Protein concentrations were determined by the BCA method (Pierce, Rockford, IL), and immunoblotting was performed as described previously (Bruchas et al., 2006, 2007) (supplemental materials, available at www.jneurosci.org as supplemental material).
Immunohistochemistry.
Intracerebroventricular cannulated mice were injected as described above and 30 min later were anesthetized with isoflurane (Sigma) and intracardially perfused with 4% paraformaldehyde in phosphate buffer (PB) (0.1 m sodium phosphate, pH 7.4). Brains were dissected and cryoprotected with solution of 30% (w/v) sucrose in PB at 4°C overnight, cut into 40 μm sections with a microtome, and placed in PB until processing. Standard immunohistochemical procedures were performed to assess phospho-KOR (KORp) staining (Bruchas et al. 2007; Xu et al., 2007) (supplemental information, available at www.jneurosci.org as supplemental material).
Data analysis.
Data are expressed as means ± SEM. Differences between groups were determined using independent t tests, one-way ANOVA followed by Dunnet's (for comparisons between relevant groups to the control group) or Neuman–Keuls post hoc test if the main effect was significant at p < 0.05. For experiments having a 2 × 2 factorial design, two-way ANOVAs followed by Neuman–Keuls post hoc tests if an interaction effect was significant at p < 0.05. Statistical analyses were conducted using GraphPad Prism (4.0) (GraphPad Software, San Diego, CA) or SPSS (11.0) (SPSS, Chicago, IL).
Results
Stress-induced aversive responses require dynorphin release
To measure the motivational effects of stress, we developed a conditioned-aversion paradigm in mice that pairs forced swim stress with a neutral odorant cue (Fig. 1A). Mice developed a significant aversion to the odorant when subsequently presented alone in the testing chamber compared with control unstressed groups, and this aversion was not evident in mice pretreated with the KOR antagonist norBNI (n = 10–12; two-way ANOVA; main effect of swim, F(1,41) = 7.41, p < 0.01; main effect of pretreatment, F(1,41) = 4.89, p < 0.05; interaction of swim times pretreatment, F(1,41) = 4.47, p < 0.05; Neuman–Keul's post hoc, p < 0.01) (Fig. 1B). Mice lacking the prodynorphin gene (Pdyn−/−) also failed to develop aversion to the stress-paired odorant (n = 9–10; t15 = 2.492; p < 0.05) (Fig. 1C). In contrast, pairing the odorant with cocaine injection caused a conditioned place preference (CPP) that was not blocked by norBNI (Fig. 1D). The lack of effect on cocaine-odorant conditioning suggests that the reduction in aversion caused by disruption of the dynorphin system did not result from impairments of olfaction or associative learning. Instead, these results suggest that the aversive quality of the stressful experience was mediated by a dynorphin-dependent mechanism. Consistent with this concept, pairing the odorant with injection of the KOR agonist U50,488 also caused subsequent dose-dependent aversion to the odorant (Fig. 1E).
Figure 1.

Stress-induced odorant aversion is dynorphin/KOR mediated. A, Schematic of the odorant-stress aversion paradigm. B, Odorant aversion scores (odorant side minus control side) show that, in the absence of stress pairing, the odorant does not elicit aversion (the odorant is neutral). Mice previously experiencing stress paired with odorant showed significant aversion to subsequent odorant exposure, and norBNI pretreated mice did not develop odorant aversion despite stress pairing. **p < 0.01, significantly different from saline/stress group. C, Pdyn(+/+), but not Pdyn(−/−), mice developed significant aversion to odorant. *p < 0.05, significantly different from Pdyn(−/−) group. D, Pairing of the neutral odorant with cocaine injection produced significant place preference for the odorant compartment that was not blocked by norBNI pretreatment. E, κ-Opioid receptor stimulation caused a dose-dependent increase in the odorant-aversion score. When odorant was paired with either 0, 2.5, 5, or 10 mg/kg of the selective κ-opioid receptor agonist U50,488, mice showed an increased aversion to the odorant when subsequently presented with the odorant alone in the T-maze (n = 4–8 animals per group).
To assess the generality of these observations, the role of dynorphin in mediating aversion to another stressor, footshock, was evaluated. Robust conditioned aversion to the footshock compartment was evident 24 h after a single 15 min episode (n = 4–7; one-way ANOVA; F(3,18) = 6.44; p < 0.01, Dunnet's post hoc compared with saline/no swim, p < 0.01); however, footshock-induced place aversion was completely blocked by pretreatment with norBNI and was absent in Pdyn (−/−) mice (Dunnet's post hoc compared with saline/no swim, p > 0.05) (Fig. 2). These data suggest that in two distinct conditioned-aversion paradigms, stress-induced aversion requires functional dynorphin/κ-opioid signaling.
Figure 2.

Footshock stress-induced aversion is mediated by the dynorphin/KOR system. Significant aversion was evident to a compartment previously paired with shock compared with unshocked controls. Unshocked controls spent nearly equal time in both compartments during the 15 min session. Wild-type (WT) mice pretreated with the κ-opioid antagonist norBNI (10 mg/kg, i.p.) did not show significant place aversion after shock training. Mice lacking dynorphin (Pdyn, −/−) also failed to develop a significant place aversion after shock training. **p < 0.01, significantly different from saline/no shock group (n = 5–8 animals per group).
CRF receptor stimulation results in KOR activation in brain structures associated with negative affect
Intracerebroventricular injection of CRF (1 μg) caused dynorphin release in the brain, as determined by an increase in phosphorylation of KOR detected by a phospho-selective antibody (KORp) (McLaughlin et al., 2003b). KOR becomes phosphorylated by G-protein receptor kinase 3 after agonist binding, and CRF injection significantly increased KORp-immunoreactivity (ir) in mouse striatum as shown by Western blot analysis (n = 4–12; one-way ANOVA, F(2,19) = 5.40, p < 0.05, Neuman–Keuls post hoc, p < 0.05) (Fig. 3A). This increase in KORp-ir was blocked by norBNI pretreatment (Neuman–Keuls post hoc; p > 0.05). Using immunohistochemistry to extend these findings, we found that KORp-ir was greatly increased in the basolateral amygdala after injection with CRF, and this increase was not evident in mice pretreated with norBNI or in Pdyn(−/−) animals (Fig. 3B). Swim stress and CRF exposure also increased KORp-ir in the dorsal raphe, nucleus accumbens, hippocampus, and several additional brain regions associated with negative affect (Carrasco and Van de Kar, 2003) (Table 1) (supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material). This increase was selective, because the caudate–putamen and central nucleus of the amygdala showed no difference in staining compared with control. These complementary biochemical and immunochemical results suggest that stress mobilizes CRF, which then acts on its receptors to induce the release of dynorphin, subsequently activating the κ-opioid system in specific stress-responsive brain regions.
Figure 3.
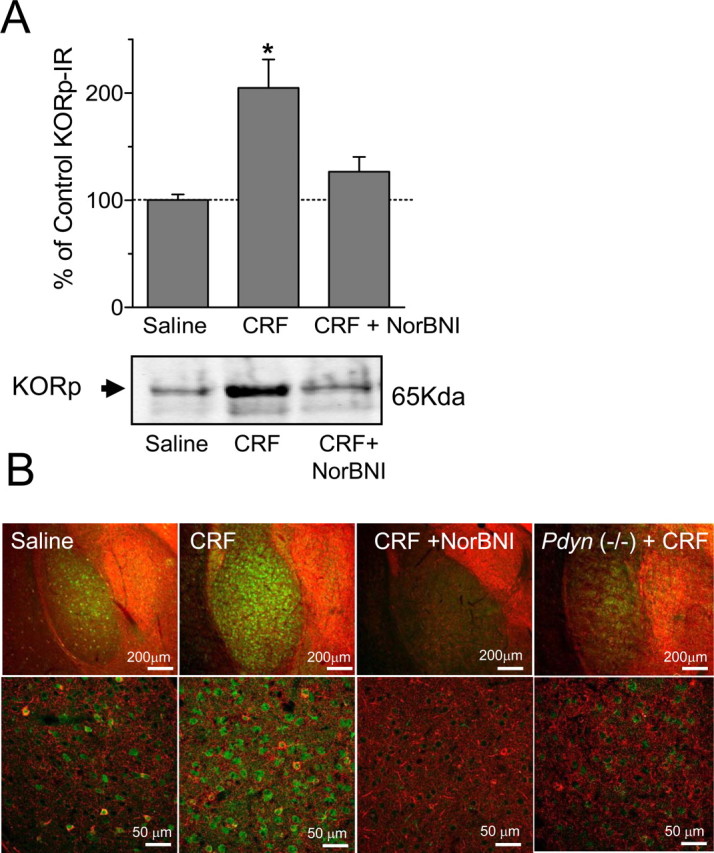
CRF injection increases KOR phosphorylation. A, Representative Western blots showing that CRF (1 μg/5 μl, i.c.v.) increased the 65 kDa KORp-ir band (lane 2) in striatal extracts compared with saline injection (lane 1). Pretreatment with norBNI (10 mg/kg, i.p.) blocked the CRF-induced increase (lane 3). The bar graph quantifies the increase in KORp-ir intensity and norBNI antagonism evident in replicate gels. *p < 0.05, significantly different from saline (n = 4–12 animals per group). B, Representative images showing a substantial increase in KORp-ir (green) in the basolateral amygdala (BLA) after injection of CRF compared with saline-injected mice. The increased KORp-ir was not evident in mice pretreated with norBNI (CRF + NorBNI) or in Pdyn(−/−) mice injected with CRF. GAD67-ir is also shown (red), and the bottom row of images is shown at higher power. Data represent three independent experiments taken from separate animals.
Table 1.
CRF injection induces KOR phosphorylation in specific mouse brain regions
| Region | CRF-induced KORp-ir | Region | CRF-induced KORp-ir |
|---|---|---|---|
| Dorsal raphe nucleus | +++ | Nucleus accumbens | ++ |
| Basolateral amygdala | +++ | Bed nucleus stria terminalis | + |
| Dorsal hippocampus | +++ | Caudate–putamen | Not detected |
| Ventral pallidum | ++ | Central nucleus amygdala | Not detected |
| Ventral tegmental area | ++ |
Compiled summary of qualitative data showing brain regions positive or negative for KORp-ir after intracerebroventricular CRF injection. Positive KORp-ir is indicated in three levels of magnitude (+++, high; ++, moderate; +, lowest but above background control).
To assess the role of dynorphin in CRF-induced aversion, mice were injected with CRF (1 μg, i.c.v.) in one compartment of a place preference apparatus and saline in the other compartment for 2 d as outlined (Fig. 4A). CRF induced CPA, and the aversion to CRF was blocked by pretreatment with norBNI (n = 8–16; two-way ANOVA; significant interaction of pretreatment times treatment, F(1,43) = 4.09; p < 0.05, Neuman–Keuls post hoc comparing all groups, p < 0.05) (Fig. 4B). Consistent with our findings in the present study, Pdyn(−/−) mice developed no place aversion to CRF compared with littermate controls (n = 5–8; t11 = 3.592; p < 0.01) (Fig. 4C). These results suggest that the aversive quality of CRF also required activation of the dynorphin/KOR system.
Figure 4.
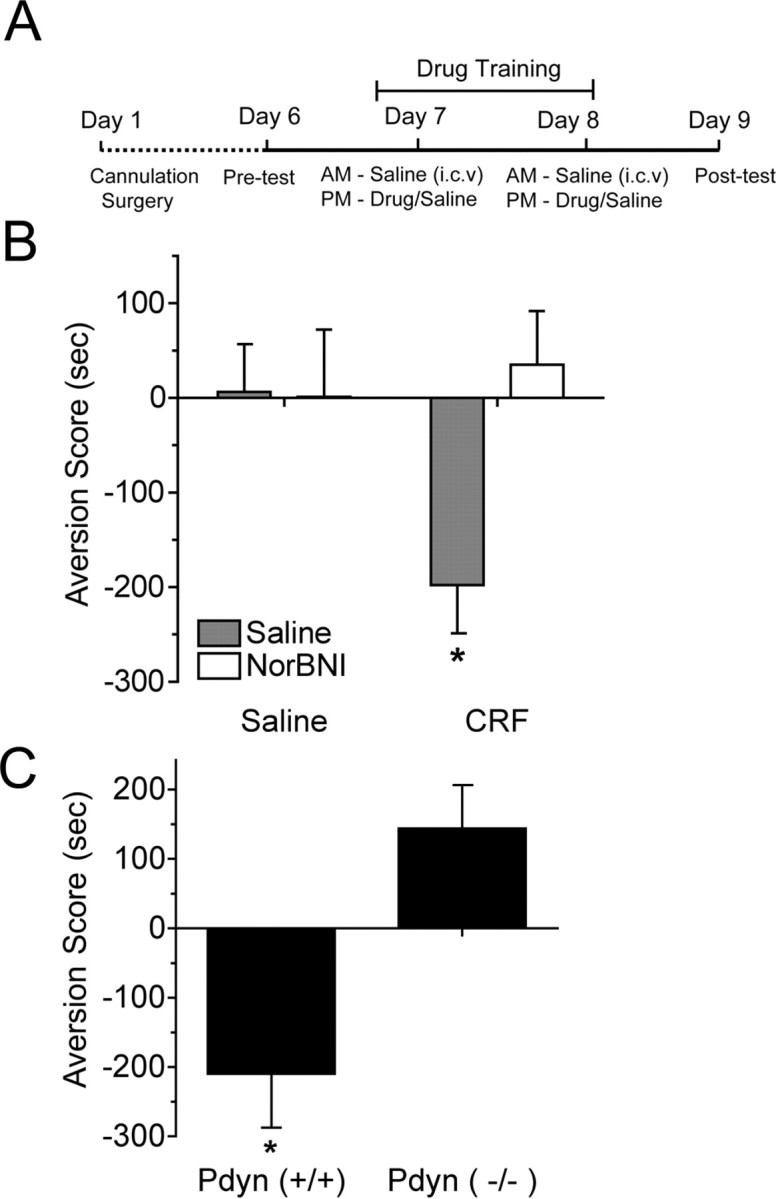
The dynorphin/κ-opioid system mediates the aversive properties of CRF. A, Schematic showing the conditioning paradigm used to assess aversion. B, CPA scores (drug compartment posttest minus pretest) showed that mice injected with saline in both compartments did not develop aversion (gray bar, left) and that norBNI (10 mg/kg, i.p.; white bars) with saline did not affect place preference. CRF (1 μg, i.c.v.) injection induced significant CPA (gray bar, right) that was blocked by norBNI pretreatment (white bar). *p < 0.05, significantly different from other groups at (n = 9–16 animals per group). C, CRF (1 μg, i.c.v.) produced significant CPA in wild-type Pdyn (+/+) mice but not in Pdyn(−/−) littermates. *p < 0.05, significantly different from Pdyn(−/−); n = 5–8 animals per group.
The CRF2-receptor mediates KOR-dependent CRF aversion
We used CRF receptor-selective agonists to establish the contributions of CRF1-R and CRF2-R to both stimulated KORp-ir and aversion. The CRF2-R selective urocortin III increased KORp-ir in a norBNI-sensitive manner (Fig. 5A). Consistent with these results, injection of urocortin III produced robust CPA that was blocked by norBNI pretreatment (n = 6; t10 = 3.015; p < 0.05) (Fig. 5B), whereas injection of stressin 1, a CRF1-R-selective agonist (0.5 μg, i.c.v.) did not cause CPA. The injection of stressin 1 was sufficient to activate CRF1-receptors, because it caused an increase in colonic motility as reported previously (Tache et al., 2001; Rivier et al., 2007). Fecal output during the two 30 min training sessions increased from 2.83 ± 0.65 boli (saline) to 7.37 ± 1.15 boli (stressin 1) (n = 6–8; t12 = 3.128; p < 0.01), demonstrating that this dose of stressin 1 was pharmacologically active and produced a physiological effect.
Figure 5.

CRF2-R activation causes dynorphin/KOR-mediated aversive behavioral responses. A, Representative Western blots showing that urocortin III (U3) injection (0.5 μg, i.c.v.) increased KORp-ir compared with saline-injected mice. The increased KORp-ir was blocked by pretreatment with norBNI (10 mg/kg, i.p.) (U3+N) (n = 4 animals per group). B, Urocortin III (UcnIII) induced significant place aversion that was blocked by norBNI, whereas Stressin 1 (0.5 μg, i.c.v.) did not induce significant aversion. *p < 0.05, significantly different from UcnIII group (n = 6 animals per group).
In a complementary experiment, we used CRF receptor-selective antagonists to identify the receptor subtype critical for mediating CRF-induced place aversion. CRF-induced CPA was blocked by preadministration of the CRF2-R selective antagonist antisauvigine-30 (ASV-30) (n = 10–11; one-way ANOVA; F(2,30) = 4.20; p < 0.05, Neuman–Keuls post hoc, p < 0.05), whereas previous administration of the CRF1-R-selective antagonist antalarmin did not block CRF-induced aversion (Neuman–Keuls post hoc; p > 0.05) (Fig. 6A). The lack of effect of antalarmin (10 mg/kg) was not caused by insufficient dosage, because CRF1-R-mediated immobility during swim stress (n = 8; t14 = 2.379; p < 0.05) and stress-induced colonic hypermotility (fecal output) were significantly blocked by antalarmin (n = 8; t14 = 4.406; p < 0.001) (Fig. 6C,D) as reported previously (Gabry et al., 2002; Griebel et al., 2002). Interestingly, CPA induced by the KOR agonist U50,488 was not blocked by ASV-30 (Fig. 6A), suggesting that CRF/CRF2-R signaling occurs upstream dynorphin-KOR signaling in the stress response (n = 5–8). Finally, antisauvigine-30 blocked CRF-induced increase of KORp-ir in the basolateral amygdala (Fig. 6B). Together, these data support the conclusion that CRF2-R activation caused by administration of CRF or urocortin III evokes dynorphin release and subsequent KOR-mediated aversion. The results support the hypothesis that the dysphoric behaviors associated with the stress experience are mediated by activation of the dynorphin/KOR system (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 6.

CRF2-R antagonism blocks CRF-induced, but not U50,488-induced, aversion. A, CRF-induced place aversion was blocked by ASV-30 (1 nmol, i.c.v.) pretreatment, whereas antalarmin (10 mg/kg, i.p.) pretreatment did not. U50,488 also induced a place aversion, and ASV-30 pretreatment did not block this aversion. Veh, Vehicle (see Materials and Methods). *p < 0.05, significantly different from ASV-30 (n = 10–11 animals per group). B, Representative images showing that the increase in KORp-ir (green) in the basolateral amygdala caused by CRF injection (middle panel) compared with saline (left panel) was blocked by pretreatment with ASV-30 (right panel). Counterstained for GAD67-ir (red). C, Injection of the CRF1-R-selective antagonist antalarmin (10 mg/kg, i.p.) before swim-stress testing significantly blocked stress-induced colonic motility (fecal output) (n = 8; ***p < 0.001; t test) as reported previously (Griebel et al., 2002). D, Injection with antalarmin (10 mg/kg, i.p.) significantly blocked swim stress-induced immobility (n = 4; *p < 0.05; t test), demonstrating that this dose of antalarmin was pharmacologically active.
Discussion
The principal findings of these experiments provide new insight into the mechanisms underlying the dysphoric response to stress. κ-Opioid receptor activation after forced swim, inescapable footshock or κ-agonist administration each produced aversion by dynorphin/KOR-dependent mechanisms. CRF receptor-selective agonists and antagonists showed that the aversion resulted from CRF2-R activation. Imaging of the brain regions mediating the response using the phospho-selective KORp antibody (McLaughlin et al., 2003b) showed that brain regions that may mediate this response correspond to those previously associated with negative affective state (Carrasco and Van de Kar, 2003). The resulting evidence strongly implicates the endogenous dynorphin system as a key mediator of the dysphoric response to stress.
In this study, we used conditioned place aversion as an objective measure of behavior and infer a relationship between the aversion measured and an underlying dysphoria. The nature of the underlying emotional state was not directly measured, but we use the term dysphoria to ascribe to a mouse a negative hedonic state having motivational qualities that can become associated with neutral cues (odorant, wall-striping patterns, flooring texture) and induce avoidance behavior to those associated cues. Dysphoria is operationally defined as the emotional response to a sustained stimulus that creates aversion. It is important to recognize that the indirect measure of the dysphoric state includes an associative learning component, and nonspecific effects of experimental manipulations on learning mechanisms need to be assessed. We found that treatments able to block stress-induced CPA did not also block the associative learning mechanisms responsible for mediating cocaine-CPP or the associative learning mediating taste aversion (Bruchas et al., 2007). In the absence of stress, mice lacking a functional dynorphin/KOR system learn normally. Thus, we infer that disruption of the dynorphin/KOR system selectively blocks the dysphoric component of stress. This concept is consistent with human reports of dysphoria after κ-agonist administration (Pfeiffer et al., 1986).
Previous reports have shown that the CRF system is involved in depression, drug-seeking behavior, and drug withdrawal behaviors (Shaham et al., 1997; Erb et al., 1998; Hammack et al., 2003; Bale and Vale, 2004; Keck et al., 2005; Koob, 2006). For example, local injection of CRF into ventral tegmental area reinstated extinguished cocaine drug seeking, and local injection of nonselective CRF receptor antagonists blocked footshock-induced reinstatement (Wang B et al., 2005). Likewise, release of dynorphin has been implicated in both prodepressant activity and drug-seeking behavior (Mague et al., 2003; Beardsley et al., 2005). The present study establishes a previously unrecognized direct link between these systems by showing that the effects of CRF through the CRF2 receptor require activation of dynorphin/KOR systems in regions known to be activated after a stressful experience (Carrasco and Van de Kar, 2003).
There are a few reports suggesting that KOR activation can elicit CRF release, establishing the potential for interaction between these systems in vivo (Nikolarakis et al., 1986; Song and Takemori, 1992). Additionally, it has been reported recently that reinstatement to drug seeking in squirrel monkeys may be mediated by KOR activation that is dependent on CRF1-R activation (Valdez et al., 2007). A role for CRF1-R activation in the bed nucleus of the stria terminalis during footshock-induced reinstatement of extinguished morphine-CPP was also suggested by local injection of the CRF1-R antagonist CP-154,526 (Wang J et al., 2005). In contrast, our results suggest that dynorphin/KOR signaling occurs downstream of CRF release and CRF2-R activation, because the KOR-antagonist norBNI attenuates the CRF-induced CPA. We also found that the CRF2-R antagonist ASV-30 does not block U50,488-induced CPA, suggesting that the effects of CRF on dynorphin/KOR activity are unidirectional. Further supporting this interpretation, Contarino and Papaleo (2005) recently reported that mice lacking CRF1 receptors are still capable of showing conditioned place aversion to U50,488. Together, these previous reports and our present study suggest that stress causes the release of CRF or urocortin III, which binds and activates the CRF2-R, eliciting a subsequent release of dynorphin and activation of KOR ultimately inducing aversion (summarized graphically in supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
We were surprised to find that the CRF1-R activation did not mediate conditioned place aversion in our study, because CRF1-R activation has been show to have a role in the aversion induced by drug withdrawal, and CRF1-R activation is involved in reinstatement of drug-seeking behavior (Shaham et al., 1998; Contarino and Papaleo, 2005). The basis for these differences is not yet clear but may reflect differences in the drug-dependent state of the animal when it experiences stress. There is also recent evidence that the role of CRF1 and CRF2 receptors in excitatory synaptic transmission can change after cocaine exposure and during withdrawal (Liu et al., 2005). In our experiments, mice had not been exposed previously to reinforcing drugs; this may represent an important difference in terms of the functional output of CRF receptor types and behavioral responses. The CRF2-R may be more important in mediating stress-dependent aversion in drug-naive mice, and this may switch to CRF1-R as animals become drug dependent. This idea is supported by Hammack et al. (2003), who found that inescapable stress in drug-naive rats causes fear conditioning that is dependent on the CRF2-R but not the CRF1-R. Additional analysis is needed to better define the roles of the CRF receptor subtypes in mediating aversion at different stages of drug experience.
In this study, we found several brain structures (including the basolateral amygdala, hippocampus, dorsal raphe, and nucleus accumbens) that showed positive KORp-ir staining after stress or CRF injection. As reported previously, phosphorylation of KOR by G-protein-coupled receptor kinase is caused by sustained activation by agonists (McLaughlin et al., 2003b). Therefore, this antibody gives us the unique ability to identify populations of cells in brain structures that respond to the endogenous KOR agonist dynorphin. The presence of activated KOR in the amygdala, hippocampus, dorsal raphe, and nucleus accumbens is consistent with other reports showing KOR-mediated effects in these structures (Wagner et al., 1993; Pliakas et al., 2001; Tao and Auerbach, 2002; Narita et al., 2006, Bruchas et al., 2007). In one important example, dynorphin in the nucleus accumbens has been shown to be involved in depression-like behavioral models, further suggesting a role for the dynorphin system in depression-related disorders (Pliakas et al., 2001). Although CRF causes an increase in KORp-ir in several brain areas, it is not likely that it is ubiquitous activation, because the caudate–putamen and central nucleus of the amygdala show negligible KORp staining after CRF injection, whereas the caudate does contain KOR that may be involved in the swim-stress immobility response, consistent with a role for caudate circuits in motor activity (Bruchas et al., 2007). Further resolution of the brain areas critical for the specific components of the dynorphin response will require additional studies using local injection approaches and circuit level analysis using electrophysiological approaches.
KORp-ir is a useful measure of sites of dynorphin action in brain, because it provides greater cellular resolution and chemical sensitivity than microdialysis. However, the method has limitations: it does not distinguish different chemical forms of dynorphin or provide fine temporal resolution of dynorphin action. Its lower sensitivity limits have not been established, and KORp-ir does not provide a strictly quantitative measure. We use it as a validated method to define cellular sites and regions of KOR activation. Nevertheless, because the lower sensitivity limits have not been ascertained, the lack of KORp staining does not prove that KOR activation did not occur.
Stress has profound effects on the risk of drug abuse and relapse of drug taking (Volkow and Li, 2004; Fox et al., 2007). Exposure to repeated forced swim stress, repeated social defeat stress, and inescapable footshock stress have been shown to enhance the rewarding properties of cocaine and reinstate cocaine self-administration by a dynorphin-dependent mechanism (McLaughlin et al., 2003a, 2006; Beardsley et al., 2005). Activation of the CRF system has been implicated in a variety of contexts and models of reinstatement to drug seeking and withdrawal. The insights gleaned from the present study suggest the stress-induced activation of the CRF system activates the KOR/dynorphin system to induce negative affective state. This negative affect may represent the driving force or motivational properties in reinstatement and potentiation of cocaine place preference. Understanding how stress increases the risk of drug addiction and relapse of drug abuse will have important implications. Results from the present study support the concept that the dynorphin/κ-system is a novel target for the treatment of addiction and other affective disorders.
Footnotes
This work was supported by the United States Public Health Service and National Institute on Drug Abuse Grants DA16898, DA07278, DA20570, and DA20430. We thank Dr. Ute Hochgeschwender for Pdyn(−/−) mice, Dan Messinger for assistance, and Dr. Paul Phillips for helpful discussion.
References
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3-and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen activated protein kinase mediates κ-opioid dependent dysphoria. J Neurosci. 2007;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cador M, Ahmed SH, Koob GF, Le Moal M, Stinus L. Corticotropin-releasing factor induces a place aversion independent of its neuroendocrine role. Brain Res. 1992;597:304–309. doi: 10.1016/0006-8993(92)91487-y. [DOI] [PubMed] [Google Scholar]
- Carey AN, Borozny K, Aldrich JV, McLaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Phamacol. 2007;569:84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- Carrasco GA, Van de Kar LD. Neuroendocrine pharmacology of stress. Eur J Pharmacol. 2003;463:235–272. doi: 10.1016/s0014-2999(03)01285-8. [DOI] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Contarino A, Papaleo F. The corticotropin-releasing factor receptor-1 pathway mediates the negative affective states of opiate withdrawal. Proc Natl Acad Sci USA. 2005;102:18649–18654. doi: 10.1073/pnas.0506999102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb S, Shaham Y, Stewart J. The role of corticotropin-releasing factor and corticosterone in stress- and cocaine-induced relapse to cocaine seeking in rats. J Neurosci. 1998;18:5529–5536. doi: 10.1523/JNEUROSCI.18-14-05529.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox HC, Bergquist KL, Hong KI, Sinha R. Stress-induced and alcohol cue-induced craving in recently abstinent alcohol-dependent individuals. Alcohol Clin Exp Res. 2007;31:395–403. doi: 10.1111/j.1530-0277.2006.00320.x. [DOI] [PubMed] [Google Scholar]
- Gabry KE, Chrousos GP, Rice KC, Mostafa RM, Sternberg E, Negrao AB, Webster EL, McCann SM, Gold PW. Marked suppression of gastric ulcerogenesis and intestinal responses to stress by a novel class of drugs. Mol Psychiatry. 2002;7:474–483. doi: 10.1038/sj.mp.4001031. [DOI] [PubMed] [Google Scholar]
- Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry. 2002;7:254–275. doi: 10.1038/sj.mp.4001032. [DOI] [PubMed] [Google Scholar]
- Griebel G, Simiand J, Steinberg R, Jung M, Gully D, Roger P, Geslin M, Scatton B, Maffrand JP, Soubrie P. 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1, 3-thiazol-2-amine hydrochloride (SSR125543A), a potent and selective corticotrophin-releasing factor(1) receptor antagonist. II. Characterization in rodent models of stress-related disorders. J Pharmacol Exp Ther. 2002;301:333–345. doi: 10.1124/jpet.301.1.333. [DOI] [PubMed] [Google Scholar]
- Hammack SE, Schmid MJ, LoPresti ML, Der-Avakian A, Pellymounter MA, Foster AC, Watkins LR, Maier SF. Corticotropin releasing hormone type 2 receptors in the dorsal raphe nucleus mediate the behavioral consequences of uncontrollable stress. J Neurosci. 2003;23:1019–1025. doi: 10.1523/JNEUROSCI.23-03-01019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry V, Vale W, Markou A. The effect of lateral septum corticotropin-releasing factor receptor 2 activation on anxiety is modulated by stress. J Neurosci. 2006;26:9142–9152. doi: 10.1523/JNEUROSCI.1494-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck ME, Ohl F, Holsboer F, Muller MB. Listening to mutant mice: a spotlight on the role of CRF/CRF receptor systems in affective disorders. Neurosci Biobehav Rev. 2005;29:867–889. doi: 10.1016/j.neubiorev.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Koob GF. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction. 2006;101:23–30. doi: 10.1111/j.1360-0443.2006.01586.x. [DOI] [PubMed] [Google Scholar]
- Koob GF, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yu B, Orozco-Cabal L, Grigoriadis DE, Rivier J, Vale WW, Shinnick-Gallagher P, Gallagher JP. Chronic cocaine administration switches corticotropin-releasing factor2 receptor-mediated depression to facilitation of glutamatergic transmission in the lateral septum. J Neurosci. 2005;25:577–583. doi: 10.1523/JNEUROSCI.4196-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. κ-Opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003a;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Xu M, Mackie K, Chavkin C. Phosphorylation of a carboxyl-terminal serine within the kappa-opioid receptor produces desensitization and internalization. J Biol Chem. 2003b;278:34631–34640. doi: 10.1074/jbc.M304022200. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Li S, Valdez J, Chavkin TA, Chavkin C. Social defeat stress-induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacology. 2006;31:1241–1248. doi: 10.1038/sj.npp.1300872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Kaneko C, Miyoshi K, Nagumo Y, Kuzumaki N, Nakajima M, Nanjo K, Matsuzawa K, Yamazaki M, Suzuki T. Chronic pain induces anxiety with concomitant changes in opioidergic function in the amygdala. Neuropsychopharmacology. 2006;31:739–750. doi: 10.1038/sj.npp.1300858. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Nikolarakis KE, Almeida OF, Herz A. Stimulation of hypothalamic beta-endorphin and dynorphin release by corticotropin-releasing factor (in vitro) Brain Res. 1986;399:152–155. doi: 10.1016/0006-8993(86)90610-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. San Diego: Elsevier; 2004. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA., Jr Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- Rivier J, Gulyas J, Kunitake K, DiGruccio M, Cantle JP, Perrin MH, Donaldson C, Vaughan J, Million M, Gourcerol G, Adelson DW, Rivier C, Tache Y, Vale W. Stressin1-A, a potent corticotropin releasing factor receptor 1 (CRF1)-selective peptide agonist. J Med Chem. 2007;50:1668–1674. doi: 10.1021/jm0613875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Funk D, Erb S, Brown TJ, Walker CD, Stewart J. Corticotropin-releasing factor, but not corticosterone, is involved in stress-induced relapse to heroin-seeking in rats. J Neurosci. 1997;17:2605–2614. doi: 10.1523/JNEUROSCI.17-07-02605.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Leung S, Buczek Y, Stewart J. CP-154,526, a selective, non-peptide antagonist of the corticotropin-releasing factor1 receptor attenuates stress-induced relapse to drug seeking in cocaine- and heroin-trained rats. Psychopharmacology (Berl) 1998;137:184–190. doi: 10.1007/s002130050608. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Herz A. Differential effects of mu and kappa opioid systems on motivational processes. NIDA Res Monogr. 1986;75:563–566. [PubMed] [Google Scholar]
- Song ZH, Takemori AE. Stimulation by corticotropin-releasing factor of the release of immunoreactive dynorphin A from mouse spinal cords in vitro. Eur J Pharmacol. 1992;222:27–32. doi: 10.1016/0014-2999(92)90458-g. [DOI] [PubMed] [Google Scholar]
- Tache Y, Martinez V, Million M, Wang L. Stress and the gastrointestinal tract III. Stress-related alterations of gut motor function: role of brain corticotropin-releasing factor receptors. Am J Physiol. 2001;280:G173–G177. doi: 10.1152/ajpgi.2001.280.2.G173. [DOI] [PubMed] [Google Scholar]
- Tao R, Auerbach SB. Opioid receptor subtypes differentially modulate serotonin efflux in the rat central nervous system. J Pharmacol Exp Ther. 2002;303:549–556. doi: 10.1124/jpet.102.037861. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Platt DM, Rowlett JK, Ruedi-Bettschen D, Spealman RD. Kappa agonist-induced reinstatment of cocaine seeking in squirrel monkeys: a role for opioid and stress-related mechanisms. J Pharmacol Exp Ther. 2007;323:525–533. doi: 10.1124/jpet.107.125484. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Li TK. Drug addiction: the neurobiology of behavior gone awry. Nat Rev Neurosci. 2004;5:963–970. doi: 10.1038/nrn1539. [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Terman GW, Chavkin C. Endogenous dynorphins inhibit excitatory neurotransmission and block LTP induction in the hippocampus. Nature. 1993;363:451–454. doi: 10.1038/363451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Shaham Y, Zitzman D, Azari S, Wise RA, You ZB. Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci. 2005;25:5389–5396. doi: 10.1523/JNEUROSCI.0955-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, You ZB, Rice KC, Wise RA. Stress-induced relapse to cocaine seeking: roles for the CRF(2) receptor and CRF-binding protein in the ventral tegmental area of the rat. Psychopharmacology (Berl) 2007;193:283–294. doi: 10.1007/s00213-007-0782-3. [DOI] [PubMed] [Google Scholar]
- Wang J, Fang Q, Liu Z, Lu L. Region-specific effects of brain corticotropin-releasing factor receptor type 1 blockade on footshock-stress- or drug-priming-induced reinstatement of morphine conditioned place preference in rats. Psychopharmacology (Berl) 2005;185:19–28. doi: 10.1007/s00213-005-0262-6. [DOI] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C. Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by κ-opioid activation of p38 mitogen-activated protein kinase. J Neurosci. 2007;27:2570–2581. doi: 10.1523/JNEUROSCI.3728-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel AW, Nickel T, Kunzel HE, Ackl N, Sonntag A, Ising M, Holsboer F. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34:171–181. doi: 10.1016/s0022-3956(00)00016-9. [DOI] [PubMed] [Google Scholar]