

Abstract
The failure of Leishmania, an intracellular pathogen, to stimulate a pro-inflammatory response following entry into macrophages has been well reported. This occurs in spite of the fact that ligands for the toll-like receptors (TLR) have been recently shown on the parasite surface and their role in disease protection well documented. The outcome of infection in leishmaniasis is determined by the Th1 versus Th2 nature of the effector response and the generation of IL-12 and IL-10 by the infected macrophages is important for this decision. We evaluated the effect of L. donovani infection of monocytes (cell line THP-1, and monocytes derived from human peripheral blood) on Pam3cys (TLR2 ligand) and lipopolysaccharide (TLR4 ligand) stimulated production of IL-12p40 and IL-10. L. donovani infection caused suppression of TLR2 and TLR4-stimulated IL-12p40, with an increase in IL-10 production. Parasites also modulated the TLR2-stimulated mitogen-activated protein kinase (MAPK) pathway by suppressing MAPK P38 phosphorylation and activating extracellular regulated kinase (ERK)1/2 phosphorylation. These effects could be reversed either by using a MAPK P38 activator, anisomycin, or ERK1/2 inhibitor, U0126. L. donovani caused modulation of TLR2-stimulated MAPK pathways in a contact-dependent mechanism. In addition parasite structural integrity but not viability was required for suppression of TLR2-stimulated IL-12p40 and activation of IL-10. These observations suggest that L. donovani has evolved survival strategies that subvert the pro-inflammatory response generated through TLRs.
Keywords: host/pathogens interactions, immune evasion, innate immunity, toll-like receptor
1. Introduction
Leishmaniasis, caused by protozoan parasites of the genus Leishmania, affects 12 million people worldwide. The parasite is transmitted by phlebotomine sandflies to several mammals, including humans in whom it causes cutaneous, muco-cutaneous and visceral forms of disease. While cutaneous leishmaniasis caused by L. major is a self-healing disease, VL, also known as Kala-azar in India, is caused by L. donovani and represents the most severe form of the disease. An estimated 350-million population is at risk of infection and approximately 100 000 cases of VL are estimated to occur annually in India alone [1,2].
Production of pro-inflammatory cytokines by antigen presenting cells is the central event for generation of a successful immune response against the intracellular pathogen, Leishmania. The control of leishmania infection requires generation of a strong Th1 response [3], which is driven by the presence of IL-12 at the initiation of the immune response [4,5]. However, it has been well established that Leishmania enter and establish infection by stealth and infection of macrophages with either the amastigote or promastigote forms of Leishmania does not activate macrophages to produce IL-12 [6–8].
An increasing number of receptor families are being implicated in the recognition of pathogen-associated molecular patterns (PAMP) by cells of the immune system. Among these, toll-like receptors (TLR) are key players that provide a link between innate and adaptive immunity in higher organisms. The TLR ligands such as LPS, glycolipids, peptidoglycans and lipopeptides are shared by large groups of microorganisms and allow recognition of a wide range of microbial pathogens [9,10]. This leads to the production of inflammatory mediators such as TNF-α and IL-12 by monocyte/macrophage lineage cells and dendritic cells [11]. The adaptor molecule, MyD88, is recruited to the cytoplasmic Toll/IL-1-like receptor domain of the TLR receptors, leading to activation of IL-1 receptor-associated kinase-1 (IRAK-1), IRAK-4 and the TNF receptor-associated factor-6 molecular complexes. Further down-stream activation of mitogen-activated protein kinase (MAPK) is important for activation of IL-12 [12–14]. Activation through TLR2 follows ligand engagement of heterodimers formed by TLR2 with either TLR1 or TLR6. TLR4 engagement causes activation through both the MyD88 dependent and non-dependent pathways [10–12].
Recent reports have implicated TLRs in the recognition of immune response to Leishmania[15]. While Myd88−/− and TLR4−/− C57BL/6 mice showed increased susceptibility to infection [16–18], monocytes from Myd88−/− mice failed to produce IL-1α on L. major infection [19]. The Leishmania surface molecule, lipophosphoglycan, has been shown to activate NK cells and monocytes through TLR2 [17,20]. However, L. donovani promastigotes appear to evade the induction of a pro-inflammatory response in naïve macrophages despite the presence of TLR2. Also, infection with L. donovani promastigotes inhibited robust extracellular regulated kinase (ERK) and Jun N-terminal kinase (JNK) phosphorlation that is stimulated by LPS, a well-established TLR4 ligand [21]. Despite recent demonstration of TLR2 ligands on Leishmania and their relevance to the establishment of infection, the strategies adopted by the parasite to silence the TLR2 stimulated pro-inflammatory responses have not been investigated.
In the present study, we have evaluated the effect of L. donovani infection on the production of the pro-inflammatory mediators, IL-12p40 and IL-10, by monocytes stimulated with the well-described ligands for TLR2 and TLR4, namely LPS and Pam3Cys-Ser-(Lys)4 (Pam3Cys). We have evaluated the effect of infection of monocytes by L. donovanipromastigotes on TLR2-stimulated phosphorylation of MAPK P38 of ERK1/2 and the effect of the MAPK P38 activator, anisomycin, or an ERK1/2 inhibitor, U0126. In addition, because recognition of the pathogen PAMP by TLR is a very early event in the infective process we assessed whether phagocytosis was required for the parasite to mediate changes in the macrophage.
2. Material and methods
2·1 Parasite
Leishmania donovani strain 2001 was maintained in culture media L-15 supplemented with 10% heat inactivated FBS (Life Technologies, Carlsbald, CA, USA) and 0·3% Tryptose phosphate broth, penicillin (100 U/ml) and streptomycin (100 µg/ml) at 25°C. Parasite virulence was maintained by periodic passage in Syrian golden hamsters at the Central Drug Research Institute, Lucknow, India. L. donovani 2001 is a strain developed from a recent clinical isolate from Bihar, India [22]. Stationary phase parasites were centrifuged at 2000 rpm for 5 min and washed with PBS, re-suspended in culture medium and used for infection of monocytes. Stationary phase parasites were centrifuged, washed and treated with 1% para-formaldehyde for 15 min to generate formaldehyde treated parasites (Ld-F). These parasites were washed five times with PBS and resuspended in culture medium before adding to monocytic cells. Stationary phase parasites were lysed by five cycles of freeze thawing to generate Ld-K and kept at 37°C temperature before adding to monocytic cells. Ld-K was used in 1:10 cell to parasite ratio in all experiments.
2·2 Cell culture and experimental protocols
Human monocytic cell line, THP-1 (National Center for Cell Science, Pune, India), was maintained in RPMI-1640 (Sigma, St Louis, MO, USA) supplemented with 10% FBS, penicillin (100 U/ml) and streptomycin (100 µg/ml). In order to induce maturation, THP-1 cells at a concentration of 5 × 105 cells/well in 0.5 ml medium were treated with 4α-Phorbol 12-myristate 13-acetate (PMA) (Sigma) in a 24-well culture plate for 12 h followed by 12 h in fresh medium prior to infection. Viable L. donovani promastigotes and Ld-F were added to THP-1 cells (0·5 × 106 cells/well) at 10:1 parasites to cell ratio and kept for 2 h at 37°C, 5% CO2 and 95% humidity. Non-ingested parasites were removed and cells were stimulated either with TLR2 ligand, Pam3cys (1 µg/ml), or TLR4 ligand, LPS (1 µg/ml), for 24 h. Human peripheral blood mononuclear cells were isolated by the density gradient sedimentation method and incubated for 1 h in FBS free medium in culture flasks in order to allow monocytes to adhere. Adherent cells were detached by incubation with Trypsin-EDTA (Life Technologies) for 1 min, flushed and washed with PBS. L. donovani promastigotes were added to monocytes (5 × 104 cells/well in 0.2 ml medium in a 96-well culture plate) at 10:1 parasites to cell ratio and stimulated with Pam3cys (10 ng/ml) or LPS (10 ng/ml) for 24 h. The dose of LPS and Pam3cys for optimal stimulation of the different cell types was standardized in preliminary experiments (data not shown). Plates were incubated at 37°C, 5% CO2 and 95% humidity for 24 h. For IL-10 neutralization assay, THP-1 cells were infected and activated with Pam3cys or LPS and IL-10 neutralization antibodies were added at a concentration of 1, 0·1, 0·01 pg/ml (BD Biosciences, Pharmingen, San Diego, CA, USA). Neutralization of IL-10 was confirmed by IL-10 ELISA in culture supernatants.
2·3 Cytokine ELISAs
Supernatants were analysed for IL-12p40 and IL-10 by commercial sandwich ELISA kits as per the manufacturer's instruction (BD Biosciences). Briefly, plates were coated with monoclonal capture antibody by overnight incubation at 4°C and blocked with 10% FBS in PBS for 1 h. One hundred µl of culture supernatant or recombinant standard was added to the plates and incubated for 2 h. Cytokines were detected by addition of biotinylated secondary antibodies and horseradish peroxidase-conjugated streptavidin labelled antibodies. Colour was developed using tetramethylbenzidine (BD Biosciences) for 15 min and absorbance was read at 570 nm.
2·4 Reverse transcriptase PCR
THP-1 cells were infected and activated with Pam3cys and LPS in six-well plates and incubated for 10 h as described above. Cells were lysed in TRIZOL reagent (Life Technologies) and total cellular RNA was extracted as per the manufacturer's instruction. One microgram of total RNA was incubated in reaction buffer containing Oligo-dT primer, 10 U of AMV reverse transcriptase (Invitrogen, Maryland, USA), RNAase inhibitor and dNTP mix for 1 h at 37°C. PCR was performed for human IL-12p40, IL-10, TLR2, TLR4 and GAPDH using gene specific primers (Table 1). PCR was performed in 25 µl reaction volume, for 30 cycles with the following cycling conditions: denaturation at 94°C for 1 min; annealing at 60–65°C (Table 1) for 1 min and extension at 72°C for 1 min. Twenty micro litres of the PCR products were resolved on 1·5% agarose gel stained with ethidium bromide. GAPDH was used as housekeeping gene.
Table 1.
Nucleotide sequences of various forward (F) and reverse (R) primers used for RT-PCR.
| Target | Primers sequences (5′-3′) | Annealing Temp (°C) | Product size (bp) |
|---|---|---|---|
| IL-12p40 | F-GGCCCAGAGCAAGATGTGTCACCA | 65 | 144 |
| R-TCTCCAGGGGCATCCGGATACCAA | |||
| IL-10 | F-ACAGCTCACCACTGCTCTGT | 60 | 327 |
| R-AGTTCACATGCGCCTTGATG | |||
| GAPDH | F-GAAGGTGAAGGTCGGAGTC | 60 | 225 |
| R-GAAGATGGTGATGGGATTTC | |||
| TLR2 | F-GCCTACTGGGTGGAGAACCT | 60 | 340 |
| R-GGCCACTCCAGGTAGGTCTT | |||
| TLR4 | F-TGCAATGGATCAAGGACCAGAGGC | 62 | 449 |
| R-GTGCTGGGACACCACAACAATCACC |
2·5 Western blot analysis
THP-1 cells were infected and activated simultaneously with Pam3cys and LPS in 40-mm culture plates and incubated for 0, 5, 15 and 30 min as described above. Cellular extracts were prepared at 4°C with ice-cold reagents. Cells were pelleted by centrifugation and re-suspended in 400 µl of ice-cold lysis buffer (1 M HEPES, 0·5 M EDTA, 0.1 M EGTA, 2 M KCl 0.1 M DTT, cocktail of protease inhibitors 5 µg/ml, and 10% NP-40). The lysates (20 µg) were resolved on 10% SDS-polyacrylamide gels and proteins were electro-transferred to nitrocellulose filter membrane (Sigma). Membranes were probed with rabbit polyclonal antibodies against phosphorylated MAPK P38 and ERK1/2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The membranes were stripped and reprobed with antibodies to non-phosphorylated P38 and ERK1/2 to ensure equal loading. Membranes were incubated with HRP-conjugated anti-rabbit goat IgG secondary antibodies (Bangalore Genei, Bangalore, India) and detection was done using chemiluminescence detection kit (Roche Diagnostics, Mannheim, Germany).
2·6 Statistics
Student's t-test was employed for inter-group comparisons and a P value of P < 0·05 was taken as significant. The study was approved by the Institutional Ethics Committee and venous blood was drawn from normal, healthy volunteers after obtaining informed consent.
3. Results
3·1 L. donovani infection had no effect on expression of TLR2 and TLR4 mRNA
In order to determine whether L. donovani infection had any effect on TLR2 and TLR4 expression, TLR2 and TLR4 mRNA levels were evaluated. THP-1 cells were infected with L. donovani and activated with Pam3cys or LPS for 10 h. Neither infection of THP-1 with L. donovani nor activation of these cells with Pam3cys or LPS had any effect on the TLR2 and TLR4 mRNA expression. The activation of L. donovani infected THP-1 cells with Pam3cys or LPS also did not have any effect on the TLR2 and TLR4 mRNA expression (Fig. 1).
Fig. 1.
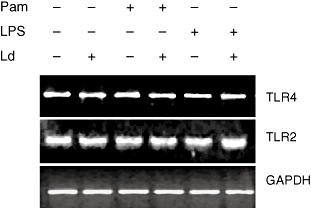
Effect of L. donovani infection on TLR2 and TLR4 mRNA expression. THP-1 cells were infected with L. donovani at 10:1 parasites to cell ratio and stimulated with Pam3cys (1 µg/ml) or LPS (1 µg/ml) for 10 h. RNA was isolated and RT-PCR performed for human TLR2, TLR4 and housekeeping gene GAPDH. Gel picture shown here is a representative of three independent experiments.
3·2 L. donovani infection suppresses TLR2-stimulated IL-12p40 production and activates IL-10 production
THP-1 cells were infected with L. donovani for 2 h and then activated with Pam3cys or LPS for 24 h. Infection of L. donovani alone had no effect on the IL-12p40 and IL-10 production; however, L. donovani infection suppressed Pam3cys-stimulated IL-12p40 production by 70% and LPS-stimulated IL-12p40 production by 35% (Fig. 2a). L. donovani infection enhanced Pam3cys and LPS-stimulated IL-10 production (Fig. 2b). The changes seen at the protein level were also seen at the mRNA transcript level (Fig. 2c and d). The observations on IL-12p40 suppression and IL-10 stimulation were similar when L. donovani and Pam3cys or LPS were added simultaneously to THP-1 cells (data not shown). L. donovani infection of human monocytes also caused suppression of Pam3cys and LPS-stimulated IL-12p40 production and activation of IL-10 production, as was seen in the THP-1 cells (Fig. 3a and b). To exclude the possibility that IL-12p40 suppression by L. donovani infection of THP-1 cells may be due to induction of IL-10 by the parasite, we added IL-10 neutralizing antibodies in culture medium. IL-10 neutralization had no effect on the IL-12p40 suppression by L. donovani infection (data not shown).
Fig. 2.
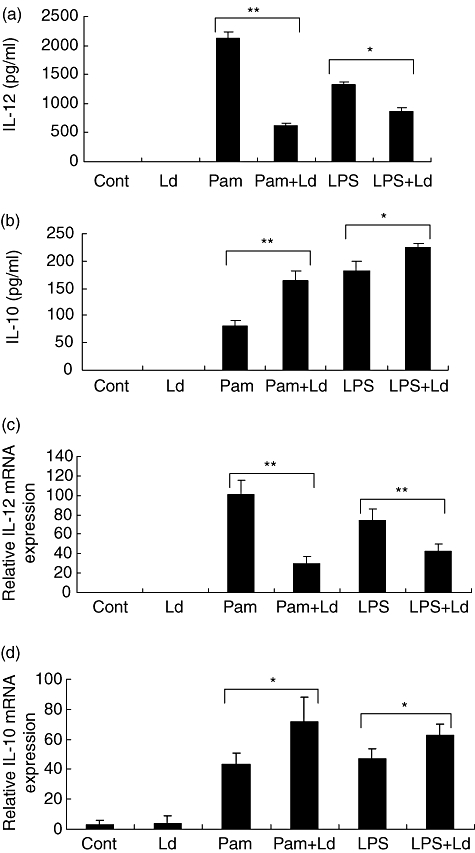
Effect of L. donovani infection on Pam3cys or LPS stimulated cytokines production by THP-1 cells. THP-1 cells were infected with L. donovani at 10:1 parasites to cell ratio and stimulated with Pam3cys (1 µg/ml) or LPS (1 µg/ml). Supernatants were collected after 24 h activations and IL-12p40 (A) and IL-10 (B) production was measured by ELISA. RNA was isolated at 10 h and RT-PCR performed by using specific primers for IL-12p40, IL-10 and housekeeping gene GAPDH. The results are expressed as ratio of densitometric readings of IL-12p40 (C) and IL-10 (D) to GAPDH mRNA. All graphs represent mean + SD of three independent experiments. Group comparison was done using student's t test P < 0·05 considered significant (*P < 0·05, **P < 0·005).
Fig. 3.
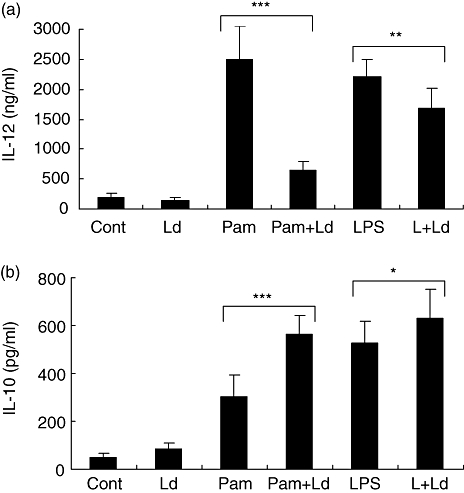
Effect of L. donovani infection on Pam3cys or LPS stimulated cytokine production by human monocytes. Isolated monocytes were infected with L. donovani at 10:1 parasites to cell ratio and stimulated with Pam3cys (10 ng/ml) or LPS (10 ng/ml). Supernatants were collected after 24 h of stimulation and IL-12p40 and IL-10 was measured by ELISA. Data represents mean + SD of five individuals. Group comparison was done using student's t test P < 0·05 considered significant (*P < 0·05, **P < 0·005, ***P < 0·0005).
3·3 L. donovani infection modulates TLR2-stimulated MAPK pathways
The MAPK are key regulators of IL-10 and IL-12 activation [23,24]. They also are important for the counter-regulation of IL-12 and IL-10, which in turn influence the Th1-Th2 balance. Because in our system, L. donovani infection caused suppression of TLR2-stimulated IL-12 with activation of IL-10, we looked at the MAPK pathways. THP-1 cells were infected and activated simultaneously with Pam3cys for 0, 5, 15 and 30 min. L. donovani alone did not cause phosphorylation of P38 and ERK1/2 (data not shown). Pam3cys-stimulated phosphorylation of MAPK P38 was seen maximally after 15 min of stimulation and this was inhibited by L. donovani infection (Fig. 4a). Pam3cys had no effect on the phosphorylation of MAPK-ERK1/2, while L. donovani infection stimulated ERK1/2 phosphorylation in Pam3cys-stimulated cells (Fig. 4b). Maximum LPS-stimulated phosphorylation of MAPK P38 was seen after 30 min of stimulation and was inhibited by L. donovani infection while ERK1/2 phosphorylation was activated in LPS-stimulated infected-cells (Fig. 4c and d). Thus L. donovani counter-regulates MAPK pathways by inhibiting the TLR2 and TLR4-stimulated phosphorylation of P38 with simultaneous activation of ERK 1/2.
Fig. 4.
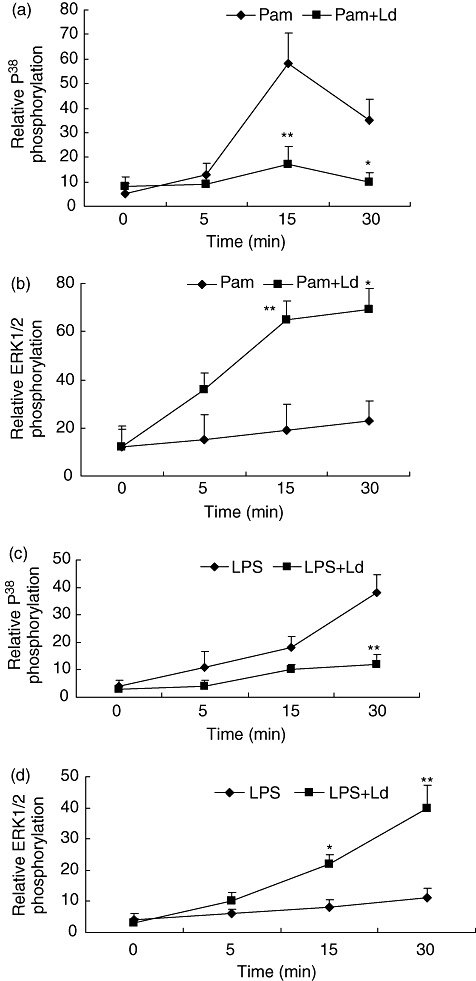
Effect of L. donovani infection on Pam3cys and LPS stimulated phosphorylation of MAPK P38 and ERK1/2. THP-1 cells were infected with L. donovani at 10:1 parasites to cell ratio and simultaneously stimulated with Pam3cys (1 µg/ml) (A, B) or LPS (1 µg/ml) (C, D) for 0, 5, 15 and 30 min. Cells lysates were subjected to electrophoresis, transferred on nitrocellulose membrane and probed with antibodies to phosphorylated and non-phosphorylated P38 and ERK1/2. The results are expressed as ratio of densitometric readings of phosphorylated P38 (A, C) and ERK1/2 (B, D) to non-phosphorylated P38 and ERK1/2. Mean + SD of three independent experiments is shown.
3·4 Effect of L. donovani infection of monocytes is reversed with pharmacological modulators
In order to assess whether the effect of L. donovani infection on MAPK phosphorylation could be modulated, we used anisomycin, a well-established activator of P38 phophorylation and ERK1/2 inhibitor U0126. THP-1 cells were infected with L. donovaniand stimulated simultaneously with Pam3cys and different doses of anisomycin (0·1, 1 and 10 ng/ml) for 15 min or THP-1 cells were pretreated with U0126 (0·1, 1 and 10 µM) followed by simultaneously infection with L. donovani and stimulation with Pam3cys. L. donovani infection showed no P38 phosphorylation by Pam3cys stimulation at 15 min; however, it was restored on activation with anisomycin or U0126 treatment in a dose-dependent manner (Fig. 5a). To demonstrate the effect of anisomycin or U0126 treatment on IL-12p40 and IL-10 levels, infected cells were incubated with anisomycin and Pam3cys for 24 h or pretreated with U0126 followed by activation with Pam3cys for 24 h. While IL-12p40 levels were increased by anisomycin or U0126 treatment in a dose-dependent manner (Fig. 5b), there was a decrease in IL-10 levels (Fig. 5c). U0126 pre-treatment had no effect on L. donovani infection and it did not cause any stimulation of IL-12p40 in infected or non-infected THP-1 cells (data not shown). These observations show that modulation of the MAPK pathway by L. donovani infection can be reverted by anisomycin and U0126.
Fig. 5.
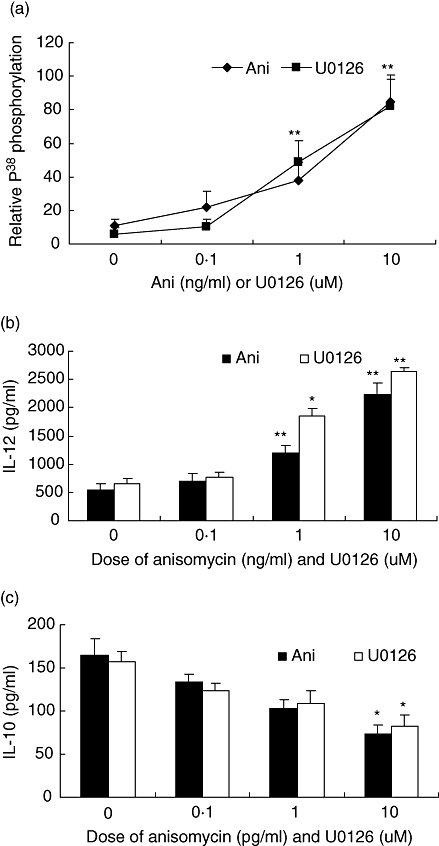
Effect of L. donovani infection on Pam3cys stimulated activation of MAPK P38 in anisomycin and U0126 treated cells. THP-1 cells were infected with L. donovani at 10:1 parasites to cell ratio and simultaneously stimulated with Pam3cys and P38 activator anisomycin (0·1, 1 and 10 ng/ml) or pretreated with ERK1/2 inhibitor U0126 (0·1, 1 and 10 µM) for 1 h followed by addition of L. donovani at 10:1 parasites to cell ratio and Pam3cys. Cell lysates were prepared after incubating for 15 min, subjected to electrophoresis, transferred on nitrocellulose membrane and probed with anti-phosphorylated P38 and non-phosphorylated P38 antibodies. The results are expressed as ratio of densitometric readings of phosphorylated P38 to non-phosphorylated P38 (A). L. donovani infected THP-1 cells were activated with anisomycin and Pam3cys (1 µg/ml) or U0126 pretreated and infected cells were activated with Pam3cys (1 µg/ml). Supernatant were collected after 24 h and IL-12p40 (B) and IL-10 (C) measurement done by ELISA. Mean + SD of three independent experiments is shown and group comparison was done using student's t test P < 0·05 considered significant (*P < 0·05, **P < 0·005).
3·5 L. donovani modulate TLR2-stimulated MAPK pathways in a contact-dependent mechanism
Our observation that P38 phosphorylation was suppressed within 15 min of L. donovaniinfection of THP-1 cells raised the question, whether parasite ingestion was required for the immuno-modulation. As 15 min appears inadequate for establishing infection, we investigated whether the suppression could be a contact-dependent phenomenon. THP-1 cells were treated with cytochalasin-B for 15 min followed by infection with different doses of parasite (2:1, 5:1 and 10:1 parasites to cell ratio) and simultaneously activated with Pam3cys for 15 min. Cytochalasin-B, an inhibitor of actin polymerization, prevents the ingestion of the parasite. Stimulation of cytochalasin-B pre-treated THP-1 cells with Pam3cys, caused phosphorylation of P38 but not ERK1/2. Addition of different doses of L. donovani with Pam3cys caused suppression of P38 phosphorylation and activated ERK1/2 phosphorylation in parasite dose-dependent manner (Fig. 6).
Fig. 6.
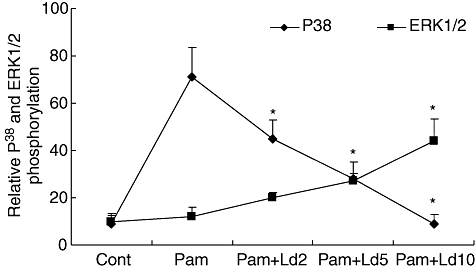
Effect of L. donovani infection on Pam3cys stimulated activation of MAPK P38and ERK1/2 in cytochalasin-B treated THP-1 cells. THP-1 cells were treated with cytochalasin-B for 15 min followed by simultaneous infection with different dose of parasite (2:1, 5:1 and 10:1 parasites to cell ratio) and activation with Pam3cys for 15 min. Cells lysates were subjected to electrophoresis, transferred on nitrocellulose membrane and probed with antibodies to phosphorylated and non-phosphorylated P38 and ERK1/2. Results show ratio of densitometic readings of phosphorylated P38 and ERK1/2 to non-phosphorylated P38 and ERK1/2. Mean + SD of three independent experiments is shown.
3·6 L. donovani structural integrity but not viability is required for modulation of TLR2-stimulated response
Because L. donovani caused modulation of the MAPK pathway without ingestion by monocytic cells, we tried to establish whether the modulation was caused by a molecule secreted by the parasite or was due to contact with parasite surface molecule. We added paraformaldehyde fixed parasites (Ld-F), which were not viable but structurally intact, and freeze-thawed parasites (Ld-K), which should have native proteins and antigens intact, to THP-1 cells. These cells were stimulated with Pam3cys for 24 h. Ld-F but not Ld-K was able to suppress Pam3cys-stimulated IL-12p40 production (Fig. 7a) and cause stimulation of IL-10 production (Fig. 7b) by THP-1 cells. Addition of paraformaldehyde fixed parasites (Ld-F) but not freeze-thawed parasites (Ld-K) was able to suppress TLR2 stimulated MAPK P38 phosphorylation and activated MAPK-ERK1/2 phosphorylation (Fig. 8a–c). These results suggest that the modulation is dependent on surface contact because killed but structurally intact parasites could cause suppression of IL-12p40, to the same degree as live parasites. This also suggests that parasite structural integrity but not viability is required for the suppression of Pam3cys-stimulated IL-12 production by monocyte [6].
Fig. 7.
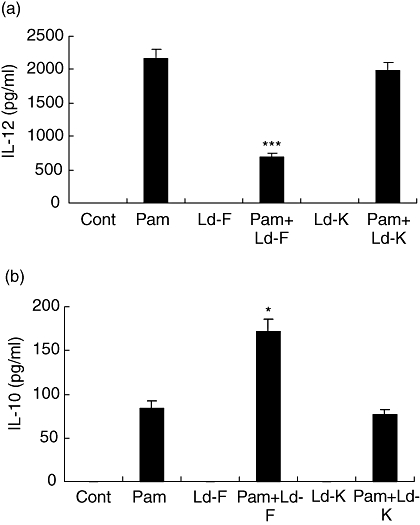
Effect of paraformaldehyde fixed parasites (Ld-F) or freeze-thawed parasites (Ld-K) infection on Pam3cys stimulated cytokines (IL-12p40 and IL-10) production and activation of MAPK pathway by THP-1 cells. THP-1 cells were infected with either Ld-F or Ld-K for 2 h, non-ingested parasites were washed and cultures were activated with Pam3cys (1 ug/ml) for 24 h. Supernatants were collected for IL-12p40 (A) and IL-10 (B) measurement. Mean + SD of three independent experiments is shown and group comparison was done using student's t test P < 0·05 considered significant (*P < 0·05, ***P < 0·005).
Fig. 8.
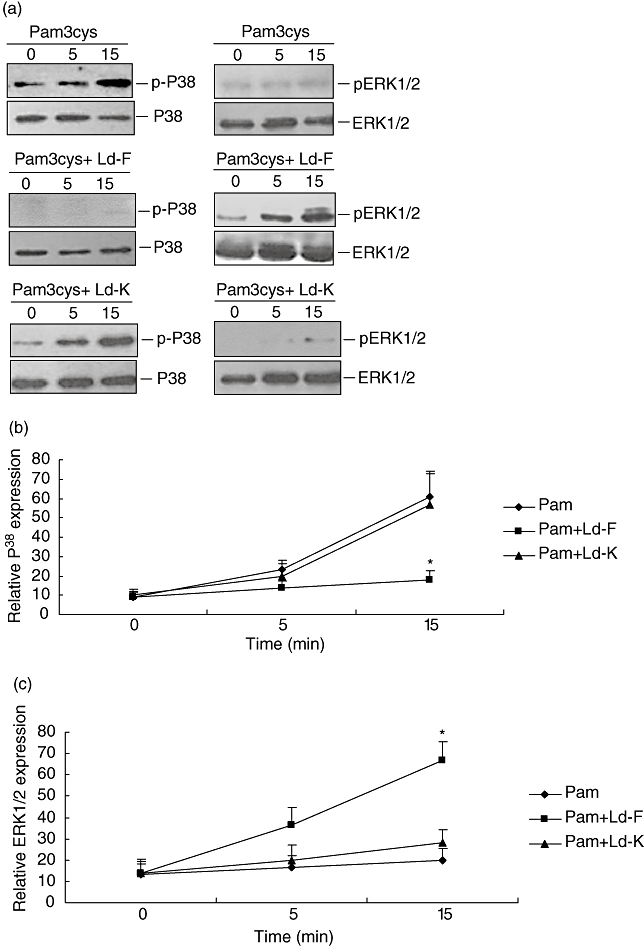
Effect of paraformaldehyde fixed parasites (Ld-F) or freeze-thawed parasites (Ld-K) infection on Pam3cys stimulated activation of MAPK pathway by THP-1 cells. THP-1 cells were infected with either Ld-F or Ld-K simultaneously activated with Pam3cys (1ug/ml) for 0, 5 and 15 min. Cells lysates were subjected to electrophoresis, transferred on nitrocellulose membrane and probed with antibodies to phosphorylated and non-phosphorylated P38 and ERK1/2. Figure shown is a representative of three independent experiments (A). Data shows ratio of densitometric reading phosphorylated P38and ERK1/2 to non-phosphorylated P38 (B) and ERK1/2 (C). Mean + SD of three independent experiments is shown and group comparison was done using student's t test P < 0·05 considered significant (*P < 0·05, ***P < 0·005).
4. Discussion
It has been previously demonstrated that the Myd88 dependent pathway is important for the protective immune response against L. major. The majority of the evidence shows that infection per se fails to stimulate an inflammatory response in monocyte/ macrophage cells [7,8,25], although there have been reports to the contrary. It has also been shown that the parasite suppresses the pro-inflammatory response that is stimulated by LPS, a TLR4 ligand [8,6,21]. In this paper, we show for the first time that L. donovanican modulate TLR2-mediated intra-cellular pathways, resulting in suppression of IL-12p40 production and activation of IL-10 production. We further show that this is a contact-mediated phenomenon that is dependent on parasite structural integrity and not viability.
As has been reported by previous workers, we too have shown that infection of monocyte cells with L. donovani promastigotes failed to stimulate a pro-inflammatory response with negligible production of IL-12. The TLR2 and TLR4 ligands stimulated production of impressive levels of IL-12 and IL-10 in THP-1 cells and primary human monocytes. However, following infection with L. donovani, there was suppression of TLR2 and TLR4 stimulated IL-12 production accompanied by simultaneous stimulation of IL-10 production. The suppression was more pronounced in TLR2-stimulated cells then in TLR4-stimulated cells. These changes were not caused by decreased expression of TLR2 or TLR4 in infected cells, because the transcript levels of TLR2 and TLR4 were unaffected by infection. While IL-12 is an important link between innate and adaptive immune responses, IL-10 promotes the Th2 immune response by down-regulating IL-12 production and during Leishmania infection plays a major role in disease susceptibility [4,26,27]. The suppression of IL-12 reported by us is, however, not caused by IL-10 secreted by the macrophages, because neutralization with anti-IL-10 antibody did not abrogate the suppression. Decreased generation of oxygen intermediate following activation of Leishmania-infected mouse macrophage cells with Zymogen (a TLR2/6 ligand) has been reported; however, cytokines generation was not studied by these workers [28].
TLR ligation leads to phosphorylation of MAPK P38 with production of IL-12 and of ERK1/2 with production of IL-10 [23,24,29]. The MAPK have been shown to play a crucial role in the control of Leishmania infection [30] and reciprocal regulation of MAPK P38 and ERK1/2 has been shown to be responsible for protection versus susceptibility to infection [31,32]. In this study, we have shown that L. donovani infection failed to induce any phosphorylation of P38 or ERK1/2 in 30′. The failure of Leishmania to activate phosphorylation of MAPK has been previously reported. While wild-type L. donovani promastigotes failed to activate MAPK in mouse bone marrow derived macrophage (BMM) they were able to do so in IFN-γ treated mouse BMM [25]. We found that in the presence of L. donovani promastigotes, TLR2 and TLR4 stimulated P38 phosphorylation was inhibited and ERK1/2, which showed very little TLR2 or TLR4 stimulated phosphorylation by 30′, showed significant increase in phosphorylation. Thus, the parasite is able to counter regulate P38 and ERK1/2 phosphorylation and this is seen as suppression of IL-12 and induction of IL-10. Counter regulation between P38 and ERK1/2 seems to be important for the decision regarding development of Th1 or Th2 type of effecter responses, which in turn has important consequences for the outcome of infection.
Further, we observed that the L. donovani-induced IL-12 suppression and IL-10 production in TLR2- and TLR4-stimulated cells could be reversed by either anisomysin, a P38 activator, or ERK1/2, specific inhibitor, U0126. Treatment of Leishmania-infected mice with anisomysin [30] or anisomysin along with CD40L as well as an ERK1/2 inhibitor has been shown to cause rapid recovery from parasitemia [31,32]. In these cases, recovery from parasitemia was accompanied by a Th1 cell expansion and restoration of CD40/CD40L mediated IL-12 production. Administration of U0126 (ERK1/2 inhibitor) to Leishmania amazonensis-infected mice was shown to delay lesion progression with reduction in parasite burden [33]. These studies emphasize the important role of these kinases in the Th1-Th2 decision and highlight the significance that the observations in the present study may have for the disease outcome.
We observed that the effects of L. donovani on the monocytes was seen irrespective of whether TLR ligands and parasites were added simultaneously or TLR ligand was added to cells with established infection. In addition, the suppression of P38 phosphorylation was seen within 15 min of adding L. donovani to monocyte cultures, which appears inadequate time for establishing infection. This suggested that surface contact between parasite and the cells is adequate to generate inhibitory signals. We found that the changes brought about by L. donovani parasites in MAPK phosphorylation could be brought about even in Cytochalasin-B pre-treated THP-1 cells within 15′. Cytochalasin-B is an inhibitor of actin polymerization and prevents phagocytosis but does not inhibit parasite attachment. These findings, for the first time, provide evidence that parasites probably modulate the host immune response within minutes of its contact with phagocytic cells. It was clear that structural integrity of the parasite was important because paraformaldehyde fixed parasites caused suppression of the TLR2-stimulated IL-12 production and activated IL-10 production while freeze-thawed parasites did not.
Thus, in the presence of activation through the TLR receptors by their ligands (as in our experiments) or through LPG during infection in vivo, how does suppression dominate over activation? One possibility is engagement of a second receptor by the parasite. It has been shown that TLR-stimulated IL-10 levels are enhanced with concomitant ERK1/2 phosphorylation by a second signal generated through FcγR on monocytes [34]. On the other hand, immunomodulatory signaling through various phagocytic receptors such as macrophage mannose receptor (MR), FcγR and complement receptor 3 (CR3), has been shown to subvert the activation of macrophages and help in the silencing of the pro-inflammatory response [33,35,36]. Of these, we cannot implicate FcγR for the changes brought about by Leishmania contact, because opsonized parasites were not used by us. The role of CR3 cannot be ruled out completely because L. major surface molecule PSA has been shown to bind with host monocyte CR3 receptor [37]. Signaling through MR has been extensively implicated in the subversion of macrophage pro-inflammatory responses in another important intracellular infection, M. tuberculosis[38]. In L. major infection, however, MR was not seen to cause suppression of CD40 stimulated IL-12 production in monocytes of MR KO mice, indicating that MR was not involved in modulation of CD40 stimulated pathways [39]. However, there is considerable variation in the results reported in various studies due to the type of cell used (primary monocytes, cell lines or DC), the type of stimulation given (IFN-γ, TNF-α, LPS) and the parasite species (L. donovani, L. major). The role of MR receptors and of CR3 receptors in the observed modulation of monocyte functions in this study needs to be further evaluated.
An alternative possibility is that receptor engagement activates the PI3K/Akt signaling pathway. Signaling through this pathway promotes anti-inflammatory responses [40] and PI3K has been shown to suppress IL-12 production triggered by TLR signaling and limit excessive TH1 polarization [41]. Leishmania promastigotes have been shown to activate this pathway to confer host cell resistance to apoptosis, another important evasion strategy adopted by the parasite [42]. The role of PI3K signaling in the dampening of the IL-12 response needs to be investigated.
Although our findings are related to the early events following promastigote-host cell contact, these may also influence the immune evasion mechanism adopted by Leishmania amastigote infection. As in the case of promastigotes, infection of monocyte or DC by amastigotes also failed to stimulate any cytokine production and inhibited LPS-stimulated IL-12 production. [43–45]. Infection with lesion-derived L. mexicanaamastigotes inhibited LPS-induced IL-12 production by mouse BMM with selective degradation of the LPS-stimulated MAPK, JNK and ERK. [45]. While L. amazonensis amastigote infection of murine peritoneal macrophages caused decrease in ERK activity as infection progressed, that of BMM caused activation of the MAPK ERK1/2 associated with parasite-induced secretion of IL-10 [33,46]. Thus the modulation of the host macrophage responses by the MAPK pathways probably plays a role at various stages of parasite entry and maturation within the host cell to maintain a favourable environment for its survival.
In summary, we describe a new contact-dependent evasion mechanism adopted by the parasite for modulation of TLR2-stimulated MAPK pathways to favour its survival. In addition, parasite structural integrity but not viability was required for the modulation of TLR2-stimulated pathways. Understanding of this mechanism will help development of new control strategies for the disease.
Acknowledgments
Dinesh Chandra is senior research fellow of the Council of Scientific and Industrial Research government of India. Dr Anuradha Dubey scientist-E (Central Drug Research Institute, Lucknow, India) provided L. donovani (strain 2001) culture.
References
- 1.Mauel J. Vaccination against Leishmaniainfection. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2:201–26. doi: 10.2174/1568008023340631. review. [DOI] [PubMed] [Google Scholar]
- 2.Singh RK, Pandey HP, Sundar S. Visceral leishmaniasis (kala-azar): challenges ahead. Indian J Med Res. 2006;12:331–44. Review. [PubMed] [Google Scholar]
- 3.Alexander J, Bryson K. T helper (h) 1/Th2 and Leishmania: paradox rather than paradigm. Immunol Lett. 2005;99:17–23. doi: 10.1016/j.imlet.2005.01.009. Review. [DOI] [PubMed] [Google Scholar]
- 4.Ghalib HW, Whittle JA, Kubin M, et al. IL-12 enhances Th1-type responses in human Leishmania donovani infections. J Immunol. 1995;154:4623–9. [PubMed] [Google Scholar]
- 5.Bacellar O, Brodskyn C, Guerreiro J, et al. Interleukin-12 restores interferon-gamma production and cytotoxic responses in visceral leishmaniasis. J Infect Dis. 1996;173:1515–18. doi: 10.1093/infdis/173.6.1515. [DOI] [PubMed] [Google Scholar]
- 6.Weinheber N, Wolfram M, Harbecke D, Aebischer T. Phagocytosis of Leishmania mexicana amastigotes by macrophages leads to a sustained suppression of IL-12 production. Eur J Immunol. 1998;28:2467–77. doi: 10.1002/(SICI)1521-4141(199808)28:08<2467::AID-IMMU2467>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Reiner SL, Zheng S, Wang ZE, Stowring L, Locksley RM. Leishmania promastigotes evade interleukin 12 (IL-12) induction bymacrophages and stimulate a broad range of cytokines from CD4+ T cells during initiation of infection. J Exp Med. 1994;179:447–56. doi: 10.1084/jem.179.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrera L, Gazzinelli RT, Badolato R, et al. Leishmania promastigotes selectively inhibit interleukin 12 induction in bone marrow-derived macrophages from susceptible and resistant mice. J Exp Med. 1996;183:515–26. doi: 10.1084/jem.183.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. Review. [DOI] [PubMed] [Google Scholar]
- 10.Akira S. Mammalian Toll-like receptors. Curr Opin Immunol. 2003;15:5–11. doi: 10.1016/s0952-7915(02)00013-4. Review. [DOI] [PubMed] [Google Scholar]
- 11.Netea MG, van der Graaf C, Van der Meer JW, Kullberg BJ. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J Leukoc Biol. 2004;75:749–55. doi: 10.1189/jlb.1103543. Review. [DOI] [PubMed] [Google Scholar]
- 12.Medzhitov R, Preston-Hurlburt P, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the Toll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–8. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 13.Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. The human toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6) J Exp Med. 1998:2097–101. doi: 10.1084/jem.187.12.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki N, Suzuki S, Duncan GS, et al. Severe impairment of interleukin-1 and Toll-like receptor signaling in mice lacking IRAK-4. Nature. 2002;416:750–6. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 15.Flandin JF, Chano F, Descoteaux A. RNA interference reveals a role for TLR2 and TLR3 in the recognition of Leishmania donovani promastigotes by interferon-gamma-primed macrophages. Eur J Immunol. 2006;36:411–20. doi: 10.1002/eji.200535079. [DOI] [PubMed] [Google Scholar]
- 16.Muraille E, De Trez C, Brait M, De Baetselier P, Leo O, Carlier Y. Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized Th2 response. J Immunol. 2003;170:4237–41. doi: 10.4049/jimmunol.170.8.4237. [DOI] [PubMed] [Google Scholar]
- 17.de Veer MJ, Curtis JM, Baldwin TM, et al. MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur J Immunol. 2003;33:2822–31. doi: 10.1002/eji.200324128. [DOI] [PubMed] [Google Scholar]
- 18.Kropf P, Freudenberg MA, Modolell M, et al. Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect Immun. 2004;72:1920–8. doi: 10.1128/IAI.72.4.1920-1928.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawn TR, Ozinsky A, Underhill DM, Buckner FS, Akira S, Aderem A. Leishmania major activates IL-1α expression in macrophages through a MyD88 dependent pathway. Microbes Infect. 2002;4:763–71. doi: 10.1016/s1286-4579(02)01596-4. [DOI] [PubMed] [Google Scholar]
- 20.Becker I, Salaiza N, Aguirre M, et al. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Mol Biochem Parasitol. 2003;130:65–74. doi: 10.1016/s0166-6851(03)00160-9. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh S, Bhattacharyya S, Sirkar M, et al. Leishmania donovani suppresses activated protein 1 and NF-kappaB activation in host macrophages via ceramide generation: involvement of extracellular signal-regulated kinase. Infect Immun. 2002;70:6828–38. doi: 10.1128/IAI.70.12.6828-6838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garg R, Gupta SK, Tripathi P, Naik S, Sundar S, Dube A. Immunostimulatory cellular responses of cured leishmania infected patients and hamsters against integral membrane proteins and non-membranous soluble proteins of a recent clinical isolate of Leishmania donovani. Clin Exp Immunol. 2005;140:149–56. doi: 10.1111/j.1365-2249.2005.02745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng GJ, Goodridge HS, Harnett MM, et al. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J Immunol. 1999;163:6403–12. [PubMed] [Google Scholar]
- 24.Suttles J, Milhorn DM, Miller RW, Poe JC, Wahl LM, Stout RD. CD40 signaling of monocyte inflammatory cytokine synthesis through an ERK1/2-dependent pathway. A target of interleukin (il)-4 and il-10 anti-inflammatory action. J Biol Chem. 1999;274:5835–42. doi: 10.1074/jbc.274.9.5835. [DOI] [PubMed] [Google Scholar]
- 25.Prive C, Descoteaux A. Leishmania donovani promastigotes evade the activation of mitogen-activated protein kinases p38, c-Jun N-terminal kinase, and extracellular signal-regulated kinase-1/2 during infection of naive macrophages. Eur J Immunol. 2000;30:2235–44. doi: 10.1002/1521-4141(2000)30:8<2235::AID-IMMU2235>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 26.Bacellar O, D’oliveira A, Jeronimo S, Carvalho EM. IL-10 and IL-12 are the main regulatory cytokines in visceral leishmaniasis. Cytokine. 2000;12:1228–31. doi: 10.1006/cyto.2000.0694. [DOI] [PubMed] [Google Scholar]
- 27.Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J Immunol. 2001;166:1141–7. doi: 10.4049/jimmunol.166.2.1141. [DOI] [PubMed] [Google Scholar]
- 28.Chakraborty R, Mukherjee S, Basu MK. Oxygen-dependent leishmanicidal activity of stimulated macrophages. Mol Cell Biochem. 1996;154:23–9. doi: 10.1007/BF00248457. [DOI] [PubMed] [Google Scholar]
- 29.Lu HT, Yang DD, Wysk M, et al. IL-12 production in mitogen-activated protein (MAP) kinase kinase 3(Mkk3)-deficient mice. EMBO J. 1999;18:1845–57. doi: 10.1093/emboj/18.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Junghae M, Raynes JG. Activation of p38 mitogen-activated protein kinase attenuates Leishmania donovani infection in macrophages. Infect Immun. 2002:5026–35. doi: 10.1128/IAI.70.9.5026-5035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Awasthi A, Mathur R, Khan A, et al. CD40 signaling is impaired in L. major-infected macrophages and is rescued by a p38MAPK activator establishing a host-protective memory T cell response. J Exp Med. 2003;197:1037–43. doi: 10.1084/jem.20022033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathur RK, Awasthi A, Wadhone P, Ramanamurthy B, Saha B. Reciprocal CD40 signals through p38MAPK and ERK-1/2 induce counteracting immune responses. Nat Med. 2004;10:540–4. doi: 10.1038/nm1045. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, Mosser DM, Zhang X. Activation of the MAPK, ERK, following Leishmania amazonensis Infection of Macrophages. J Immunol. 2007;178:1077–85. doi: 10.4049/jimmunol.178.2.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Edwards JP, Mosser DM. Dynamic and transient remodeling of the macrophage IL-10 promoter during transcription. J Immunol. 2006;177:1282–8. doi: 10.4049/jimmunol.177.2.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sutterwala FS, Noel GJ, Clynes R, Mosser DM. Selective suppression of interleukin-12 induction after macrophage receptor ligation. J Exp Med. 1997;185:1977–85. doi: 10.1084/jem.185.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marth T, Kelsall BL. Regulation of interleukin-12 by complement receptor 3 signaling. J Exp Med. 1997;185:1987–95. doi: 10.1084/jem.185.11.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kedzierski L, Montgomery J, Bullen D, et al. A leucine-rich repeat motif of Leishmania parasite surface antigen 2 binds to macrophages through the complement receptor 3. J Immunol. 2004;172:4902–6. doi: 10.4049/jimmunol.172.8.4902. [DOI] [PubMed] [Google Scholar]
- 38.Chieppa M, Bianchi G, Doni A, et al. Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J Immunol. 2003;171:4552–60. doi: 10.4049/jimmunol.171.9.4552. [DOI] [PubMed] [Google Scholar]
- 39.Akilov OE, Kasuboski RE, Carter CR, McDowell MA. The role of mannose receptor during experimental leishmaniasis. J Leukoc Biol. 2007;81:1188–96. doi: 10.1189/jlb.0706439. [DOI] [PubMed] [Google Scholar]
- 40.Ruse M, Knaus UG. New players in TLR-mediated innate immunity: PI3K and small Rho GTPases. Immunol Res. 2006;34:33–48. doi: 10.1385/IR:34:1:33. Review. [DOI] [PubMed] [Google Scholar]
- 41.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–63. doi: 10.1016/s1471-4906(03)00139-x. Review. [DOI] [PubMed] [Google Scholar]
- 42.Ruhland A, Leal N, Kima PE. Leishmania promastigotes activate PI3K/Akt signaling to confer host cell resistance to apoptosis. Cell Microbiol. 2007;9:84–96. doi: 10.1111/j.1462-5822.2006.00769.x. [DOI] [PubMed] [Google Scholar]
- 43.Bennett CL, Misslitz A, Colledge L, Aebischer T, Blackburn CC. Silent infection of bone marrow-derived dendritic cells by Leishmania mexicana amastigotes. Eur J Immunol. 2001;31:876–83. doi: 10.1002/1521-4141(200103)31:3<876::aid-immu876>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 44.Belkaid Y, Butcher B, Sacks DL. Analysis of cytokine production by inflammatory mouse macrophages at the single-cell level: selective impairment of IL-12 induction in Leishmania-infected cells. Eur J Immunol. 1998;28:1389–400. doi: 10.1002/(SICI)1521-4141(199804)28:04<1389::AID-IMMU1389>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 45.Cameron P, McGachy A, Anderson M, et al. Inhibition of lipopolysaccharide- induced macrophage IL-12 production by Leishmania mexicana amastigotes: the role of cysteine peptidases and the NF-kB signaling pathway. J Immunol. 2004;173:3297–304. doi: 10.4049/jimmunol.173.5.3297. [DOI] [PubMed] [Google Scholar]
- 46.Martiny A, Meyer-Fernandes JR, de Souza W, Vannier-Santos MA. Altered tyrosine phosphorylation of ERK1 MAP kinase and other macrophage molecules caused by Leishmania amastigotes. Mol Biochem Parasitol. 1999;1021:1–12. doi: 10.1016/s0166-6851(99)00067-5. [DOI] [PubMed] [Google Scholar]