

Summary
With the increasing prevalence of obesity, research has focused on the molecular mechanism(s) linking obesity and skeletal muscle insulin resistance. Metabolic alterations within muscle, such as changes in the cellular location of fatty acid transporter proteins, decreased mitochondrial enzyme activity and defects in mitochondrial morphology, likely contribute to obesity and insulin resistance. These defects are thought to play a role in the reduced skeletal muscle fatty acid oxidation (FAO) and increased intramuscular lipid (IMCL) accumulation that is apparent with obesity and other insulin resistant states, such as type 2 diabetes. Intramuscular triacylglycerol (IMTG) does not appear to be a ubiquitous marker of insulin resistance, although specific IMCL intermediates such as long-chain fatty acyl-CoAs (LCFA-CoAs), ceramide and diacylglycerol (DAG) may inhibit insulin signal transduction. In this review, we will briefly summarize the defects in skeletal muscle lipid metabolism associated with obesity, and discuss proposed mechanisms by which these defects may contribute to insulin resistance.
INTRODUCTION
The prevalence of obesity throughout the world has reached epidemic proportions. The World Health Organization classified at least 400 million people as obese (body mass index or BMI ≥ 30 kg/m2) in 2005 and projected this number to reach 700 million by 2015. A number of comorbidities have been attributed to obesity including cardiovascular disease and type 2 diabetes (1). Based on the link between obesity and type 2 diabetes, it is not surprising that the rate of diabetes has also escalated. It is estimated that by the year 2010 over 220 million people world-wide will be affected by type 2 diabetes, an increase of 46% in the past decade (2). With the increased prevalence of obesity and insulin resistant disorders, research has attempted to elucidate the potential mechanisms behind these diseases, with the hope of ultimately providing effective interventions.
Research examining skeletal muscle is attractive based on this tissue’s integral role in regulating whole-body homeostasis. Approximately 70–80% of ingested glucose is taken up by skeletal muscle and is either stored as glycogen or oxidized for energy (3). Skeletal muscle also plays an important role in lipid metabolism. In the rested state, skeletal muscle fatty acid oxidation (FAO) comprises approximately 90% of the energy requirements for this tissue (4, 5).
Insulin-resistant conditions such as obesity and type 2 diabetes typically have circulating free fatty acid (FFA) concentrations double that of their lean, healthy counterparts (6). Early speculation was that the increased availability of FFA resulted in substrate competition within the muscle (ie., Randle Cycle) and as a consequence glucose metabolism was inhibited (7). However, lipid infusion studies report a 2 to 4 hour lag between the increase in plasma FFA and the onset of insulin resistance (8, 9) suggesting that elevated FFA is not directly responsible for insulin resistance. Instead, a current theory is that obesity induced insulin resistance is a multifactorial process. Research suggests that with obesity, skeletal muscle is faced with increasing amounts of lipid which it is unable to oxidize. As a consequence, lipids accumulate within the muscle cell; this stored intramuscular lipid (IMCL) is hypothesized to play a role in the development of insulin resistance. The intent of this paper is to provide a brief overview of skeletal muscle lipid metabolism, followed by a brief description of the effects of obesity on metabolic processes within the muscle, and finally a discussion on how these metabolic decrements may contribute to insulin resistance.
SKELETAL MUSCLE LIPID METABOLISM
Fatty Acid Transport
Plasma FFA circulate bound to albumin (10) and enter the myocyte through either diffusion or protein-mediated transport (11, 12). Research has focused on the latter based on the mounting evidence supporting the role of protein transporters in regulating fatty acid entry into metabolically active tissue such as skeletal muscle (11, 13–15). The membrane-associated proteins in skeletal muscle have been identified as the 88-kDa heavily glycosylated fatty acid translocase (FAT/CD36) (11), the 40-kDa fatty acid binding protein (FABPpm) (16), and a family of ~70-kDa fatty acid transport protein (FATP1–6) (17–19).
FAT/CD36 is a class B scavenger receptor protein that is critical to long chain fatty acid (LCFA) transport and intracellular metabolism (20). For instance, FAT/CD36 overexpression has been shown to increase the rate of FAO during contraction (11), whereas FAT/CD36 knockout mice have a significant reduction in LCFA uptake suggesting a functional role in lipid metabolism (21). Skeletal muscle fatty acid transport is regulated by FAT/CD36 acutely via translocation from an intracellular pool to the plasma membrane and chronically via changes in gene expression. The signaling pathway responsible for insulin-stimulated transport involves the activation of phosphatidylinositol (PI) 3 kinase (22), whereas emerging evidence seems to indicate that the signaling pathway responsible for contraction-induced FAT/CD36 translocation is AMP kinase (23). FABPpm is located on the outer leaflet of the plasma membrane and may be co-localized with FAT/CD36 (6). Electrotransfection of FABPpm resulted in elevated plasmalemmal FABPpm protein overexpression and an increased rate of palmitate transport into skeletal muscle signifying the role of this transporter in coordinating LCFA entry into the cell (24). FATP1 has been shown to enhance insulin-sensitive fatty acid transport in skeletal muscle (25). In addition, overexpression of FATP1 in rat muscle results in elevated rates of LCFA transport (26). Collectively, these data support the critical function of protein-mediated transport in the regulation of FA uptake into the cell.
Mitochondrial Oxidation
Once FFAs enter the myocyte, they are activated by the enzyme acyl-CoA synthethase to form an fatty acyl-CoA (FA-CoA) complex, which then enters the mitochondria for oxidation or is partitioned towards the synthesis of intramyocellular lipid (IMCL) (27). Skeletal muscle FAO involves the coordinated action of three main metabolic pathways [β-oxidation, Krebs Cycle and Electron Transport Chain (ETC)] located within the mitochondria. For a more detailed review, the reader is referred to reviews by Jeukendrup (27) and Kiens (28).
While short and medium chain FA-CoA are believed to diffuse into the mitochondria, LCFA-CoA require modification before crossing the inner mitochondrial membrane for oxidation. The LCFA-CoA is transported across the inner mitochondrial membrane as an acyl-carnitine complex which is synthesized via carnitine palmitoyltransferase 1 (CPT-1). Overexpression of CPT 1 in skeletal muscle has been reported to repartition fatty acids towards FAO at the expense of storage (29), highlighting the importance of this enzyme in lipid metabolism regulation. Once across the inner mitochondrial membrane, the acyl-carnitine is converted back to the original FA-CoA and released into the matrix. Within the mitochondria, the fatty acid is degraded into two carbon fragments (Acetyl-CoA) during β-oxidation. β-Hydroxy acyl CoA dehydrogenase (β-HAD) is a key enzyme during β-oxidation and has been reported to be highly correlated with the rate of FAO in skeletal muscle (30). Acetyl-CoA produced during β-oxidation undergoes additional processes within the mitochondria matrix and inner mitochondrial membrane (Krebs Cycle and ETC, respectively), which ultimately leads to ATP production. The oxidation of fatty acids is thus a complex and highly regulated process which warrants research to determine if defects at any, if not several of these metabolic steps, are evident in the obese state.
ALTERATIONS IN SKELETAL MUSCLE METABOLISM WITH OBESITY
Obesity has been associated with a number of alterations in the transport and metabolic pathways. It remains unclear what specific mechanism(s) are responsible for obesity induced insulin resistance, however, a number of contributing factors have been suggested. The following discussion highlights some possible mechanisms.
Increased Plasma Membrane Transporter Proteins
The cellular location of the transporter proteins, FABP and FAT/CD36, may play a critical role in the increased fatty acid uptake, accumulation of IMCL, and impaired insulin action observed with obesity and type 2 diabetes. Plasmalemmal FABPpm protein expression has been reported to be elevated in skeletal muscle during conditions of increased plasma FFA availability, such as fasting (31) and a high fat diet (32). In addition, this protein is elevated in the skeletal muscle of individuals with type 2 diabetes (33), and obesity (34). Bonen and colleagues (35) showed that LCFA transport into giant sarcolemmal vesicles, prepared from obese and type 2 diabetic individuals, was approximately 4-fold higher than their lean counterparts. This increased fatty acid uptake was associated with increased intramuscular triglyceride (IMTG) accumulation and an increase in sarcolemmal but not total FAT/CD36 (35). Therefore, it appears that in conditions of high FFA availability, such as obesity, muscle cells respond by maintaining a higher concentration of transporter proteins on the cell membrane and as a consequence, fatty acid uptake is increased and ultimately stored as IMCL, a potential contributor to insulin resistance.
Potential Defects Within the Metabolic Pathway(s)
While fatty acid uptake appears to be upregulated with obesity, the ability to oxidize the increased flux of lipid is not matched. Obesity has been associated with decrements at a number of key regulatory steps in the abovementioned metabolic pathways, including reduced enzyme activity. For example, CPT-1 (fatty acid transfer into mitochondria), β-HAD (β-oxidation), citrate synthase (Krebs Cycle), and cyctochrome oxidase (ETC), have all been reported to have diminished activity in the skeletal muscle from obese individuals (34, 36). Early research by Simoneau and Kelley (37) reported that the skeletal muscle of type 2 diabetics, and to a lesser extent, obese individuals, had an increased ratio of glycolytic to oxidative enzyme capacity. This ratio (hexokinase to citrate synthase activity) was negatively correlated with insulin sensitivity, providing further evidence that decreased oxidative enzyme activity may contribute to insulin resistance in type 2 diabetic and obese populations. A decreased level of CPT-1 in the skeletal muscle of obese individuals has also received considerable attention. In vitro research has demonstrated that overexpressing CPT-1 in L6 cells increased β-oxidation and protected the cells against fatty acid-induced insulin resistance (38). ETC activity has also been found to be depressed in the skeletal muscle of type 2 diabetics and obese individuals compared to healthy, lean individuals, even when adjusting for differences in mitochondrial content (39).
Emerging evidence suggests that obesity is linked with an accumulation of intramuscular lipid, which may induce a lipid burden on mitochondria and create a possible disconnect between the metabolic pathways. For instance, metabolic profiling studies by Koves et al. (40) demonstrated that obesity-induced increases in the rates of β-oxidation overwhelmed the capacity of the Kreb’s Cycle, allowing only partial degradation of fatty acids. As a consequence, mitochondrial derived by-products, such as acylcarnitine derivatives, accumulated and potentially contributed to lipid-induced impairments in insulin action. Although the precise connection between an increased rate of β-oxidation and insulin resistance is not clear, it has been demonstrated that the increased accumulation of β-oxidative by-products is linked with impaired insulin action in obese individuals (40). Taken together, these findings suggest that the diminished activity of mitochondrial enzymes or inefficient coupling between the metabolic pathways in the skeletal muscle of obese individuals may contribute to insulin resistance.
Reduced Mitochondrial Content and Altered Mitochondrial Morphology
Since the primary metabolic pathways involved in lipid metabolism are located within the mitochondria, many speculate that a defect in this organelle could contribute to reduced lipid oxidation and insulin resistance. In addition to the reduced enzyme activity within key metabolic pathways discussed previously, the diminished enzyme activity could reflect a functional defect or reduced mitochondrial content. The reduced activity of citrate synthase and CPT-1 in obese muscle has been used to imply reduced mitochondrial content (36). More recently, mitochondrial DNA (mtDNA) has been used as a marker of mitochondrial content, and has been found to be lower in the skeletal muscle of obese individuals compared to their lean counterparts (39).
In addition to the decreased mitochondrial content observed with obesity, there are also reports of alterations in mitochondria morphology. Research using transmission electron microscopy has reported that the mitochondria of obese and type 2 diabetics were 35% smaller than the mitochondria of healthy lean individuals, and that the size of the mitochondria were significantly correlated with insulin action (41). Additionally, this study noted that a number of mitochondria in obese and type 2 diabetics were ‘fractured’, suggesting an increased rate of mitochondrial apoptosis.
In summary, it is unlikely that there is a sole contributor responsible for the impairment of lipid metabolism observed with obesity. The following sections will discuss the consequences of these metabolic defects, such as reduced skeletal muscle FAO and IMCL accumulation. In addition, evidence will be provided suggesting a link between these events and the onset of insulin resistance.
FACTORS CONTRIBUTING TO OBESITY AND INSULIN RESISTANCE
Reduced Skeletal Muscle FAO
Researchers have determined that the preferential oxidation of glucose over lipid in the rested state can predispose an individual to weight gain. Whole-body respiratory quotient (RQ) is a frequently used measure to estimate substrate oxidation. Marra et al. (42) and others (43) have reported that a high whole-body RQ (indicating a diminished capacity to oxidize fat), is a significant predictor of weight gain. As skeletal muscle plays a significant role in lipid oxidation, it is not surprising that the decrement in whole body lipid oxidation exhibited with obesity, is also observed in skeletal muscle. It has been suggested that skeletal muscle oxidative capacity may be a key predictor of whole-body insulin action (33). In agreement, Kelley et al. (44) reported that obese individuals had a greater leg RQ and lower fat oxidation levels in the rested state compared to their lean counterparts. Fasting leg RQ values were negatively correlated with insulin sensitivity, providing further evidence that a connection exists between obesity, reduced skeletal muscle FAO, and insulin resistance. These findings, in addition to the health consequences associated with obesity, justify further research to examine the association between reduced skeletal muscle FAO and obesity.
Our laboratory has reported reduced skeletal muscle FAO in extremely obese individuals under several experimental conditions (ie., muscle strips, muscle homogenate, and primary cell culture). An initial study by Hulver et al. (45) compared FAO between lean (BMI ~ 24 kg/m2), moderately obese (BMI ~ 30 kg/m2), and extremely obese individuals (BMI ~ 38 kg/m2) and reported that FAO in extremely obese individuals was 58% and 83% lower than muscle strips from lean and moderately obese subjects, respectively (45). In support, we have also reported a similar depression in palmitate oxidation (~50%) using vastus lateralis muscle homogenate from extremely obese individuals comared to lean individuals (36). Validation in different muscle groups is critical due to the heterogenous properties of muscle tissue, including differences in contractile activity, as well as, fiber type composition.
Skeletal muscle fiber type may play a role in the diminished lipid oxidation and insulin resistance observed with obesity. Type I or red oxidative fibers are phenotypically more oxidative and insulin sensitive than Type II white or glycolytic fibers (46, 47). It has been reported that morbidly obese subjects have a lower percentage of Type I fibers compared to their lean counterparts (i.e. approximately 42% vs 55%) (47, 48), which could contribute to the lower skeletal muscle oxidation and insulin resistance observed with obesity. The percentage of Type I fibers may also predict the amount of weight loss an individual can achieve in response to interventions. Tanner and colleagues (48) reported that in a morbidly obese group that underwent gastric bypass surgery, those with the greatest percentage of Type I fibers lost the greatest amount of weight (48). However, it was interesting to note that dramatic weight loss alone did not increase the number of Type I fibers in this group (49) or change skeletal muscle FAO (50). These findings collectively provide evidence that Type I fibers may play a role in regulating skeletal muscle FAO, but also indicate that weight loss is not strictly dependent on changes in skeletal muscle FAO.
Our group has also utilized primary human skeletal cells to further investigate the impact of obesity on skeletal muscle metabolism. This method involves isolating satellite cells from muscle biopsies and subsequently treating the cells so that they proliferate into myoblasts and ultimately differentiate into myotubes (51). We reported that the capacity for lipid oxidation in myotubes was depressed in cells derived from extremely obese donors (52). The magnitude of the decrement in FAO was similar to our in-vivo (53, 54) and in-vitro (45) findings comparing lean and extremely obese individuals, indicating that this model is relevant to the in-vivo condition. Others (42) have reported that myotubes derived from patients with type 2 diabetes have a reduced capacity to oxidize fat compared to the myotubes cultured from healthy controls. These data indicate that the reduced FAO phenotype observed with obesity and type 2 diabetes is retained in cell culture, which suggests a possible genetic or epigenetic origin.
While reduced lipid oxidation may play a role in obesity and insulin resistance, studies on weight loss indicate that improved insulin sensitivity is not always dependent on changes in FAO. Berggren et al. (50) reported that dramatic weight loss (~55 kg) by means of gastric bypass surgery significantly improved insulin sensitivity without changes in skeletal muscle FAO. This finding suggests that factors other than FAO may play a role in obesity and insulin resistance. The potential role of IMCL in the regulation of obesity induced insulin resistance is discussed in the following section.
Accumulation of IMCL
The increased entry of FFA into the myocyte without a corresponding increase in lipid oxidation likely contributes to the accumulation of IMCL. A negative relationship between IMCL and insulin sensitivity has been reported in non-obese adults (55), high fat feeding models (56) and the lean offspring of type 2 diabetics (57). Recently, studies investigating the impact of low calorie diets in type 2 diabetics have reported decreased IMCL along with improved insulin sensitivity (58, 59). Collectively, these studies point to IMCL accumulation, as a prominent marker in the development of insulin resistance.
Despite studies reporting a link between IMCL and insulin resistance (55, 57, 60), some findings suggest that this is not a simple cause and effect relationship. For example, endurance athletes are extremely insulin sensitive, yet have elevated IMCL levels (61). Also, Type I fibers are more insulin sensitive then Type II fibers, yet contain higher IMCL stores. Research has attempted to shed light on this apparently conflicting relationship by examining the relationships between intramyocellular triglyceride (IMTG) and other lipid intermediate metabolites. The triglyceride (TG) synthesis enzyme diacylglycerol (DAG) acyltransferase 1 (DGAT1) has been shown to increase in muscle cells in response to exercise (62). DGAT1 catalyzes the last step in the glycerol phosphate pathway of TG synthesis and produces TG from DAG and FA-CoA (63, 64). Thus, this enzyme has dual significance in that it promotes TG storage but also decreases fatty acid substrates (65). Exercise-induced increases in DGAT1 in humans (62), as well as, the overexpression of DGAT1 in the skeletal muscle of mice (66), has been reported to increase both IMTG and insulin sensitivity (62, 66). Further, overexpression of steroyl CoA desaturase 1 (SCD1), an enzyme that converts saturated fatty acids to monounsaturated fatty acids, has been reported to protect L6 myotubes from fatty acid-induced insulin resistance despite increased TG esterfication (67). These authors (67) suggest that the accumulation of TG provides a protective effect within the cell by limiting the accumulation of other lipid metabolites such as DAG and ceramide, which are known to have an inhibitory effect on insulin signaling. Collectively, these indings support earlier work conducted in vitro demonstrating that the promotion of fatty acids into IMTG reduced lipotoxicity in the cell (68) and substantiate the notion that IMTG should not be regarded as a ubiquitous marker of insulin resistance. Instead, metabolites such as long-chain fatty acyl-CoAs (LCFA-CoAs), DAG and ceramide may play a more active role in insulin resistance. The following sections will explain how each of these lipid metabolites may be responsible for inducing insulin resistance.
Insulin Signaling and the Effects of Lipid Accumulation
The insulin signaling pathway is responsible for the translocation of the GLUT4 protein to the plasma membrane and ultimately insulin-mediated glucose transport into the cell. Insulin binds to and phosphorylates the insulin receptor on tyrosine residues which results in a cascade of events including the phosphorylation of insulin receptor substrate-1/2 and subsequently the activation of phosphatidylinositol 3-kinase (PI3-K) (69). Insulin activated PI3-K regulates the activation of atypical protein kinase C (aPKC), as well as Akt which is responsible for the activation of Akt substrate160 kD (AS160) (70, 71). The current model for insulin signaling suggests that AS160 and aPKC coordinate the translocation of GLUT4 to the cell membrane, resulting in the facilitated diffusion of glucose into the myocyte (70, 72).
Whereas insulin mediated glucose uptake is initiated with the tyrosine phosphorylation of the insulin receptor, research has demonstrated that serine and threonine phosphorylation of the insulin receptor diminishes insulin action (73). As many as 12 isoforms of PKC have been identified (74), and while the activation of certain isoforms may augment insulin signaling (ie., aPKC), others are speculated to evoke insulin resistance (ie., PKCθ) (75). Therefore, the accumulation of lipid intermediates (ie., LCFA-CoAs, DAG and ceramide) may be responsible for impairing the insulin signaling pathway via PKC and other inhibitory mechanisms (76).
LCFA-CoAs
LCFA-CoAs are a metabolically active form of intracellular fatty acids and have been cited as a better predictor of insulin resistance than IMTG (77). Impaired insulin-mediated glucose disposal and elevated LCFA-CoAs has been reported in both moderately and morbidly-obese individuals (45, 78). Studies utilizing lipid infusion (79) and high fat diets (77) have demonstrated increased skeletal muscle LCFA-CoAs in conjunction with insulin resistance. Additionally, pharmacological intervention of type 2 diabetics with Acipmox (a potent inhibitor of lipolysis) has been reported to significantly improve whole body glucose disposal with a parallel decrease in total muscle LCFA-CoA (80). Collectively, these data support a link between LCFA-CoA and insulin resistance.
It remains unclear through which mechanism(s) LCFA-CoAs may affect insulin action, but some data suggests that LCFA-CoAs may interfere with insulin signaling through the activation of PKC isozymes (81). Although a direct link between skeletal muscle LCFA-CoAs and PKC has yet to be identified, it is worth noting that during instances of high lipid exposure (ie., lipid infusion) both intramyocellular fatty acyl-CoA and PKC are elevated (81). It is also possible that LCFA-CoAs act indirectly by acting as a precursor to other lipid intermediates such as DAG and ceramide.
Ceramides
Ceramides can accumulate in skeletal muscle either by the hydrolysis of sphingomyelin (a phosholipid located in the lipid bylayer of the cell) (82), or by de novo synthesis from long-chain saturated fatty acids (83). Since ceramide synthesis is primarily dependent on the availability of fatty acids, it is not surprising that muscle ceramide content has been found to be significantly correlated with basal plasma FFA concentrations (84). Elevated levels of ceramide have been observed in the skeletal muscle of insulin-resistant animals (85), lipid infused humans (86), obese, insulin resistant humans (84) and the lean offspring of Type 2 diabetics (87). Others have reported no difference in skeletal muscle ceramide levels between obese and lean individuals with similar levels of insulin sensitivity (88).
Recently, Holland et al. (89) reported that different fatty acids antagonize insulin stimulated glucose uptake through distinct intracellular mechanisms. Whereas, ceramide levels are typically unchanged when unsaturated fatty acids (ie., linoleate) are used to induce insulin resistance, ceramide appears to be a primary regulator of saturated fatty acid (ie., palmitate) induced insulin resistance. Since saturated fatty acids are required for ceramide synthesis, it is not surprising that models utilizing unsaturated fatty acids have found no change in ceramide accumulation (89). In addition, these authors (89) demonstrated that pharmacological inhibition of ceramide synthesis was capable of improving glucose tolerance and preventing diabetes in obese rodents (89), further supporting the inhibitory role this lipid metabolite may have on insulin action.
Although not completely understood, it has been speculated that ceramides may inhibit insulin signaling via inhibition at the level of Akt (84, 90). In addition to observing elevated ceramide levels in obese individuals, Adams and colleagues (84) reported that insulin-induced Akt phosphorylation was reduced. In vitro studies also support this inhibitory link as incubation of L6 cells with a ceramide analogue resulted in a loss of Akt activity and the complete absence of insulin-mediated glucose uptake (90). In addition, blocking ceramide synthesis has restored insulin-mediated Akt activation in previously insulin-resistant myotubes (91).
Recent findings suggest that the effect of intramuscular ceramides on obesity induced insulin resistance may not be as significant as once thought. Skovbro and co-workers (92) investigated skeletal muscle ceramide levels in individuals with a range of insulin sensitivities. These authors reported no differences in ceramide levels between type 2 diabetics, glucose intolerant individuals, healthy control subjects, and endurance trained athletes (92). Although reduced Akt activity is characteristic of obese individuals (84) it is also known that these individuals have marked abnormalities in more proximal insulin signaling markers such as the IR and IRS-1 tyrosine phosphorylation, as well as, IRS-1-associated PI 3-kinase activity (93). This suggests that ceramide alone is not responsible for the reduced insulin sensitivity in the muscle and highlights the potential role of other lipid intermediates such as LCFA-CoA or DAG.
DAG
DAG acts as a key intermediate in both triacylglycerol and phospholipid metabolism and acts as an important second messenger in the regulation of intracellular signaling. Formation of DAG can be generated by the breakdown of phospholipids via phospholipases (ie., Phospholipase C), or through de novo synthesis via the esterfication of two LCFA-CoAs to glycerol-3-phosphate (94). The latter process is likely the more important source responsible for lipid induced insulin resistance (94). Increased skeletal muscle DAG content muscle has been reported with fasting (95) and lipid infusion (9, 81).
The majority of research supports the notion that DAG promotes insulin resistance through the activation of PKC isoforms. In rats, Yu and co-workers (81) demonstrated that lipid infusion resulted in a three-fold increase in intracellular DAG mass, which was associated with PKCθ activation. It is believed that PKCθ activation results in the serine phosphorylation of upstream molecules in the insulin signaling cascade, which subsequently inhibits this pathway. In support of this relationship, Yu et al. (81) observed a 30% reduction in IRS-1 tyrosine phosphorylation and an approximate 50% reduction in IRS-1 associated PI3 kinase activity, in response to the increased DAG mass and PKCθ activation. In humans, Itani et al. (96) reported that PKCθ content and activity were elevated in the skeletal muscle of obese, diabetic patients compared to obese, non-diabetics. These authors later demonstrated fatty acid induced insulin resistance in normal volunteers was related with increased DAG mass and membrane translocation of PKC isoforms β2 and δ (9).
It is speculated that increased shuttling of fatty acid into the mitochondria for oxidation would decrease the accumulation of lipid and thus, protect the cell from insulin resistance. In support of this notion, Sebastian et al. (97) demonstrated that the overexpression of CPT-1 in L6 cells protected the cells against fatty acid-induced insulin resistance by inhibiting the build up of lipid by-products such as DAG and ceramide, and also decreasing the activation of PKCθ. Additionally, exercise intervention studies which typically produce increased skeletal muscle FAO, have been reported to increase insulin sensitivity and decrease levels of DAG and ceramide (98, 99) despite either no change (98) or increases in IMTG (99). These findings support the importance of skeletal muscle FAO in alleviating the muscle from the accumulation of harmful lipid intermediates.
SUMMARY
There is accumulating evidence that obesity and type 2 diabetes are associated with a defect in the ability of skeletal muscle to oxidize lipid. As a consequence, the intracellular environment within the muscle cell may be conducive to the partitioning of lipids towards storage. While it was originally speculated that IMTG was linked with inducing insulin resistance, current research suggest other mechanisms such as the accumulation of intramuscular lipid intermediates (LCFA-CoA, ceramide and DAG) promote the onset of insulin resistance by inhibiting key proteins within the insulin signaling pathway (Figure 1). However, additional research is needed to fully elucidate the relationship between obesity and the accumulation of these lipid intermediates, and the impact these lipid species may have on insulin action.
Figure 1.
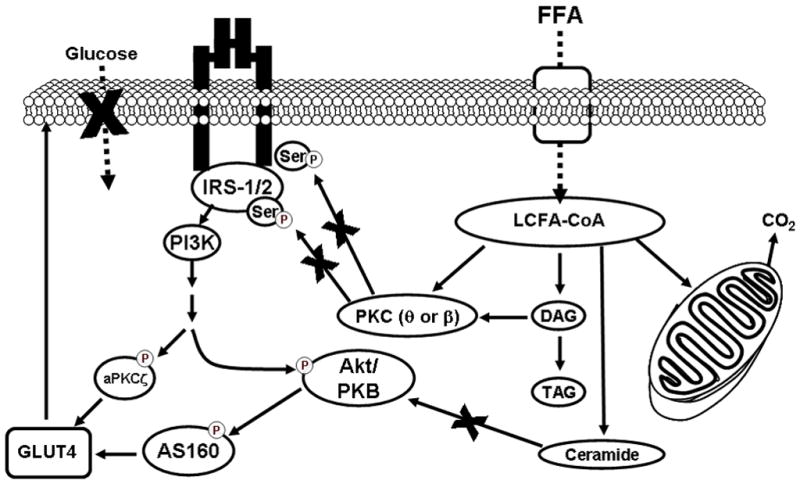
Proposed model of obesity-induced skeletal muscle insulin resistance. Increased plasma free fatty acids (FFA) can result in elevations in the rate of skeletal muscle long chain fatty acid (LCFA) uptake, which may lead to the accumulation of intramuscular long-chain fatty acyl-CoAs (LCFA-CoAs), diacylglycerol (DAG), triacylglycerol (TAG), and ceramide. Both LCFA-CoAs and DAG can impair the insulin signaling cascade via the action of protein kinase C (PKC). PKC increases serine phosphorylation resulting in a decrease in the association between insulin receptor substrate 1 (IRS1) and phosphatidylinositol 3-kinase (PI3-K) and subsequent reduction of insulin mediated glucose uptake. Intracellular accumulation of ceramide can also attenuate insulin mediated glucose uptake, via decreased phosphorylation of Akt.
Acknowledgments
Supported by National Institutes of Health grants AG 025205 and DK 56112 (to Joseph A. Houmard).
Contributor Information
Leslie A. Consitt, Department of Exercise and Sport Science and Human Performance Laboratory, 363 Ward Sports Medicine Building, East Carolina University, Greenville, NC 27858, Phone: 252-744-3465, Fax: 252-737-4689, Email: consittl@ecu.edu
Jill A. Bell, Department of Physiology, 600 Moye Boulevard, East Carolina University, Greenville, NC 27834, Phone: 252-744-3465, Email: bellj@ecu.edu
Joseph A. Houmard, Department of Exercise and Sport Science and Human Performance Laboratory, 363 Ward Sports Medicine Building, East Carolina University, Greenville, NC 27858, Phone: 252-737-4617, Fax: 252-737-4689, Email: houmardj@ecu.edu
References
- 1.Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999–2000. Jama. 2002;288:1723–1727. doi: 10.1001/jama.288.14.1723. [DOI] [PubMed] [Google Scholar]
- 2.Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med. 1997;14(Suppl 5):S1–85. [PubMed] [Google Scholar]
- 3.DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- 4.Dagenais GR, Tancredi RG, Zierler KL. Free fatty acid oxidation by forearm muscle at rest, and evidence for an intramuscular lipid pool in the human forearm. J Clin Invest. 1976;58:421–431. doi: 10.1172/JCI108486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelley DE, Mokan M, Simoneau JA, Mandarino LJ. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J Clin Invest. 1993;92:91–98. doi: 10.1172/JCI116603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belfort R, Mandarino L, Kashyap S, Wirfel K, Pratipanawatr T, Berria R, Defronzo RA, Cusi K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes. 2005;54:1640–1648. doi: 10.2337/diabetes.54.6.1640. [DOI] [PubMed] [Google Scholar]
- 7.Oakes ND, Cooney GJ, Camilleri S, Chisholm DJ, Kraegen EW. Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes. 1997;46:1768–1774. doi: 10.2337/diab.46.11.1768. [DOI] [PubMed] [Google Scholar]
- 8.Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest. 1994;93:2438–2446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 10.Campbell J, Martucci AD, Green GR. Plasma albumin as an acceptor of free fatty acids. Biochem J. 1964;93:183–189. doi: 10.1042/bj0930183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abumrad N, Harmon C, Ibrahimi A. Membrane transport of long-chain fatty acids: evidence for a facilitated process. J Lipid Res. 1998;39:2309–2318. [PubMed] [Google Scholar]
- 12.Hamilton JA. Fatty acid transport: difficult or easy? J Lipid Res. 1998;39:467–481. [PubMed] [Google Scholar]
- 13.Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. 2000;275:14501–14508. doi: 10.1074/jbc.275.19.14501. [DOI] [PubMed] [Google Scholar]
- 14.Chabowski A, Coort SL, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Bonen A. Insulin stimulates fatty acid transport by regulating expression of FAT/CD36 but not FABPpm. Am J Physiol Endocrinol Metab. 2004;287:E781–789. doi: 10.1152/ajpendo.00573.2003. [DOI] [PubMed] [Google Scholar]
- 15.Coburn CT, Knapp FF, Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- 16.Schwieterman W, Sorrentino D, Potter BJ, Rand J, Kiang CL, Stump D, Berk PD. Uptake of oleate by isolated rat adipocytes is mediated by a 40-kDa plasma membrane fatty acid binding protein closely related to that in liver and gut. Proc Natl Acad Sci U S A. 1988;85:359–363. doi: 10.1073/pnas.85.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gimeno RE, Ortegon AM, Patel S, Punreddy S, Ge P, Sun Y, Lodish HF, Stahl A. Characterization of a heart-specific fatty acid transport protein. J Biol Chem. 2003;278:16039–16044. doi: 10.1074/jbc.M211412200. [DOI] [PubMed] [Google Scholar]
- 18.Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79:427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 19.Hirsch D, Stahl A, Lodish HF. A family of fatty acid transporters conserved from mycobacterium to man. Proc Natl Acad Sci U S A. 1998;95:8625–8629. doi: 10.1073/pnas.95.15.8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 22.Luiken JJ, Koonen DP, Willems J, Zorzano A, Becker C, Fischer Y, Tandon NN, Van Der Vusse GJ, Bonen A, Glatz JF. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes. 2002;51:3113–3119. doi: 10.2337/diabetes.51.10.3113. [DOI] [PubMed] [Google Scholar]
- 23.Luiken JJ, Coort SL, Willems J, Coumans WA, Bonen A, van der Vusse GJ, Glatz JF. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes. 2003;52:1627–1634. doi: 10.2337/diabetes.52.7.1627. [DOI] [PubMed] [Google Scholar]
- 24.Clarke DC, Miskovic D, Han XX, Calles-Escandon J, Glatz JF, Luiken JJ, Heikkila JJ, Bonen A. Overexpression of membrane-associated fatty acid binding protein (FABPpm) in vivo increases fatty acid sarcolemmal transport and metabolism. Physiol Genomics. 2004;17:31–37. doi: 10.1152/physiolgenomics.00190.2003. [DOI] [PubMed] [Google Scholar]
- 25.Wu Q, Ortegon AM, Tsang B, Doege H, Feingold KR, Stahl A. FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol Cell Biol. 2006;26:3455–3467. doi: 10.1128/MCB.26.9.3455-3467.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nickerson J, Bonen A. Defining a role for skeletal muscle fatty acid transport proteins. 2nd Northern Lights Conference. Canadian Federation of Biological Societies; Guelph, Ontario, Canada. 2005. p. F47. [Google Scholar]
- 27.Jeukendrup AE. Regulation of fat metabolism in skeletal muscle. Ann N Y Acad Sci. 2002;967:217–235. doi: 10.1111/j.1749-6632.2002.tb04278.x. [DOI] [PubMed] [Google Scholar]
- 28.Kiens B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev. 2006;86:205–243. doi: 10.1152/physrev.00023.2004. [DOI] [PubMed] [Google Scholar]
- 29.Bruce CR, Brolin C, Turner N, Cleasby ME, van der Leij FR, Cooney GJ, Kraegen EW. Overexpression of carnitine palmitoyltransferase I in skeletal muscle in vivo increases fatty acid oxidation and reduces triacylglycerol esterification. Am J Physiol Endocrinol Metab. 2007;292:E1231–1237. doi: 10.1152/ajpendo.00561.2006. [DOI] [PubMed] [Google Scholar]
- 30.Kiens B. Effect of endurance training on fatty acid metabolism: local adaptations. Med Sci Sports Exerc. 1997;29:640–645. doi: 10.1097/00005768-199705000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Turcotte LP, Srivastava AK, Chiasson JL. Fasting increases plasma membrane fatty acid-binding protein (FABP(PM)) in red skeletal muscle. Mol Cell Biochem. 1997;166:153–158. doi: 10.1023/a:1006846907394. [DOI] [PubMed] [Google Scholar]
- 32.Roepstorff C, Helge JW, Vistisen B, Kiens B. Studies of plasma membrane fatty acid-binding protein and other lipid-binding proteins in human skeletal muscle. Proc Nutr Soc. 2004;63:239–244. doi: 10.1079/PNS2004332. [DOI] [PubMed] [Google Scholar]
- 33.Bruce CR, Anderson MJ, Carey AL, Newman DG, Bonen A, Kriketos AD, Cooney GJ, Hawley JA. Muscle oxidative capacity is a better predictor of insulin sensitivity than lipid status. J Clin Endocrinol Metab. 2003;88:5444–5451. doi: 10.1210/jc.2003-030791. [DOI] [PubMed] [Google Scholar]
- 34.Simoneau JA, Veerkamp JH, Turcotte LP, Kelley DE. Markers of capacity to utilize fatty acids in human skeletal muscle: relation to insulin resistance and obesity and effects of weight loss. Faseb J. 1999;13:2051–2060. doi: 10.1096/fasebj.13.14.2051. [DOI] [PubMed] [Google Scholar]
- 35.Bonen A, Parolin ML, Steinberg GR, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Heigenhauser GJ, Dyck DJ. Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. Faseb J. 2004;18:1144–1146. doi: 10.1096/fj.03-1065fje. [DOI] [PubMed] [Google Scholar]
- 36.Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279:E1039–1044. doi: 10.1152/ajpendo.2000.279.5.E1039. [DOI] [PubMed] [Google Scholar]
- 37.Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol. 1997;83:166–171. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- 38.Perdomo G, Commerford SR, Richard AM, Adams SH, Corkey BE, O’Doherty RM, Brown NF. Increased beta-oxidation in muscle cells enhances insulin-stimulated glucose metabolism and protects against fatty acid-induced insulin resistance despite intramyocellular lipid accumulation. J Biol Chem. 2004;279:27177–27186. doi: 10.1074/jbc.M403566200. [DOI] [PubMed] [Google Scholar]
- 39.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 40.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 41.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 42.Marra M, Scalfi L, Covino A, Esposito-Del Puente A, Contaldo F. Fasting respiratory quotient as a predictor of weight changes in non-obese women. Int J Obes Relat Metab Disord. 1998;22:601–603. doi: 10.1038/sj.ijo.0800612. [DOI] [PubMed] [Google Scholar]
- 43.Zurlo F, Lillioja S, Esposito-Del Puente A, Nyomba BL, Raz I, Saad MF, Swinburn BA, Knowler WC, Bogardus C, Ravussin E. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol. 1990;259:E650–657. doi: 10.1152/ajpendo.1990.259.5.E650. [DOI] [PubMed] [Google Scholar]
- 44.Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol. 1999;277:E1130–1141. doi: 10.1152/ajpendo.1999.277.6.E1130. [DOI] [PubMed] [Google Scholar]
- 45.Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, MacDonald KG, Cline GW, Shulman GI, Dohm GL, Houmard JA. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. 2003;284:E741–747. doi: 10.1152/ajpendo.00514.2002. [DOI] [PubMed] [Google Scholar]
- 46.Essen B, Jansson E, Henriksson J, Taylor AW, Saltin B. Metabolic characteristics of fibre types in human skeletal muscle. Acta Physiol Scand. 1975;95:153–165. doi: 10.1111/j.1748-1716.1975.tb10038.x. [DOI] [PubMed] [Google Scholar]
- 47.Hickey MS, Carey JO, Azevedo JL, Houmard JA, Pories WJ, Israel RG, Dohm GL. Skeletal muscle fiber composition is related to adiposity and in vitro glucose transport rate in humans. Am J Physiol. 1995;268:E453–457. doi: 10.1152/ajpendo.1995.268.3.E453. [DOI] [PubMed] [Google Scholar]
- 48.Tanner CJ, Barakat HA, Dohm GL, Pories WJ, MacDonald KG, Cunningham PR, Swanson MS, Houmard JA. Muscle fiber type is associated with obesity and weight loss. Am J Physiol Endocrinol Metab. 2002;282:E1191–1196. doi: 10.1152/ajpendo.00416.2001. [DOI] [PubMed] [Google Scholar]
- 49.Gray RE, Tanner CJ, Pories WJ, MacDonald KG, Houmard JA. Effect of weight loss on muscle lipid content in morbidly obese subjects. Am J Physiol Endocrinol Metab. 2003;284:E726–732. doi: 10.1152/ajpendo.00371.2002. [DOI] [PubMed] [Google Scholar]
- 50.Berggren JR, Boyle KE, Chapman WH, Houmard JA. Skeletal muscle lipid oxidation and obesity: influence of weight loss and exercise. Am J Physiol Endocrinol Metab. 2008;294:E726–732. doi: 10.1152/ajpendo.00354.2007. [DOI] [PubMed] [Google Scholar]
- 51.Berggren JR, Tanner CJ, Houmard JA. Primary cell cultures in the study of human muscle metabolism. Exerc Sport Sci Rev. 2007;35:56–61. doi: 10.1249/JES.0b013e31803eae63. [DOI] [PubMed] [Google Scholar]
- 52.Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, Hoffman EP, Thyfault JP, Stevens R, Dohm GL, Houmard JA, Muoio DM. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005;2:251–261. doi: 10.1016/j.cmet.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guesbeck NR, Hickey MS, MacDonald KG, Pories WJ, Harper I, Ravussin E, Dohm GL, Houmard JA. Substrate utilization during exercise in formerly morbidly obese women. J Appl Physiol. 2001;90:1007–1012. doi: 10.1152/jappl.2001.90.3.1007. [DOI] [PubMed] [Google Scholar]
- 54.Thyfault JP, Kraus RM, Hickner RC, Howell AW, Wolfe RR, Dohm GL. Impaired plasma fatty acid oxidation in extremely obese women. Am J Physiol Endocrinol Metab. 2004;287:E1076–1081. doi: 10.1152/ajpendo.00177.2004. [DOI] [PubMed] [Google Scholar]
- 55.Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 56.Dobbins RL, Szczepaniak LS, Bentley B, Esser V, Myhill J, McGarry JD. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes. 2001;50:123–130. doi: 10.2337/diabetes.50.1.123. [DOI] [PubMed] [Google Scholar]
- 57.Jacob S, Machann J, Rett K, Brechtel K, Volk A, Renn W, Maerker E, Matthaei S, Schick F, Claussen CD, Haring HU. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes. 1999;48:1113–1119. doi: 10.2337/diabetes.48.5.1113. [DOI] [PubMed] [Google Scholar]
- 58.Jazet IM, Schaart G, Gastaldelli A, Ferrannini E, Hesselink MK, Schrauwen P, Romijn JA, Maassen JA, Pijl H, Ouwens DM, Meinders AE. Loss of 50% of excess weight using a very low energy diet improves insulin-stimulated glucose disposal and skeletal muscle insulin signalling in obese insulin-treated type 2 diabetic patients. Diabetologia. 2008;51:309–319. doi: 10.1007/s00125-007-0862-2. [DOI] [PubMed] [Google Scholar]
- 59.Lara-Castro C, Newcomer BR, Rowell J, Wallace P, Shaughnessy SM, Munoz AJ, Shiflett AM, Rigsby DY, Lawrence JC, Bohning DE, Buchthal S, Garvey WT. Effects of short-term very low-calorie diet on intramyocellular lipid and insulin sensitivity in nondiabetic and type 2 diabetic subjects. Metabolism. 2008;57:1–8. doi: 10.1016/j.metabol.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Greco AV, Mingrone G, Giancaterini A, Manco M, Morroni M, Cinti S, Granzotto M, Vettor R, Camastra S, Ferrannini E. Insulin resistance in morbid obesity: reversal with intramyocellular fat depletion. Diabetes. 2002;51:144–151. doi: 10.2337/diabetes.51.1.144. [DOI] [PubMed] [Google Scholar]
- 61.Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. 2001;86:5755–5761. doi: 10.1210/jcem.86.12.8075. [DOI] [PubMed] [Google Scholar]
- 62.Schenk S, Horowitz JF. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest. 2007;117:1690–1698. doi: 10.1172/JCI30566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bell RM, Coleman RA. Enzymes of glycerolipid synthesis in eukaryotes. Annu Rev Biochem. 1980;49:459–487. doi: 10.1146/annurev.bi.49.070180.002331. [DOI] [PubMed] [Google Scholar]
- 64.Lehner R, Kuksis A. Biosynthesis of triacylglycerols. Prog Lipid Res. 1996;35:169–201. doi: 10.1016/0163-7827(96)00005-7. [DOI] [PubMed] [Google Scholar]
- 65.Bagnato C, Igal RA. Overexpression of diacylglycerol acyltransferase-1 reduces phospholipid synthesis, proliferation, and invasiveness in simian virus 40-transformed human lung fibroblasts. J Biol Chem. 2003;278:52203–52211. doi: 10.1074/jbc.M305760200. [DOI] [PubMed] [Google Scholar]
- 66.Liu L, Zhang Y, Chen N, Shi X, Tsang B, Yu YH. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J Clin Invest. 2007;117:1679–1689. doi: 10.1172/JCI30565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinnamaneni SK, Southgate RJ, Febbraio MA, Watt MJ. Stearoyl CoA desaturase 1 is elevated in obesity but protects against fatty acid-induced skeletal muscle insulin resistance in vitro. Diabetologia. 2006;49:3027–3037. doi: 10.1007/s00125-006-0427-9. [DOI] [PubMed] [Google Scholar]
- 68.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Folli F, Saad MJ, Backer JM, Kahn CR. Insulin stimulation of phosphatidylinositol 3-kinase activity and association with insulin receptor substrate 1 in liver and muscle of the intact rat. J Biol Chem. 1992;267:22171–22177. [PubMed] [Google Scholar]
- 70.Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65–71. doi: 10.2119/2005-00029.Saltiel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frosig C, Rose AJ, Treebak JT, Kiens B, Richter EA, Wojtaszewski JF. Effects of endurance exercise training on insulin signaling in human skeletal muscle: interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes. 2007;56:2093–2102. doi: 10.2337/db06-1698. [DOI] [PubMed] [Google Scholar]
- 72.Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem. 2006;281:31478–31485. doi: 10.1074/jbc.M605461200. [DOI] [PubMed] [Google Scholar]
- 73.Itani SI, Zhou Q, Pories WJ, MacDonald KG, Dohm GL. Involvement of protein kinase C in human skeletal muscle insulin resistance and obesity. Diabetes. 2000;49:1353–1358. doi: 10.2337/diabetes.49.8.1353. [DOI] [PubMed] [Google Scholar]
- 74.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 75.Cortright RN, Azevedo JL, Jr, Zhou Q, Sinha M, Pories WJ, Itani SI, Dohm GL. Protein kinase C modulates insulin action in human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;278:E553–562. doi: 10.1152/ajpendo.2000.278.3.E553. [DOI] [PubMed] [Google Scholar]
- 76.Schmitz-Peiffer C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann N Y Acad Sci. 2002;967:146–157. doi: 10.1111/j.1749-6632.2002.tb04272.x. [DOI] [PubMed] [Google Scholar]
- 77.Ellis BA, Poynten A, Lowy AJ, Furler SM, Chisholm DJ, Kraegen EW, Cooney GJ. Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am J Physiol Endocrinol Metab. 2000;279:E554–560. doi: 10.1152/ajpendo.2000.279.3.E554. [DOI] [PubMed] [Google Scholar]
- 78.Dohm GL, Tapscott EB, Pories WJ, Dabbs DJ, Flickinger EG, Meelheim D, Fushiki T, Atkinson SM, Elton CW, Caro JF. An in vitro human muscle preparation suitable for metabolic studies. Decreased insulin stimulation of glucose transport in muscle from morbidly obese and diabetic subjects. J Clin Invest. 1988;82:486–494. doi: 10.1172/JCI113622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chalkley SM, Hettiarachchi M, Chisholm DJ, Kraegen EW. Five-hour fatty acid elevation increases muscle lipids and impairs glycogen synthesis in the rat. Metabolism. 1998;47:1121–1126. doi: 10.1016/s0026-0495(98)90287-6. [DOI] [PubMed] [Google Scholar]
- 80.Bajaj M, Suraamornkul S, Romanelli A, Cline GW, Mandarino LJ, Shulman GI, DeFronzo RA. Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty Acyl-CoAs and insulin action in type 2 diabetic patients. Diabetes. 2005;54:3148–3153. doi: 10.2337/diabetes.54.11.3148. [DOI] [PubMed] [Google Scholar]
- 81.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 82.Hannun YA, Obeid LM. The Ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem. 2002;277:25847–25850. doi: 10.1074/jbc.R200008200. [DOI] [PubMed] [Google Scholar]
- 83.Merrill AH, Jr, Jones DD. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- 84.Adams JM, 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53:25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 85.Turinsky J, O’Sullivan DM, Bayly BP. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem. 1990;265:16880–16885. [PubMed] [Google Scholar]
- 86.Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I, Baranowski M, Zendzian-Piotrowska M, Brzezinska Z, Gorski J. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes. 2004;53:1215–1221. doi: 10.2337/diabetes.53.5.1215. [DOI] [PubMed] [Google Scholar]
- 87.Straczkowski M, Kowalska I, Baranowski M, Nikolajuk A, Otziomek E, Zabielski P, Adamska A, Blachnio A, Gorski J, Gorska M. Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia. 2007;50:2366–2373. doi: 10.1007/s00125-007-0781-2. [DOI] [PubMed] [Google Scholar]
- 88.Serlie MJ, Meijer AJ, Groener JE, Duran M, Endert E, Fliers E, Aerts JM, Sauerwein HP. Short-term manipulation of plasma free fatty acids does not change skeletal muscle concentrations of ceramide and glucosylceramide in lean and overweight subjects. J Clin Endocrinol Metab. 2007;92:1524–1529. doi: 10.1210/jc.2006-2347. [DOI] [PubMed] [Google Scholar]
- 89.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 90.Hajduch E, Balendran A, Batty IH, Litherland GJ, Blair AS, Downes CP, Hundal HS. Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia. 2001;44:173–183. doi: 10.1007/s001250051596. [DOI] [PubMed] [Google Scholar]
- 91.Chavez JA, Summers SA. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys. 2003;419:101–109. doi: 10.1016/j.abb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 92.Skovbro M, Baranowski M, Skov-Jensen C, Flint A, Dela F, Gorski J, Helge JW. Human skeletal muscle ceramide content is not a major factor in muscle insulin sensitivity. Diabetologia. 2008 doi: 10.1007/s00125-008-1014-z. [DOI] [PubMed] [Google Scholar]
- 93.Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. doi: 10.1172/JCI7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Timmers S, Schrauwen P, de Vogel J. Muscular diacylglycerol metabolism and insulin resistance. Physiol Behav. 2008;94:242–251. doi: 10.1016/j.physbeh.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 95.Turinsky J, Bayly BP, O’Sullivan DM. 1,2-Diacylglycerol and ceramide levels in rat liver and skeletal muscle in vivo. Am J Physiol. 1991;261:E620–627. doi: 10.1152/ajpendo.1991.261.5.E620. [DOI] [PubMed] [Google Scholar]
- 96.Itani SI, Pories WJ, Macdonald KG, Dohm GL. Increased protein kinase C theta in skeletal muscle of diabetic patients. Metabolism. 2001;50:553–557. doi: 10.1053/meta.2001.22512. [DOI] [PubMed] [Google Scholar]
- 97.Sebastian D, Herrero L, Serra D, Asins G, Hegardt FG. CPT I overexpression protects L6E9 muscle cells from fatty acid-induced insulin resistance. Am J Physiol Endocrinol Metab. 2007;292:E677–686. doi: 10.1152/ajpendo.00360.2006. [DOI] [PubMed] [Google Scholar]
- 98.Bruce CR, Thrush AB, Mertz VA, Bezaire V, Chabowski A, Heigenhauser GJ, Dyck DJ. Endurance training in obese humans improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid content. Am J Physiol Endocrinol Metab. 2006;291:E99–E107. doi: 10.1152/ajpendo.00587.2005. [DOI] [PubMed] [Google Scholar]
- 99.Dube JJ, Amati F, Stefanovic-Racic M, Toledo FG, Sauers SE, Goodpaster BH. Exercise-induced alterations in intramyocellular lipids and insulin resistance: the athlete’s paradox revisited. Am J Physiol Endocrinol Metab. 2008;294:E882–888. doi: 10.1152/ajpendo.00769.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]