

Abstract
Co-chaperonin protein 10 (cpn10, GroES in Escherichia coli) is a ring-shaped heptameric protein that facilitates substrate folding when in complex with cpn60 (GroEL in E. coli). The cpn10 from the hyperthermophilic, ancient bacterium Aquifex aeolicus (Aacpn10) has a 25-residue C-terminal extension in each monomer not found in any other cpn10 protein. Earlier in vitro work has shown that this tail is not needed for heptamer assembly or protein function. Without the tail, however, the heptamers (Aacpn10del-25) readily aggregate into fibrillar stacked rings. To explain this phenomenon, we performed binding experiments with a peptide construct of the tail to establish its specificity for Aacpn10del-25 and used cryo-electron microscopy to determine the three-dimensional (3D) structure of the GroEL–Aacpn10–ADP complex at an 8-Å resolution. We found that the GroEL–Aacpn10 structure is similar to the GroEL–GroES structure at this resolution, suggesting that Aacpn10 has molecular interactions with cpn60 similar to other cpn10s. The cryo-electron microscopy density map does not directly reveal the density of the Aacpn10 25-residue tail. However, the 3D statistical variance coefficient map computed from multiple 3D reconstructions with randomly selected particle images suggests that the tail is located at the Aacpn10 monomer–monomer interface and extends toward the cis-ring apical domain of GroEL. The tail at this location does not block the formation of a functional co-chaperonin/chaperonin complex but limits self-aggregation into linear fibrils at high temperatures. In addition, the 3D variance coefficient map identifies several regions inside the GroEL–Aacpn10 complex that have flexible conformations. This observation is in full agreement with the structural properties of an effective chaperonin.
Keywords: co-chaperonin, cryo-electron microscopy, hyperthermophile, conformational heterogeneity, variance coefficient
Introduction
Co-chaperonin protein 10 (cpn10) is a ring-shaped heptameric protein found in all bacterial and eukaryotic organisms.1 The well-known in vivo function of cpn10s is to assist cpn60 in the folding of nonnative proteins.2,3 The cpn10 acts as a cap to the cpn60 chamber, sequestering nonnative proteins from the surrounding environment by cycling on and off the cpn60 oligomer in an ATP-dependent process.4,5 Structures of different forms of cpn10–cpn60 complexes, mostly from the Escherichia coli GroES–GroEL system but also from Thermus thermophilus, have been described using X-ray crystallography,6,7 NMR (nuclear magnetic resonance) spectroscopy,8,9 and cryo-electron microscopy (cryo-EM).10,11 Structures of cpn10 heptamers alone have been reported for GroES, human mitochondrial cpn10 (hmcpn10), Mycobacterium leprae, and Mycobacterium tuberculosis cpn10 proteins.12-16 The cpn10 heptamer consists of seven identical subunits that adopt irregular β-barrel structures. The predominant subunit–subunit interaction in the heptamer is an antiparallel pairing of the last β-strand of one monomer with the first β-strand of the next monomer.12 Biophysical studies on hmcpn1013 GroES14-16 have shown that isolated monomers can be folded when the protein is diluted to very low concentrations, but the thermodynamic stability of these monomers is low. Much of cpn10s' overall heptamer stability comes from the subunit–subunit interface interactions.17
Biophysical work on cpn10 proteins has focused on hmcpn10, GroES, and cpn10 from Aquifex aeolicus (Aacpn10).14-18 A. aeolicus is a hyperthermophilic bacterium isolated at 95 °C, for which the complete genome has been sequenced.19 Although A. aeolicus lives at the upper thermal limit of known life, genomic data provide few clues for the mechanisms by which the organism increases the thermostability of its proteins. One novel feature seems to be the presence of residue insertions or deletions in proteins in the Aquificae family as compared with their mesostable homologs.20 Interestingly, Aacpn10 was found to follow this trend.20 While most other insertions and deletions detected in the Aquificae proteins are only a few residues in length, Aacpn10 contains a 25-residue C-terminal extension in each monomer. An extension of this type is not found in any other known cpn10 protein, and the sequence of the C-terminal tail bears no similarity to any other known sequence in the SWISS-PROT database.18
Stability studies in vitro, using a variety of denaturants, and equilibrium, kinetic, and calorimetric experiments have been reported for hmcpn10,17,21,22 GroES,14-16 and Aacpn10,18,21,22 as well as for a mutant of Aacpn10 where the C-terminal tail has been removed (Aacpn10del-25).18 Comparative biophysical studies on Aacpn10 and Aacpn10del-25 have shown that the presence of the tail does not affect heptamer assembly, thermal or chemical stability, folding/assembly mechanisms, or co-chaperonin function.18,22 Both with and without the tail, Aacpn10 was found to function as well as GroES in an ATP-dependent GroEL refolding assay.18 The presence of the tail, however, was shown to prevent the formation of fibrillar Aacpn10 heptamer aggregates at high temperatures and at high protein concentrations.18 To gain structural information on how the tail interacts with the rest of the Aacpn10 heptamer, we assessed properties of the tail by two approaches: (1) using binding experiments with the tail added in trans as a synthetic peptide and (2) determining the three-dimensional (3D) structure of the GroEL–Aacpn10–ADP complex by single-particle cryo-EM and assessing the regions of the cryo-EM map with high structural variability by statistical variance coefficient (VC) analysis. We found that while the tail peptide binds specifically to Aacpn10del-25 heptamers, binding is weak. This result agrees with our cryo-EM structure of the complex, which demonstrates that the tails are heterogeneously and flexibly positioned on the heptamer surface near the Aacpn10 subunit–subunit interfaces and extend onto the surface of the GroEL cis-ring.
Results and Discussion
Aacpn10del-25 aggregation at high temperature is buffer dependent
Previous protein incubation studies showed that Aacpn10del-25 is more prone than Aacpn10 to aggregation at high temperatures.18 Given the negative charges on both the top and bottom surfaces of Aacpn10del-25, we proposed that bridging buffer molecules are involved in the observed ordered aggregation.18,23 To test this idea, we performed aggregation experiments with Aacpn10del-25 in a variety of buffer, salt, and pH conditions at varying temperatures (Table 1). Inspection of the data revealed that Aacpn10del-25 is more prone to aggregation at a lower pH condition, where the amino group of Tris is protonated, providing a positive charge to promote electrostatic aggregation. Above the Tris pKa, however, the neutral amino group does not induce aggregation. In addition, the presence of salt (i.e., 250 mM NaCl) further reduces the amount of aggregated protein in neutral and high pH conditions; however, salt has little effect on Aacpn10del-25 aggregation at an acidic pH condition. Phosphate, instead of Tris buffer, reduces Aacpn10del-25 aggregation at all pH levels. The results show that Aacpn10del-25 aggregation is strongly dependent on buffer composition and pH conditions: the combination of low pH, no salt, and Tris buffer results in the greatest aggregation. This result supports the idea that stacking/aggregation of Aacpn10del-25 involves electrostatic interactions with surrounding buffer ions.
Table 1.
Buffer dependence on co-chaperonin (Aacpn10 and Aacpn10del-25) aggregation at different temperatures (see Materials and Methods)
| Buffer | pH | Salt concentration (NaCl) |
Aggregation at 80 °C (%) |
Aggregation at 100 °C (%) |
|---|---|---|---|---|
| Aacpn10del-25 | ||||
| 25 mM | 6.5 | 0 M | 40±7 | 90±5 |
| Tris–HCl | 6.5 | 250 mM | 25±1 | 91±7 |
| 7.5 | 0 M | 14±6 | 27±2 | |
| 7.5 | 250 mM | 9±2 | 23±3 | |
| 8.5 | 0 M | 19±4 | 40±4 | |
| 8.5 | 250 mM | 3±1 | 5±1 | |
| 9.5 | 0 M | 15±2 | 25±1 | |
| 9.5 | 250 mM | 11±2 | 9±2 | |
| 5 mM Na2PO4 | 6 | 0 M | 7±4 | 16±7 |
| 7 | 0 M | 6±3 | 16±6 | |
| 7 | 250 mM | 8±1 | 14±4 | |
| 8 | 0 M | 8±6 | 11±6 | |
| 25 mM | 7.5 | 0 M | 1±4 | 10±3 |
| Tris–HCl+T | ||||
| 25 mM | 7.5 | 0 M | 24±4 | 44±6 |
| Tris–HCl+T-Scramble | ||||
| Aacpn10 | ||||
| 25 mM Tris–HCl | 6.5 | 0 M | 1±2 | 5±2 |
| 7.5 | 0 M | 2±3 | 3±1 | |
| 7.5 | 250 mM | 3±2 | 7±4 | |
| 8.5 | 0 M | 5±4 | 7±3 | |
| 9.5 | 0 M | 3±2 | 8±4 | |
| 5 mM Na2PO4 | 6 | 0 M | 5±3 | 6±2 |
| 7 | 0 M | 6±4 | 8±3 | |
| 7 | 250 mM | 2±1 | 6±3 | |
| 8 | 0 M | 2±2 | 4±1 |
All data were taken from at least four independent experiments. T and T-Scramble are synthetic peptides added in trans.
We also performed correlating negatively stained EM experiments to confirm that the buffer-dependent high-temperature aggregation involves ordered stacking of Aacpn10del-25 heptamers (data not shown). As expected, stacked-ring aggregates were the dominant species observed in negatively stained images for the samples where a high percentage of the protein aggregated upon heating. In buffer conditions with mostly soluble protein at high temperatures, however, individual heptamers were primarily observed in the negatively stained images. As a control, full-length Aacpn10 was also incubated at high temperatures in all tested buffer/pH conditions. In none of the conditions did we find Aacpn10 to aggregate similar to Aacpn10del-25 (Table 1).
Inhibition of aggregation with tail added in trans
To test if the peptide tail added in trans affects Aacpn10del-25 ring–ring stacking, we performed temperature-induced aggregation experiments using Aacpn10del-25 and two peptides—one with the correct tail sequence (T) and one with a scrambled version (T-Scramble). Aacpn10del-25 and T or T-Scramble (in 10-fold excess) were incubated at a range of fixed temperatures between 20 and 100 °C (25 mM Tris–HCl, pH 7.5). The fraction of soluble protein at each condition was determined by SDS-PAGE, as in the abovementioned buffer-dependent experiments. We found that the addition of T decreases Aacpn10del-25 aggregation at high temperatures, whereas T-Scramble does not (Fig. 1a; Table 1). This suggests that T can bind to Aacpn10del-25 and, like in full-length Aacpn10, can disrupt the interactions that cause heptamer–heptamer aggregation. Since T-Scramble had no effect on Aacpn10del-25 aggregation, it appears that T binding to Aacpn10del-25 is specific.
Fig. 1.

(a) Aggregation of Aacpn10del-25 as a function of temperature and peptides. The fraction of soluble protein after 20 min of incubation is plotted versus incubation temperature. Circles indicate Aacpn10del-25;triangles, Aacpn10del-25+T; squares, Aacpn10del-25+T-Scramble. (b, c) Negatively stained images of Aacpn10del-25 with T (b) and T-Scramble (c). The scale bar represents 50 nm. At these conditions, full-length Aacpn10 does not stack into fibrillar aggregates (not shown).
Aacpn10del-25 mixtures with either T or T-Scramble (in 10-fold excess) were characterized by negatively stained EM (Fig. 1b and c) to directly test if the peptide added in trans abolishes the ordered Aacpn10del-25 aggregates. While negatively stained EM does not resolve free peptides and is not sensitive enough to reveal if peptides are bound to Aacpn10del-25, these experiments demonstrate that the presence of the T peptide inhibits ring–ring stacking of Aacpn10del-25. Negatively stained images of Aacpn10del-25 mixed with T-Scramble, in contrast, still contain the fibrillar aggregates (Fig. 1c). These experiments again support that T binds specifically to Aacpn10del-25 and thereby disrupts electrostatic ring–ring interactions.
Peptide added in trans binds only to Aacpn10del-25
The ability of T to bind to Aacpn10del-25 and other cpn10 variants was examined by analytical size-exclusion chromatography. The difference in molecular weight between Aacpn10 and Aacpn10del-25 heptamers (96,000 versus 70,000) allows for the detection of peptide binding to Aacpn10del-25 based on size. If each monomer binds one peptide, the Aacpn10del-25+T mixture should appear at the molecular weight correlating to full-length Aacpn10. If the C-terminal tails of the Aacpn10 monomers interact specifically with the heptamer ring, T will bind to Aacpn10del-25 but not to Aacpn10, hmcpn10, or GroES. If binding is more generic, however, both hmcpn10 and GroES may interact with the peptide, as they both lack this extension and are homologous to Aacpn10del-25. Since Aacpn10 already has tails present, the peptide should not bind in either scenario.
Our chromatograms show that in accord with their size, Aacpn10 elutes at 13.2 mL and Aacpn10del-25 elutes at 14.8 mL (Fig. 2). Both T and T-Scramble elute in three distinct peaks, with volumes of 21, 22.5, and 23.8 mL, corresponding to approximate molecular weights of 9000, 5000, and 2000, respectively. These results suggest some peptide aggregation. The self-association of the peptides is reversible, however, since reinjection of the peak at 21 mL shows a redistribution into all three elution volumes (data not shown).
Fig. 2.

Peptide binding to Aacpn10del-25 monitored by size-exclusion chromatography. Elution proteins are depicted as follows: red, Aacpn10; blue, Aacpn10del-25; green, T; gold, T-Scramble; purple, Aacpn10del-25+T; and black, Aacpn10del-25+T-Scramble (200-μL sample—100 μM protein, 1 mM peptide; 4 °C). Molecular weight standards confirm the assignment of the peaks (not shown). Inset: Expansion of the 12- to 16-mL region.
When Aacpn10del-25 is incubated with T, the sample elutes at 13.5, 14.8, 21.2, 22.7, and 24 mL. The appearance of a peak for the Aacpn10del-25+T mixture sample at a position similar to that for full-length Aacpn10 (i.e., 13.5 mL) indicates binding of peptides to the Aacpn10del-25 heptamers. Since only one distinct peak appears, it suggests that seven peptides bind to each heptamer (i.e., one peptide per monomer). Isolation and subsequent analysis of the peak corresponding to this complex reveal the same distribution of species as the original mixture (data not shown). This supports a thermodynamic equilibrium between all species. We integrated the peak intensities in the chromatogram for the Aacpn10del-25+T mixture, correcting for the extinction coefficients, to estimate an apparent peptide–protein dissociation constant (Kd)of ∼0.2±0.05 mM (4 °C, pH 7.5). For this measurement, we assumed one peptide binding to each monomer (i.e., seven identical and independent binding sites in each heptamer) without cooperative or peptide self-association effects. We note that the appearance of an “all-or-none” type of binding indicates some degree of cooperativity or some form of interaction between binding sites. Incubation of Aacpn10del-25 with T-Scramble does not produce any new peaks in the chromatogram at volumes lower than 14.5 mL (Fig. 2). In addition, we found no evidence for any complex formation when T or T-Scramble is incubated with Aacpn10, hmcpn10, or GroES (data not shown). Thus, T binding appears specific for Aacpn10del-25.
Cryo-EM structure of GroEL–Aacpn10–ADP
To visualize the Aacpn10 structure and thereby localize the tail, we collected cryo-EM data on Aacpn10 complexed with GroEL and ADP. Previous cryo-EM studies of the GroEL–GroES complex have shown that it can adopt two major distinct forms10,24: a fraction of transient football-like structures when ATP is present and a majority of stable “bullet”-like structures with ADP. Since the ADP complexes are stable25, we studied GroEL–Aacpn10–ADP by cryo-EM. Figure 3a is a typical image for the GroEL–Aacpn10–ADP complexes taken in a 300-kV cryo-electron microscope. Some particle images with the bullet shape could be observed (white arrows). Our initial data analysis indicated that the GroEL–Aacpn10–ADP complex is heterogeneous, containing several particle subpopulations (data not shown): GroEL–Aacpn10–ADP (bullet-like); GroEL alone; single-ring GroEL11; and single-ring GroEL complexed with Aacpn10. This type of heterogeneity has been reported previously in both GroELD398A–GroES–ATP and GroEL–GroES– cryo-EM ADP studies.10
Fig. 3.

(a) A typical cryo-image of GroEL complexed with Aacpn10 and ADP taken in a 300-kV cryo-electron microscope. Two side-view particle images with a bullet shape are pointed by white arrows. The defocus for this image is about 3 μm. The scale bar represents 15 nm. (b) Representative 2D class averages for the GroEL–Aacpn10–ADP complex.
The GroEL–Aacpn10–ADP subpopulation with the bullet shape was computationally isolated from the entire data set of 90,699 particle images. Figure 3b shows representative two-dimensional (2D) class averages from the selected data set (10,772 particle images). The first row shows top views or slightly tilted top views with the seven subunits detectable; rows 2 through 5 show side views or slightly tilted side views for the bullet-like particles of GroEL–Aacpn10–ADP. No football-like particle image was obviously detected based on our 2D class average analysis. The final 3D structure of the GroEL–Aacpn10–ADP complex was reconstructed using EMAN26 with a C7-symmetry constraint from the computationally sorted particles to a resolution of ∼8 Å(Fig. 4a).
Fig. 4.
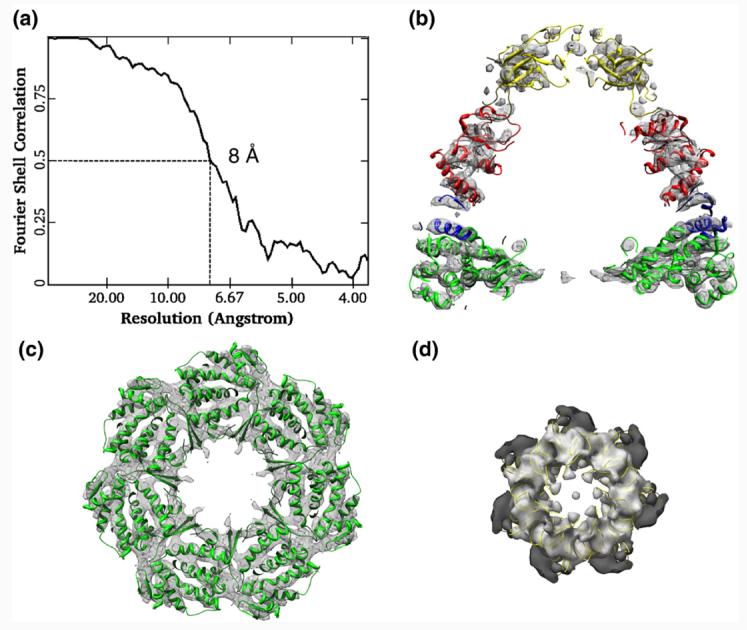
The GroEL–GroES–ADP crystal structure (PDB code 1AON; in ribbons) was docked into the 8-Å cryo-EM density map of the GroEL–Aacpn10–ADP complex using domain-as-rigid-body fitting. (a) Resolution assessment of 8 Å for the GroEL–Aacpn10–ADP cryo-EM structure with the use of a criterion of Fourier shell correlation at 0.5. (b) A central cross-section of the cis-ring along a 7-fold symmetry axis showing the overall good fitting of the cis-ring. (c) A cross-section through the cis-ring equatorial domain of GroEL showing the excellent correspondence of α-helices. (d) Fitting between the GroES structure (in yellow ribbon) and the dome-shaped cap of Aacpn10 showing that the roof loops fit well with the densities of Aacpn10 from the GroEL–Aacpn10–ADP complex. The ribbons from different domains of the crystal structure of GroEL–GroES–ADP in (b) are colored differently.
Rigid-body fitting of seven domains from the GroEL–GroES–ADP crystal structure [Protein Data Bank (PDB) code 1AON] into the cryo-EM density map of the GroEL–Aacpn10–ADP complex was performed using UROX27; thus, the pseudo-atomic model for the GroEL–Aacpn10–ADP complex based on the domain-as-rigid-body fitting was obtained. The domain-as-rigid-body fitting approach was chosen because the flexibility between domains is well known in this type of molecular machine.6,10 Figure 4b shows that each domain of cis-ring matches well between the cryo-EM density map of the GroEL–Aacpn10–ADP complex and the pseudo-atomic model. In particular, the cis-ring equatorial domain of the cryo-EM density map has the best fit with the pseudo-atomic model as evidenced in the excellent match of the α-helix positions (Fig. 4c). The structural matches are generally better visually in the cis-ring than in the trans-ring probably due to the flexibility of the trans-ring without the bound cap, and such less match on the trans-ring has already been seen previously in other cryo-EM studies of the GroEL–GroES complex.10 Overall, a correlation coefficient value of 0.93 was found using Chimera28 between the cryo-EM map of the GroEL–Aacpn10–ADP complex and the derived pseudo-atomic model.
The Aacpn10 cap region also agrees well with the structure of GroES, including the roof loops that form the top of the dome-shaped cap (Fig. 4d). Furthermore, we also performed an additional structural comparison between our pseudo-atomic model and that derived from the GroEL–GroES–ADP cryo-EM structure10 and found no significant difference (data not shown). Hence, when bound to GroEL, Aacpn10 might adopt a heptameric structure similar to GroES. Moreover, GroEL may adopt a similar chaperonin/co-chaperonin complex with the hyperthermostable Aacpn10 as with its natural partner, GroES.
Statistical VC analysis of GroEL–Aacpn10–ADP locates the tail
From the abovementioned comparisons, no significant density was observed around the C-terminus of each Aacpn10 monomer that could directly be identified as the 25 C-terminal residues of Aacpn10. One possibility for the lack of C-terminal tail density in the cryo-EM structure is that the 25 residues in the C-terminus of each Aacpn10 subunit are flexible or heterogeneously interact with the rest of each monomer in the GroEL–Aacpn10–ADP complex. To test this hypothesis, we calculated the VC map using the adapted statistical bootstrap technique.29 The physical plausibility of the VC map calculated from the multiple 3D reconstructions based on randomly selected particle images can be attributed either to a computational error or to an actual density variation among thousands of particle images used in the reconstructions. Since our subnanometer-resolution cryo-EM density map has an excellent match with the pseudo-atomic model in terms of α-helix positions (Fig. 4c), we interpret that the region with the highest VC density is caused by the high conformational flexibility and/or variations instead of any computational error. Our 3D VC map, indeed, shows that the cis-ring equatorial domain in the cryo-EM density map, known as the least flexible region in GroEL–GroES complex, has the relatively least density. A threshold was set such that the VC density from the cis-ring equatorial domain was minimized in order to evaluate the 3D VC map. With this threshold criterion, we noted that four major distinct regions have the higher variation. In order to display their locations (numbers 1–4 in Fig. 5), we superimposed the 3D VC map on the part of the pseudo-atomic model.
Fig. 5.

3D VC map of the GroEL–Aacpn10–ADP complex locating the 25-residue C-terminal tail (labeled as region 4) and other major flexible regions (numbers 1–3) in the molecule. (a) Front half of the 3D VC map superimposed on the three GroEL subunits and three GroES subunits from the pseudo-atomic structure for the GroEL–Aacpn10–ADP complex. The VC map was set at a threshold to minimize the VC density in the least flexible cis-ring equatorial domain. The pound sign points to the positions of the mobile helical hairpin on the tip of the orange ribbons. (b) With the front half of the VC map removed, this image shows the internal flexibility of the molecule. Four major flexible regions in (a) and (b) are labeled by numbers 1–4. The green scale bar at the bottom of (b) represents 20 Å.
Simulations (data not shown) ruled out the possibility of reconstruction artifacts of the symmetry axis contributing to regions 1 and 2 (Fig. 5b). The only flexibility in these two regions is from the C-termini, which are not resolved in the crystal structure due to their flexibility.6 Therefore, regions 1 and 2 (Fig. 5b) correspond to the last 23 residues on the C-terminus of the GroEL equatorial domain in the cis-ring and the trans-ring, respectively. Region 3 (Fig. 5a and b, bottom) shows the region having high structural variation on the trans-ring apical domain that contains the flexible helical hairpin (fitted orange ribbon pointed by a pound sign in Fig. 5a) formed by helices K and L in the crystal structure of GroEL–GroES–ADP and is not capped by co-chaperonin. This flexibility can also account for the region's less agreement between the cryo-EM density map and the pseudo-atomic model when compared with the good cis-ring fitting (Fig. 4b).
Most interesting in this 3D VC map analysis is region 4 (Fig. 5a and b, top), which has a long rod shape (∼35 Å in length) over the cis-ring apical domain, which starts from the predicted area for the C-terminus of Aacpn10 (Fig. 6) and extends from the Aacpn10 monomer–monomer interface downward over the surface of the cis-ring apical domain of GroEL. The flexibility in this region is not likely caused by local expansion or shrinkage of the cis-ring apical domain, since GroEL conformational changes generally occur along the hinges, and each domain can be generally considered a rigid body.10 In addition, it is not possibly contributed by the misalignment of particle images during reconstruction because the cis-ring equatorial domain of our cryo-EM density map has an excellent fit with the pseudo-atomic model (Fig. 4b). A reasonable interpretation for the large variation in region 4 is that it originates from the extra 25-residue tail on the C-terminus of Aacpn10. Thus, it appears that the unique tails extend on the outer surface of the complex, from the Aacpn10 subunit–subunit interface to the GroEL cis-ring apical domain.
Fig. 6.

Close-up top view of the 3D VC map of region 4 as shown in Fig. 5. 3D VC map (in magenta) overlaid on the cryo-EM density map of the GroEL–Aacpn10–ADP complex (in gray), which is displayed at a low isosurface threshold. Two GroES yellow ribbons from the pseudo-atomic model for the GroEL–Aacpn10–ADP complex are shown in the cap, and the C-termini are indicated. The cryo-EM density map is displayed with a transparency.
We point out that despite the presence of the tail in Aacpn10, there is no major difference between these two pseudo-atomic models derived from the GroEL–Aacpn10–ADP and the GroEL–GroES–ADP10 cryo-EM structures at an ∼8-Å resolution level (data not shown), based on the domain-as-rigid-body fitting. This agrees with the reported biophysical data of GroES-like co-chaperonin activity for Aacpn10 both with and without the tail.18 Moreover, the external location of the tail in the Aacpn10 heptamer explains the earlier finding of the tail having no effect on heptamer assembly or Aacpn10 stability and folding. Finally, the presence of the tail on the surface gives a physical explanation, in terms of possible altered charge–charge pattern and steric repulsion, for how the tail limits Aacpn10 heptamer–heptamer aggregation in vitro.
Conclusions
Our goal with this study was to explain how the unique C-terminal tail in Aacpn10 can act as an antiaggregation domain but have few other effects. The collected data on the trans peptides and various cpn10 homologs demonstrate that (1) the tail peptide binds to Aacpn10del-25 when added in trans; (2) binding is specific, as T-Scramble cannot bind, and no other cpn10 interacts with the tail peptide; and (3) when T is bound in trans, the same favorable antiaggregation effects as for the full-length protein are seen. The cryo-EM data and the 3D statistical VC analysis of the GroEL–Aacpn10–ADP complex suggest that the 25-residue tail in the complex is positioned on the subunit–subunit interface of the Aacpn10 heptamer, extending to the outer surface of the cis-ring apical domain of GroEL. Upon comparison with the GroES–GroEL structure, we conclude that Aacpn10 may adopt a similar tertiary conformation as GroES and that its complex with GroEL matches that of GroES with GroEL. We found significant variations in several other regions of the GroEL–Aacpn10 molecule by calculation of the statistical VC map. This GroEL–Aacpn10 plasticity may be revealing a common feature of cpn10–cpn60 complexes as required for substrate-refolding activity.
A. aeolicus is a hyperthermophile that is thought to have diverged early from other bacteria.30 This suggests that the C-terminal tail in Aacpn10 is an ancient remnant. Evolution may have adopted the residues on the cpn10 surface so as to prevent aggregation at high temperatures. In agreement, GroES, hmcpn10, and other cpn10 proteins have fewer charged residues in their sequences.16,22 Unneeded for prevention of aggregation and of no benefit to heptamer stability, subsequent diverging organisms likely eliminated the C-terminal tail from cpn10 proteins to reduce protein length.31
Materials and Methods
Proteins and peptides
Aacpn10, Aacpn10del-25, hmcpn10, GroES, and GroEL were expressed in E. coli and purified as described previously.16,18,32 Protein concentrations were determined (1) using the Bio-Rad protein assay for Aacpn10 and GroES and (2) spectroscopically via ε280 values of 4460, 4200, and 9800 M−1 cm−1 for Aacpn10del-25, hmcpn10, and GroEL, respectively. All protein concentrations are reported per monomer. Two synthetic peptides, one corresponding to the C-terminal sequence (T; YSSLIGGEVRWQQRQLSTTRKQGQN) and another with a scrambled combination of the same amino acids (T-Scramble; RVGYLSSEQQQGKSGNTTQWIRQRL), were obtained from GenScript (Scotch Plains, NJ). Both peptides were of >95% purity. Peptide concentrations were determined by ε280 of 7017 M−1 cm−1.
Biophysical assays
For high-temperature aggregation studies, protein samples (∼200 μM) were incubated for 20 min at specific temperatures between 20 and 100 °C. Buffer conditions and peptide additions were varied as indicated in the text and tables. Samples were then centrifuged at 14,000 rpm for 1 min to remove aggregated/precipitated protein, followed by visualization of the soluble fractions by SDS-PAGE. Quantification of the extent of aggregation was derived via the integrated density of the soluble protein on the gel (FluorChem 5500 MultiImage Light Cabinet; software by Alph Innotech). As a control for the accuracy of these gel experiments, the time course of protein aggregation at selected specific temperatures was monitored in solution by light scattering at 500 nm.
Peptide binding to various cpn10 proteins was tested by analytical gel filtration (Superdex 200 10/300 GL, GE Healthcare). The column was equilibrated with 50 mM Tris–HCl, pH 7.5, for Aacpn10 and GroES; 25 mM Tris–HCl, pH 7.5, for Aacpn10del-25; and 5 mM sodium phosphate, pH 7, for hmcpn10. The column and samples (200 μL) were kept at 4 °C for each run, and 100 mM KCl was included in the solvent to prevent binding of the protein to the column resin. Proteins and peptides were mixed in 1:10 ratios for all experiments: 100 μM protein and 1 mM peptide.
EM and data processing
For negatively stained EM, the final protein concentration was 0.05 mg/mL; buffer conditions and peptide additions were as indicated in Table 1. Samples were absorbed onto fresh thin carbon film and were washed in distilled water. The proteins were then stained with 0.75% uranyl formate. The negatively stained images were taken using a JEM2010 electron microscope on a Gatan 2k×2k charge-coupled device camera at a nominal magnification of 50,000×.
For cryo-EM, Aacpn10 was incubated with GroEL in 50 mM Tris–HCl, 30 mM MgCl2, and 2 mM ADP, pH 7.5, at 37 °C for 1 h prior to freezing. The complex (∼894 kDa) was diluted to a final concentration of 4 mg/mL. The ratio of GroEL monomers to Aacpn10 monomers was ∼1:1.1. Using the pretreated33 and freshly glow-discharged quantifoil grids (R1.2/1.3, Quantifoil Micro Tools GmbH, Jena, Germany), 3 μL of the sample solution was applied on each grid and then blotted for 2.5 s before submersion in liquid ethane cooled by liquid nitrogen using an automated vitrification device, Vitrobot (FEI Company†). Frozen grids were stored in liquid nitrogen until insertion into the microscope column. Images were collected on SO-163 Kodak films at a nominal magnification of 50,000× with a dose of ∼36 e/Å2 on each image in a 300-kV JEOL cryo-electron microscope, JEM3000SFF, cooled with liquid helium to ∼4.2 K for the specimen temperature.33 The films were developed for 12 min in Kodak D19 and fixed for 10 min in Kodak fixer and scanned into a digital format using a Nikon Super Coolscan 9000 scanner at a 6.35-μm step size, yielding a pixel size of 1.27 Å on the specimen level. The scanned images were then averaged 1.5 times to produce data at 1.91 Å/pixel. Particle selection was performed semiautomatically from ∼720 micrographs using the EMAN26 program boxer, followed by a rough and manual screening out of the bad contaminants, to generate a data set with 90,699 particle images in total. The contrast transfer function fitting for each micrograph was performed automatically using the program fitctf.py (C. Yang, W. Jiang, D.-H. Chen, U. Adiga, E.G. Ng, W. Chiu, unpublished data) and then fine-tuned manually using the EMAN program ctfit.
The methodology of multiple-model refinement for the compositionally and conformationally heterogeneous complex, which has been described previously,11 was used to sort out different particle subpopulations from the whole data set. In this work, however, two EMAN programs, multirefine and refine2d.py,11 were used to do the particle sorting. Finally 10,772 particle images that correspond to the subpopulation of GroEL–Aacpn10–ADP complexes with a bullet shape were separated from the complete data set. The particle orientation refinement and 3D reconstruction for the GroEL–Aacpn10–ADP complex were performed using the regular single-model refinement EMAN program refine, assuming a 7-fold symmetry. The final resolution was assessed using a criterion of Fourier shell correlation34 cutoff at 0.5 from two independent half sets of data. The EMAN program refine2d.py11 was also used for 2D class average analysis, in which no symmetry was imposed. Chimera28 was used for the surface representations of the cryo-EM density maps.
Atomic structure fitting and VC map calculation
UROX27 was used to perform the domain-as-rigid-body fitting of the GroEL–GroES–ADP crystal structure (PDB code 1AON)6 into the cryo-EM density map of the GroEL–Aacpn10–ADP complex. Three domains, equatorial, intermediate, and apical, were split from one GroEL subunit of the GroEL–GroES–ADP crystal structure. Then, six domains of GroEL (three from the cis-ring and three from the trans-ring) and one GroES subunit from the crystal structure (PDB code 1AON) were fitted into the cryo-EM density map of the GroEL–Aacpn10–ADP complex as rigid bodies, imposing a 7-fold symmetry. The pseudo-atomic model for the GroEL–Aacpn10–ADP complex was compared with that derived from the GroEL–GroES–ADP cryo-EM structure10 using Chimera.28
In the 3D VC map calculation for the GroEL–Aacpn10–ADP complex, the bootstrap technique29 was implemented in the EMAN program calculateMapVariance.py. Briefly, 100 independent 3D reconstructions were obtained from the randomly sampled particle images with the assumption of 7-fold symmetry. Then, the 3D standard deviation map and the 3D average map were calculated among those 100 reconstructions. The final 3D VC map was calculated from the normalized standard deviation map against the average map and further low-pass filtered to ∼15 Å.
Acknowledgements
This research was supported by the National Institutes of Health (grants P01GM064692, P41RR02250, and PN2EY016525 through the National Institutes of Health Roadmap for Medical Research and grant 5R90DK071054 through the Nanobiology Training Grant administered by the Gulf Coast Consortia) and by the Welch Foundation (C-1588). This work was supported in part by the Rice Computational Research Cluster funded by the National Science Foundation under grant CNS-0421109 and a partnership between Rice University and AMD and Cray. We thank Michael Perham for preparation of hmcpn10 protein, Megan Guelker for assistance with negative staining, and Yunyun Jiang for assistance with negatively stained EM imaging. We also acknowledge the help on the UROX program provided by Dr. Xavier Siebert from the Institute for Structural Biology (Grenoble, France).
Abbreviations used
- 3D
three dimensional
- Aacpn10
Aquifex aeolicus cpn10
- cpn10
co-chaperonin protein 10
- EM
electron microscopy
- hmcpn10
human mitochondrial cpn10
- T
tail peptide
- T-Scramble
peptide with scrambled sequence
- VC
variance coefficient
Footnotes
EM Data Bank accession code
The cryo-EM density map of GroEL-Aacpn10-ADP has been deposited in the EM data bank with accession code EMD-1531.
References
- 1.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 2.Fenton WA, Horwich AL. Chaperoninmediated protein folding: fate of substrate polypeptide. Q. Rev. Biophys. 2003;36:229–256. doi: 10.1017/s0033583503003883. [DOI] [PubMed] [Google Scholar]
- 3.Kawata Y, Nosaka K, Hongo K, Mizobata T, Nagai J. Chaperonin GroE and ADP facilitate the folding of various proteins and protect against heat inactivation. FEBS Lett. 1994;345:229–232. doi: 10.1016/0014-5793(94)00456-0. [DOI] [PubMed] [Google Scholar]
- 4.Rye H, Roseman A, Chen S, Furtak K, Fenton W, Saibil H, Horwich A. GroEL–GroES cycling: ATP and nonnative polypeptide direct alternation of folding-active rings. Cell. 1999;97:325–338. doi: 10.1016/s0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- 5.Weissman J, Hohi C, Kovalenko O, Kashi Y, Chen S, Braig K, et al. Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES. Cell. 1995;83:577–587. doi: 10.1016/0092-8674(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 6.Xu Z, Horwich AL, Sigler PB. The crystal structure of the asymmetric GroEL–GroES–(ADP)7 chaperonin complex. Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 7.Shimamura T, Koike-Takeshita A, Yokoyama K, Masui R, Murai N, Yoshida M, et al. Crystal structure of the native chaperonin complex from Thermus thermophilus revealed unexpected asymmetry at the cis-cavity. Structure. 2004;12:1471–1480. doi: 10.1016/j.str.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 8.Fiaux J, Bertelsen EB, Horwich AL, Wuthrich K. NMR analysis of a 900K GroEL–GroES complex. Nature. 2002;418:207–211. doi: 10.1038/nature00860. [DOI] [PubMed] [Google Scholar]
- 9.Nishida N, Motojima F, Idota M, Fujikawa H, Yoshida M, Shimada I, Kato K. Probing dynamics and conformational change of the GroEL–GroES complex by 13C NMR spectroscopy. J. Biochem. (Tokyo) 2006;140:591–598. doi: 10.1093/jb/mvj188. [DOI] [PubMed] [Google Scholar]
- 10.Ranson N, Clare D, Farr G, Houldershaw D, Horwich A, Saibil H. Allosteric signaling of ATP hydrolysis in GroEL–GroES complexes. Nat. Struct. Mol. Biol. 2006;13:147–152. doi: 10.1038/nsmb1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen DH, Song JL, Chuang DT, Chiu W, Ludtke SJ. An expanded conformation of single-ring GroEL–GroES complex encapsulates an 86 kDa substrate. Structure. 2006;14:1711–1722. doi: 10.1016/j.str.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Hunt JF, Weaver AJ, Landry SJ, Gierasch L, Deisenhofer J. The crystal structure of the GroES co-chaperonin at 2.8 Å resolution. Nature. 1996;379:37–45. doi: 10.1038/379037a0. [DOI] [PubMed] [Google Scholar]
- 13.Guidry JJ, Wittung-Stafshede P. Low stability for monomeric human chaperonin protein 10: interprotein interactions contribute majority of oligomer stability. Arch. Biochem. Biophys. 2002;405:280–282. doi: 10.1016/s0003-9861(02)00406-x. [DOI] [PubMed] [Google Scholar]
- 14.Boudker O, Todd MJ, Freire E. The structural stability of the co-chaperonin GroES. J. Mol. Biol. 1997;272:770–779. doi: 10.1006/jmbi.1997.1263. [DOI] [PubMed] [Google Scholar]
- 15.Seale JW, Gorovits BM, Ybarra J, Horowitz PM. Reversible oligomerization and denaturation of the chaperonin GroES. Biochemistry. 1996;35:4079–4083. doi: 10.1021/bi953087n. [DOI] [PubMed] [Google Scholar]
- 16.Luke K, Wittung-Stafshede P. Folding and assembly pathways of co-chaperonin proteins 10: origin of thermostability. Arch. Biochem. Biophys. 2006;456:8–18. doi: 10.1016/j.abb.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Guidry JJ, Moczygemba C, Steede NK, Landry S, Wittung-Stafshede P. Reversible denaturation of the oligomeric human chaperonin 10: denatured state depends on chemical denaturant. Protein Sci. 2000;9:2109–2117. doi: 10.1110/ps.9.11.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luke K, Apiyo D, Wittung-Stafshede P. Role of the unique peptide tail in hyperthermostable Aquifex aeolicus co-chaperonin protein 10. Biochemistry. 2005;44:14385–14395. doi: 10.1021/bi051131l. [DOI] [PubMed] [Google Scholar]
- 19.Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, et al. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 20.Guidry JJ, Wittung-Stafshede P. First characterization of co-chaperonin protein 10 from hyper-thermophilic Aquifex aeolicus. Biochem. Biophys. Res. Commun. 2004;317:176–180. doi: 10.1016/j.bbrc.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 21.Luke K, Apiyo D, Wittung-Stafshede P. Dissecting homo-heptamer thermodynamics by isothermal titration calorimetry: entropy-driven assembly of co-chaperonin protein 10. Biophys. J. 2005;89:3332–3336. doi: 10.1529/biophysj.105.067223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luke K, Perham M, Wittung-Stafshede P. Kinetic folding and assembly mechanisms differ for two homologous heptamers. J. Mol. Biol. 2006;363:729–742. doi: 10.1016/j.jmb.2006.08.058. [DOI] [PubMed] [Google Scholar]
- 23.Xu D, Lin SL, Nussinov R. Protein binding versus protein folding: the role of hydrophilic bridges in protein associations. J. Mol. Biol. 1997;265:68–84. doi: 10.1006/jmbi.1996.0712. [DOI] [PubMed] [Google Scholar]
- 24.Ranson NA, Farr GW, Roseman AM, Gowen B, Fenton WA, Horwich AL, Saibil HR. ATP-bound states of GroEL captured by cryo-electron microscopy. Cell. 2001;107:869–879. doi: 10.1016/s0092-8674(01)00617-1. [DOI] [PubMed] [Google Scholar]
- 25.Roseman AM, Chen S, White H, Braig K, Saibil HR. The chaperonin ATPase cycle: mechanism of allosteric switching and movements of substrate-binding domains in GroEL. Cell. 1996;87:241–251. doi: 10.1016/s0092-8674(00)81342-2. [DOI] [PubMed] [Google Scholar]
- 26.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semi-automated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 27.Navaza J, Lepault J, Rey FA, Alvarez-Rua C, Borge J. On the fitting of model electron densities into EM reconstructions: a reciprocal-space formulation. Acta Crystallogr., Sect. D.: Biol. Crystallogr. 2002;58:1820–1825. doi: 10.1107/s0907444902013707. [DOI] [PubMed] [Google Scholar]
- 28.Goddard TD, Huang CC, Ferrin TE. Software extensions to UCSF Chimera for interactive visualization of large molecular assemblies. Structure (Camb.) 2005;13:473–482. doi: 10.1016/j.str.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Penczek PA, Yang C, Frank J, Spahn CM. Estimation of variance in single-particle reconstruction using the bootstrap technique. J. Struct. Biol. 2006;154:168–183. doi: 10.1016/j.jsb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Bocchetta M, Gribaldo S, Sanangelantoni A, Cammarano P. Phylogenetic depth of the bacterial genera Aquifex and Thermotoga inferred from analysis of ribosomal protein, elongation factor, and RNA polymerase subunit sequences. J. Mol. Evol. 2000;50:366–380. doi: 10.1007/s002399910040. [DOI] [PubMed] [Google Scholar]
- 31.Jaenicke R, Bohm G. The stability of proteins in extreme environments. Curr. Opin. Struct. Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 32.Perham M, Chen M, Ma J, Wittung-Stafshede P. Unfolding of heptameric co-chaperonin protein follows “fly casting” mechanism: observation of transient nonnative heptamer. J. Am. Chem. Soc. 2005;127:16402–16403. doi: 10.1021/ja055574o. [DOI] [PubMed] [Google Scholar]
- 33.Chen DH, Jakana J, Chiu W. Single-particle cryo-EM data collected on a 300-kV liquid helium-cooled electron cryomicroscope. J. Chinese Elec. Microsc. Soc. 2007;26:473–479. [Google Scholar]
- 34.Harauz G, van Heel M. Exact filters for general geometry three dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]