

Abstract
The structure of a novel tetradehydrocorrin, factor IV, isolated from Propionibacterium shermanii has been established by multidimensional NMR spectroscopy. Incorporation of radiolabeled factor IV into cobyrinic acid established the biointermediacy of this cobalt complex, whose structure has implications for the mechanisms of the anaerobic pathway to B12.
Keywords: tetradehydrocorrin, biosynthetic intermediate, NMR spectroscopy
In the aerobic bacterium Pseudomonas denitrificans, the biosynthetic pathway to vitamin B12, featuring an oxygen-requiring step, has been established (for reviews, see refs. 1 and 2), but it is now clear that a second, anaerobic route to corrin has existed in nature for ca. 4 billion years (2, 3). Thus, the obligate anaerobes, such as methanogenic bacteria, and the semianaerobic Propionibacterium shermanii are able to synthesize B12 by processes that differ at several pivotal steps from the aerobic pathway. The Ps. denitrificans route uses a two-stage ring contraction sequence on the metal-free substrate precorrin-3, using O2 (4) to fashion γ-lactone and tertiary hydroxyl functions at C-1 and C-20 in precorrin-3x (Scheme SI) as a prelude to a pinacol-like rearrangement, whereas cobalt is inserted late, i.e., not until all S-adenosylmethionine (AdoMet)-derived methyl groups are in place and amidation of the carboxylates has begun (1, 2).
Scheme I.
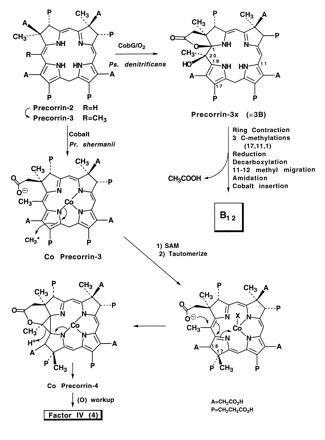
In contrast, it has been found (5) that in Pr. shermanii cobalt is inserted early into precorrin-3, which is ring-contracted without O2 as cofactor (Scheme SI), and at a later, unknown stage, a unique exchange of carbonyl oxygen at the C-27 (ring A) acetate occurs (6, 7); this is another event that is not paralleled in aerobic metabolism (8). In this report, we describe the isolation of a new B12 intermediate from Pr. shermanii whose surprising structure provides circumstantial but compelling evidence for those mechanisms operating during anaerobic biosynthesis that mediate ring contraction and loss of the 2-carbon fragment from the western side.
MATERIALS AND METHODS
Preparation of Factor IV from epi-Factor II.
A cell-free extract of Pr. shermanii (from 50 g of wet cells) was prepared by sonication (50 min at 0–4°C) in phosphate buffer (pH 7.6; 50 ml). Glutathione (5 mg), ATP (22 mg), NAD (5 mg), NADH (10 mg), AdoMet (75 μmol), EDTA (3.5 μmol), Co2+ (4 μmol), and Co3+ (1.8 μmol) were added to the supernatant of the centrifuged suspension (17,000 × g; 25 min at O°C). For preparative isolation, 3-epi; 8-epi factor II (Fig. 1; 500 μmol) was added, and the incubation was carried out at 31°C for 16 hr. Sodium cyanide (5 mg) was then added, and the tetrapyrrolic mixture was adsorbed on DEAE–Sephadex A-25 (Pharmacia) followed by extraction and esterification by MeOH/H2SO4 (95:5) for 16 hr at 20°C. The mixture was extracted with CH2Cl2 and chromatographed on silica gel in the solvent mixture toluene/EtOAc/MeOH (150:96:4; vol/vol/vol) in the presence of NaCN. Rechromatography of the baseline fraction in CH2Cl2/MeOH (95:5) afforded factor IV methylester (8 nmol) (Rf 0.46) λmax(CH2Cl2)-760 (0.47); 702 (0.50) 486 (0.39) 387 (1.00) 341 (0.67) 299 (0.66) nm (rel. intensities) FABMS C51H63N4O16 Co: m/z 1046.
Figure 1.
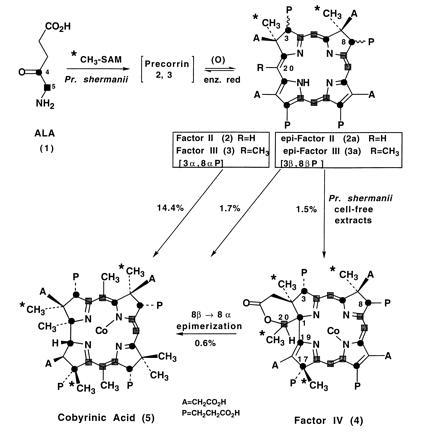
Bioconversion (% radiochemical yields) of factors II and III, (and their 3 and 8 epimers) derived from labeled ALA and AdoMet and of factor IV into cobyrinic acid (5) using cell-free extracts of Pr. shermanii. Labeling patterns were derived from either stable isotopes (•, [4-13C]ALA; ▪, [5-13C]ALA; ∗, [methyl-13C]AdoMet) or radioisotopes (•, [4-14C]ALA; ∗, [methyl-3H]AdoMet) as described in the text. The intermediates are isolated as methyl esters and reincubated (after hydrolysis) in their oxidized forms (factors II–IV) but it is assumed that the biotransformations occur at the reduced (precorrin) level since precorrin-2 can be isolated anaerobically from Pr. shermanii (9).
Isolation of 13C-Enriched Factor IV.
The above incubation was carried out in the presence of [4-13C]δ-aminolevulinic acid (ALA), [5-13C] ALA, and 13CH3-AdoMet to afford specimens of pure factor IV (200–250 μg), which were used for NMR structural determination by the methods previously described for precorrin-3x (8).
Bioconversion of Factor II, (3β,8β-epi) Factor II, and Factor IV to Cobyrinic Acid.
Doubly labeled factors were prepared as described previously (5) from [5-14C]ALA and C3H3-AdoMet. Cobester was isolated from incubation of cell-free extract with the radiolabeled factors and chromatographed to constant radioactivity. The radio incorporations were determined by 14C/13H scintillation counting, and the results are summarized in Fig. 1.
NMR Correlations of Factor IV.
13C-chemical shifts, heteronuclear multiple bond, and heteronuclear multiple quantum correlations were acquired exactly as described for the structure determination of precorrin-3x (8). The assignments are summarized in Table 1.
Table 1.
13C NMR assignments of factor IV heptamethyl ester in benzene-d6
| Position enriched | δ13C (Jcc Hz) |
|---|---|
| From [5-13C]ALA | |
| C4 | 178.0 (68) |
| C5 | 96.6 (69) |
| C9 | 177.5 (65) |
| C10 | 95.2 (65) |
| C14 | 151.8 (75) |
| C15 | 99.8 (75, 69) |
| C16 | 173.0 (69) |
| C20 | 81.9 (38) |
| From [4-13C]ALA | |
| C1 | 83.4 (57.8) |
| C3 | 60.16 |
| C6 | 171.76 |
| C8 | 55.7 |
| C11 | 157.86 |
| C13 | 146.68 |
| C17 | 64.0 (33) |
| C19 | 151.4 (58.2) |
| Methyl groups from 13CH3-AdoMet | |
| C2 | 20.13 |
| C7 | 27.27 |
| C17 | 21.26 (33) |
| C20 | 18.77 (38) |
RESULTS AND DISCUSSION
Factor IV (4) was isolated from cell-free extracts of Pr. shermanii by esterification (MeOH/H2SO4) and multiple thin-layer chromatography. The presence of four methyl groups was deduced by the 3H/14C ratios of specimens derived from [methyl-3H3]-AdoMet and either 14C factor II (2) or 14C factor III (3) (Fig. 1, ∗ = 3H; • = 14C). Mass spectral analysis of the esters prepared from both MeOH (m/z 1046) and EtOH (m/z = 1144) revealed that seven ester groups were present (M + 98 for the ethyl analog); the eighth carboxylic function was a δ-lactone (FTIR: 1740 cm-1). When factor IV was isolated from incubations using [4-13C]ALA (1) and [methyl-13C]AdoMet as substrates, one-dimensional 13C-NMR analysis (see Table 1) of the resultant 13C-enriched heptamethyl ester (Fig. 1, factor IV 4: A = CH2CO2Me; P = CH2CH2CO2Me; • = 13C) showed characteristic C-C coupling (J = 58 Hz) between C-19 and C-1, which is evidence that ring contraction had occurred, whereas coupling (J = 33 Hz) between one of the four AdoMet-derived methyl groups and a propionate terminus (δ 64.0) suggested C-methylation at C-17. The observation of coupled doublets (J = 38 Hz) at δ 81.9 and 18.8 ppm in factor IV enriched from [5-13C]ALA and 13C-AdoMet (Fig. 1, ▪ and ∗ = 13C) indicates that C-20 and its attached methyl group are still present in the molecule. Further NMR analyses (distortionless enhancement for polarization transfer, heteronuclear multiple quantum correlation, heteronuclear multiple bond correlation) disclose that C-20 is appended to C-1, and bears a proton, a methyl group, and the oxygen terminus of the δ-lactone as the fourth substituent. The assignment of methyl resonances at δ 20.1 and δ 27.3 ppm to C-2 and C-7, respectively, reveals that while C-3 is in the natural (α) configuration, the C-8 propionate is β-oriented, thereby releasing the γ-effect on the adjacent C-7 methyl, a well-known phenomenon in such epimerizations (10, 11).
Incubation of 14C-labeled factor IV [biosynthesized from a mixture of 3-epi and 8-epi factor II (2a) with Pr. shermanii extracts] affords cobyrinic acid (5, isolated as cobester; A = CH2CO2Me; P = CH2CH2CO2Me) in 0.6% radiochemical yield [Fig. 1, (4) → (5)]. Epimerization at C-8 of factor II has obviously caused derailment, thereby allowing isolation of 8β-configured factor IV, which is partially reepimerized to the natural (3α, 8α) isomer to allow synthesis of (5), confirmed by conversion of a mixture of 3-epi and 8-epi factor II (2a) directly to cobyrinic acid (5) in a comparable 1.7% radiochemical yield, together with a substantial amount of 8-epi factor IV. Indirect evidence for inversion of stereochemistry at C-8 had previously been observed by the exchange of H for D at C-3, C-8, and C-13 when vitamin B12 was isolated from Pr. shermanii cells grown in 50% 2H2O (12). Factor IV, whose absolute stereochemistry at C-2, C-7, and C-17 follows from bioconversion to cobyrinic acid, has been oxidized during isolation, and like factor II must be reduced by enzyme(s) present in the cell-free extract (1, 2, 9) to the precorrin-4 oxidation level (Scheme SII) and then epimerized at C-8 to allow incorporation to proceed. It should be noted that precorrin-2 and -3 were first isolated in their oxidized forms, factors II and III, respectively, and are reduced back to the corresponding precorrins by reductive enzyme(s) present in the cell-free extracts of Pr. shermanii and thus reenter the biosynthetic pathway (9).
Scheme II.
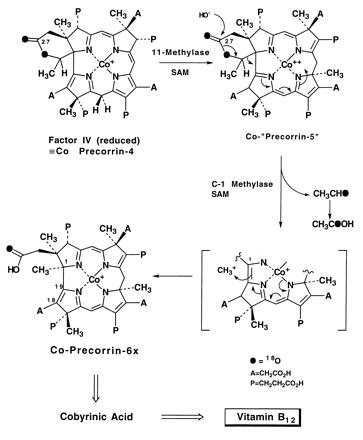
A mechanism for the formation of factor IV is proposed in Scheme SI and features δ-lactone formation at C-20 that delivers the oxygen required for subsequent loss of the “C2” unit. Formation of a δ-lactone from the ring A acetate to the C-20 position was first promulgated by Eschenmoser (13) as a means of functionalizing C-20 prior to ring contraction and as part of a scaffolding device for the ring contraction process. The discovery of the “aerobic” mechanism in which molecular oxygen serves as the source of the C-20 hydroxyl in precorrin-3x (=3B) (Scheme SI) led Eschenmoser to suggest recently (14) that involvement of the δ-lactone, however attractive as a chemical model, would probably require an oxidative step. The new structure of factor IV, together with the experimental demonstration of the transfer of 18O from the ring A carboxylate to the acetic acid isolated in the anaerobic pathway (15) and the loss of 18O from the same carboxylate function (6, 7), now provide strong support for the renaissance of the nonoxidative “δ-lactone” hypothesis, although not quite in its original form. The ring contraction takes place by a mechanism that may involve a cyclopropane intermediate followed by ring opening to forge the A-D ring linkage, leaving the pendant C-20 at the oxidation level of acetaldehyde. Thirty years ago, before the origin (AdoMet) of the C-1 methyl group of B12 was discovered, Eschenmoser and colleagues (16, 17) had envisaged a cyclopropane intermediate formed by proton catalyzed reduction at the 20-meso position as part of the ring contraction process. Again, Eschenmoser (14) has referred to the “death of this cherished hypothesis” when the origin of the C-1 methyl group of B12 was found to be methionine (2) rather than the C-20 meso carbon. The biosynthesis of factor IV may indeed involve just such a cyclopropane intermediate (Scheme SI), which subsequently loses C-20 as acetaldehyde (→ acetic acid), thereby reviving one of the earliest concepts for ring contraction. Several alternatives remain, including proton catalyzed ring closure of a seco-corrinoid (18, 19). Thus, in cobalt precorrin-4 (Scheme SII) the ring contraction is orchestrated not by molecular oxygen, but by internal delivery of oxygen functionality that in turn could be mediated by remote interaction of cobalt with the carboxylate function in ring A and/or addition of the fourth C-methyl group in ring D at position 17 or 18. The latter position is favored stereoelectronically but would require subsequent migration of the methyl to C-17, just as is found for C-11 → C-12 methyl shift in the aerobic pathway. Subsequent hydrolysis of the δ-lactone rationalizes the specific exchange of 18O at C-27 (6, 7) and the transfer of 18O to C-20 (15) and is coupled with retro-aldol release of acetaldehyde [from the putative Co-precorrin-5 (Scheme SII)]. The excision of acetaldehyde requires revision of a previous mechanism (20, 21) based on loss of acetic acid, and would also necessitate a subsequent enzyme-catalyzed oxidation of the liberated acetaldehyde to acetic acid, which was isolated from Pr. shermanii extracts fed with 14C-labeled precursors (20, 21). Conclusive evidence for this scenario is provided in the accompanying paper by Wang et al. (22).
The suggested anaerobic pathway to B12 is shown in Scheme SII where it is assumed that the two pathways converge at precorrin -6x(23). Verification must await the discovery of the appropriate biosynthetic enzymes by analysis of a genomic library of Pr. shermanii from which the first methyl transferase (CobA) has recently been isolated (24). The unique structure of factor IV adds strength to the postulate that all of the anaerobic intermediates from precorrin -2 onward are cobalt complexes (5, 25) and explains why attempts to elicit the appropriate enzyme activities from cloned Salmonella genes (26) that are responsible for corrin synthesis have so far been unsuccessful using cobalt-free substrates, although some crossover between the aerobic, metal-free intermediates precorrin -6x and -8x was observed when these were incorporated (albeit in low yield) into cobyrinic acid in cell-free extracts of Pr. shermanii (23).
Acknowledgments
We thank Prof. A. Eschenmoser for an enlightening discussion of the proposed mechanism. This work was supported by the National Institutes of Health, the Robert A. Welch Foundation (grant to A.I.S.), and the Deutsche Forschungsgemeinschaft (grant to G.M.).
Footnotes
Abbreviations: ALA, δ-aminolevulinic acid; AdoMet, S-adenosylmethionine.
References
- 1.Blanche F, Cameron B, Crouzet J, Debussche L, Thibaut D, Vuilhorgne M, Leeper F J, Battersby A R. Angew Chem Intl Ed Engl. 1995;34:383–411. [Google Scholar]
- 2.Scott A I. Tetrahedron. 1994;50:13315–13334. [Google Scholar]
- 3.Scott A I. Heterocycles. 1994;39:471–476. [Google Scholar]
- 4.Spencer J B, Stolowich N J, Roessner C A, Min C, Scott A I. J Am Chem Soc. 1993;115:11610–11611. [Google Scholar]
- 5.Müller G, Zipfel F, Hlineny K, Savvidis E, Hertle R, Traub-Eberhard V, Scott A I, Williams H J, Stolowich N J, Santander P J, Warren M J, Blanche F, Thibaut D. J Am Chem Soc. 1994;113:9893–9895. [Google Scholar]
- 6.Scott A I, Stolowich N J, Atshaves B P, Karuso P, Warren M J, Williams H J, Kajiwara M, Kuramaya K, Okazaki T. J Am Chem Soc. 1991;113:9891–9892. [Google Scholar]
- 7.Vishwakarma, R. A., Balachandran, S., Alanine, A. I. D., Stamford, N. P. J., Kuichi, F., Leeper, F. J. & Battersby, A. R. (1993) J Chem. Soc. Perkin Trans. 1 2893–2898.
- 8.Stolowich N J, Wang J, Spencer J B, Roessner C A, Santander P J, Scott A I. J Am Chem Soc. 1996;118:1657–1662. [Google Scholar]
- 9.Battersby, A. R., Frobel, K., Hammerschmidt, F. & Jones, C. (1982) J. Chem. Soc. Chem. Commun. 455–458.
- 10.Scott A I, Townsend C A, Cushley R J. J Am Chem Soc. 1973;95:5759–5761. doi: 10.1021/ja00798a053. [DOI] [PubMed] [Google Scholar]
- 11.Battersby, A. R., McDonald, E., Neier, R. & Thompson, M. (1979) J. Chem. Soc. Chem. Commun. 960–962.
- 12.Scott A I, Kajiwara M, Santander P J. Proc Natl Acad Sci USA. 1987;84:6616–6620. doi: 10.1073/pnas.84.19.6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eschenmoser A. Angew Chem Int Ed Engl. 1988;27:5–39. [Google Scholar]
- 14.Eschenmoser A. CIBA Found Symp. 1994;180:310–323. [Google Scholar]
- 15.Arigoni D. CIBA Found Symp. 1994;180:280–281. [Google Scholar]
- 16.Wehrli, P. (1967) Ph.D. dissertation (Eidgenössiche Technische Hochschule, Zurich).
- 17.Muller P M, Farooq S, Hardegger B, Salmond W S, Eschenmoser A. Angew Chem Int Ed Engl. 1973;12:914–916. [Google Scholar]
- 18.Yamada Y, Miljkovic D, Wehrli P, Golding B, Löliger P, Keese R, Müller K, Eschenmoser A. Angew Chem Int Ed Engl. 1969;8:343–348. doi: 10.1002/anie.196903431. [DOI] [PubMed] [Google Scholar]
- 19.Ofner S, Rasetti V, Zehnder B, Eschenmoser A. Helv Chim Acta. 1981;64:1431–1443. [Google Scholar]
- 20.Mombelli L, Nussbaumer C, Weber H, Müller G, Arigoni D. Proc Natl Acad Sci USA. 1981;78:9–10. doi: 10.1073/pnas.78.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battersby A R, Bushell M J, Jones C, Lewis N G, Pfenninger A. Proc Natl Acad Sci USA. 1981;78:13–15. doi: 10.1073/pnas.78.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Stolowich N I, Santander P J, Scott A I. Proc Natl Acad Sci USA. 1996;93:14320–14322. doi: 10.1073/pnas.93.25.14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanche F, Thibaut D, Debussche L, Hertle R, Zipfel F, Müller G. Angew Chem Int Ed Engl. 1993;32:1651–1653. [Google Scholar]
- 24.Sattler I, Roessner C A, Stolowich N J, Hardin S H, Harris-Haller L W, Yokubaitis N T, Murooka Y, Hashimoto Y, Scott A I. J Bacteriol. 1995;177:1564–1569. doi: 10.1128/jb.177.6.1564-1569.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balachandran, S., Vishwakarma, R. A., Monaghan, S. M., Prelle, A., Stamford, N. P. J., Leeper, F. J. & Battersby, A. R. (1994) J. Chem. Soc. Perkin Trans. 1 487–491.
- 26.Roessner C A, Warren M J, Santander P J, Atshaves B P, Ozaki S-I, Stolowich N J, Iida K, Scott A I. FEBS Lett. 1992;301:73–78. doi: 10.1016/0014-5793(92)80213-z. [DOI] [PubMed] [Google Scholar]