

Abstract
It has been proved that, during anaerobic biosynthesis of the corrin macrocycle, the two-carbon fragment excised from the precursor, precorrin-3, is acetaldehyde, which originates from C-20 and its attached methyl group. This apparently contradictory finding is rationalized in terms of the subsequent enzymatic oxidation of acetaldehyde to acetic acid, which was previously regarded as the volatile fragment released by the action of the biosynthetic enzymes of Propionibacterium shermanii. The observation that acetaldehyde (rather than acetic acid) is extruded during anaerobic B12 synthesis is in full accord with the structure of factor IV, a new intermediate on the pathway.
Keywords: acetaldehyde, 13C-NMR spectroscopy, Propionibacterium shermanii
During an investigation of vitamin B12 biosynthesis in Propionibacterium shermanii, we discovered a new intermediate, factor IV, whose structure (Scheme SI) contains several important clues pertaining to the sequence of corrin biosynthesis and to the loss of a two-carbon fragment during B12 formation (1).
Scheme I.
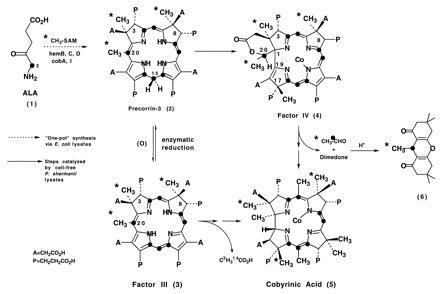
Factor IV is a cobalt complex of a tetradehydrocorrin (corresponding to ring-contracted precorrin-3) that still retains the two-carbon unit embedded in a δ-lactone and that has undergone C-methylation at C-17. The presence of a proton on the C-20 carbon, which also bears a methyl group and serves as the terminus of the δ-lactone function, requires that subsequent hydrolysis of the δ-lactone would release a two-carbon fragment as acetaldehyde. However, in two earlier studies (2, 3), it was shown that incubation of the 14C/3H labeled precursor, factor III (≡ oxidized precorrin-3), with a P. shermanii extract afforded ca. 0.75 equivalents of doubly labeled acetic acid after a 16-hr incubation, together with 1 equivalent of radiolabeled cobyrinic acid. To reconcile these apparently contradictory findings, the identity of the “two-carbon fragment” has been reinvestigated by a trapping experiment in which the excised species is isolated as soon as it is liberated by the enzymes of the cell-free extract.
MATERIALS AND METHODS
Materials.
Dimedone was a product of Aldrich. Acetaldehyde ([1,2-13C], 99%) and l-[13CH3]methionine were purchased from Cambridge Isotope Laboratories (Cambridge, MA). All other biochemicals were purchased from Sigma. [5-13C]5-aminolevulinic acid was prepared as described (4). [13CH3]S-adenosylmethionine (AdoMet) was synthesized from [13CH3]methionine as described (5). Precorrin-3 was prepared by a recently described modification (6) of the original procedure (7).
Preparation of Cell-Free Extract of P.shermanii.
Freshly harvested wet cells (100 g) of P. shermanii were suspended in potassium phosphate buffer (100 ml; 0.01 M; pH 7.6) and sonicated for 50 min at 0–10°C. After centrifugation at 17,000 × g for 25 min at 0°C, the supernatant was used for the following experiments.
Incubation of Precorrin-3 with Cell-Free Extracts of P. shermanii.
Precorrin-3 (30 mg) prepared from [5-13C]5-aminolevulinic acid (4), and [13CH3]AdoMet was added to degassed cell-free homogenate (120 ml, pH 7.6) containing the following ingredients: 80 mg AdoMet/80 mg MgCl2·6H2O/50 mg NADH/25 mg NADPH/35 mg CoCl2·6H2O/100 mg dimedone. After incubation overnight in the dark, air was admitted to the flask and the pH was adjusted to 4.5 with HCl (2 M). The resulting mixture was extracted with CH2Cl2 (400 ml three times). The combined organic phase was dried over anhydrous Na2SO4 and evaporated. The mixture of unlabeled formaldehyde–dimedone, acetaldehyde–dimedone, and labeled acetaldehyde–dimedone adducts was isolated by chromatography on silica gel using n-hexane-ethyl acetate (2:1). Acetic acid (5 ml) was then added to the mixture and the resulting solution was refluxed for 6 hr. After evaporation of solvent in vacuo, labeled and unlabeled acetaldehyde–dimedone–anhydride adducts were purified by preparative thin-layer chrmoatography (silica) using n-hexane-ethyl acetate (2:1). The adducts were used directly for the NMR determinations as described in Fig. 1.
Figure 1.
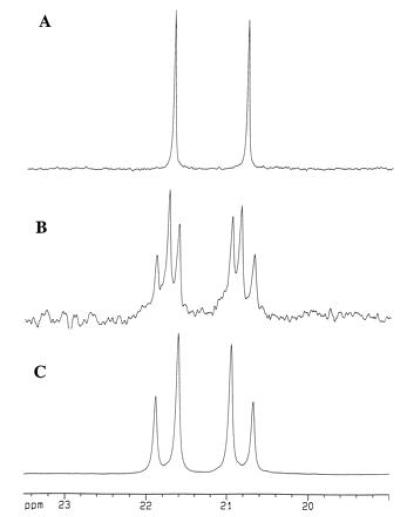
Selected region of the 125 MHz 13C NMR spectra (CDCl3) of anhydro-dimedone adducts of acetaldehyde detailing the methyl and methine carbons derived from acetaldehyde. Spectrum A is that of the adduct derived from P. shermanii extracts using unlabeled precorrin-3 as a control. Spectrum B resulted from an overnight incubation of doubly labeled precorrin-3 (as shown in Scheme SI) with P. shermanii extracts as described in the text. Spectrum C represents a fully 13C-enriched standard derived nonenzymatically from pure [1,2-13C]acetaldehyde.
RESULTS AND DISCUSSION
A sample of 13C-enriched precorrin-3 (2) prepared from [5-13C]5-aminolevulinic acid (Scheme SI, •) and [13CH3]AdoMet (Scheme SI, ∗) and labeled as shown in Scheme SI was incubated anaerobically in a cell-free extract of P. shermanii containing NADH, NADPH, CoCl2, and dimedone (pH = 7.6). After 16 hr, the pH was adjusted to 4.5, and a mixture of formaldehyde and acetaldehyde–dimedone derivatives (8) was extracted into CH2Cl2. Conversion of this mixture to the corresponding anhydro derivatives with acetic acid and chromatographic separation (9) afforded the pure acetaldehyde adduct 6. Inspection of the 13C-NMR spectrum (Fig. 1B) revealed that both methyl (∗) and methine (•) carbons (see 6, Scheme SI) were enriched above natural abundance in 13C and furthermore were coupled (J = 34.3 Hz, Fig. 1B), thereby providing unequivocal evidence for the extrusion of acetaldehyde during the bioconversion of cobalt precorrin-3 to cobyrinic acid. The spectrum of the acetaldehyde adduct at natural abundance (Fig. 1A) was obtained from a control experiment using unlabeled precorrin-3 and shows that the incubation generates “background” acetaldehyde (ca. 1–2 mg per 100 ml) from a source other than precorrin-3, whose concentration at natural abundance would be insufficient to detect any acetaldehyde formed from the unlabeled precorrin. The coupled doublets display a slanted “AB” coupling pattern due to their close chemical shift proximity, confirmed by comparison with the spectrum of an authentic sample of doubly labeled acetaldehyde–dimedone adduct (Fig. 1C).
Comparison of the 13C signal heights in the NMR spectrum of the acetaldehyde adduct 6 and that of cobester (5; Scheme SI, A = CH2CO2Me; P = CH2CH2CO2Me) isolated from the same experiment established that approximately 0.8–0.9 equivalents of acetaldehyde is formed. In the control experiment (without enzymes), the amount of detectable acetaldehyde fell below the sensitivity of the NMR experiment (<0.1 equivalent).
To explain the isolation of 0.6–0.8 equivalents of acetic acid reported previously (2, 3), we postulate that redox enzyme(s) present in the cell-free extract are capable of oxidizing acetaldehyde to acetic acid, since the only difference between the two approaches lies in the timing of the trapping of product. Experimental support for this concept was obtained when doubly labeled acetaldehyde (13CH313CHO) was added to the cell-free extract, and the 13C NMR time course of the incubation was observed. Initially, only signals originating from both the aldehyde (δ 208 ppm) and hydrate (δ 90 ppm) forms of acetaldehyde were observed. After 3 hr, a small amount (<10%) of doubly labeled ethanol had been produced, which increased after 6 hr (13CH313CH2OH: δ 18.5, 60 ppm; J = 37 Hz). The signals for doubly labeled acetate (13CH313CO2H: δ 25, 182 ppm; J = 51 Hz) were also apparent at 6 hr, whereas at the end of the incubation (16 hr) all of the acetaldehyde had been converted to a mixture of ethanol/acetic acid (≈1:2 ratio). In the previous analysis of the bioconversion of precorrin-3 to cobyrinic acid, the amount of labeled acetic acid isolated was normally less than 1 equivalent, the remaining radioactivity presumably being in the ethanol, i.e., both C-2 species are formed from the liberated acetaldehyde (10).
As previously noted (2, 3), the identity of the volatile two-carbon fragment assumes major importance in any mechanism proposed for B12 biosynthesis, particularly for the ring contraction, the subsequent hydrolysis step (which extrudes the C-2 unit), and the overall oxidation level maintained throughout the pathway.
By analogy with the aerobic route to B12 in Pseudomonas denitrificans in which the 2-carbon unit is indeed liberated as acetic acid (11), we have suggested (1) that an intermediate of the latter pathway, precorrin-5 (12), has a cobalt-containing counterpart in the anaerobic series, and that it is from this or a closely related species that the hydrolytic loss of acetaldehyde takes place, with subsequent C-methylation (at C-1) leading to cobalt precorrin-6x, which has been promulgated as an intermediate in the anaerobic pathway (13). The endogenous formation of both formaldehyde and acetic acid in P. shermanii has for many years complicated the analysis of this system and has contributed to the complexity of the problem. Thus, the enzymatic conversion of acetaldehyde to acetic acid in the cell-free system could not have been foreseen and turned out to be a “hidden trap” for acetaldehyde. Similarly, in an experiment performed almost 20 years ago (14), a dimedone adduct, believed to be that of formaldehyde, was isolated from incubations of P. shermanii with 14C precursors. At that time, we were unaware that the adducts of formaldehyde and acetaldehyde cannot be separated chromatographically unless they are first converted (9) to the corresponding anhydro forms (as 6). The explanation for our earlier observation therefore resides in the co-crystallization of the (carrier) formaldehyde–dimedone adduct with the 14C-labeled acetaldehyde adduct. Now that the true identity of the two-carbon unit has been discovered, all of the earlier observations can be reinterpreted and thus are no longer mutually exclusive.
A new theory for anaerobic corrin biosynthesis, based on the structure of factor IV and requiring the liberation of acetaldehyde, has been presented (1) and finds experimental support in this work. The salient features of the theory include mechanistic proposals for the exchange of 18O at the C-27 carbonyl (15), the transfer of 18O from the ring A acetate carboxyl (C-27) to the acetic acid isolated after prolonged incubation (16), and the role of cobalt in orchestrating the C-methylation at C-17 or C-18 prior to ring contraction (1).
Acknowledgments
We thank Prof. G. Müller (Stuttgart) for advice on the trapping and analysis of acetaldehyde. This work was suppported by the National Institutes of Health and the Robert A. Welch Foundation.
Footnotes
Abbreviation: AdoMet, S-adenosylmethionine.
References
- 1.Scott A I, Stolowich N J, Wang J, Gawatz O, Fridrich E, Müller G. Proc Natl Acad Sci USA. 1996;93:14316–14319. doi: 10.1073/pnas.93.25.14316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mombelli L, Nussbaumer C, Weber H, Müller G, Arigoni D. Proc Natl Acad Sci USA. 1981;78:11–12. doi: 10.1073/pnas.78.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Battersby A R, Bushell M J, Jones C, Lewis N G, Pfenninger A. Proc Natl Acad Sci USA. 1981;78:13–15. doi: 10.1073/pnas.78.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfaltz A, Anwar S. Tetrahedron Lett. 1984;25:2977–2981. [Google Scholar]
- 5.Park J, Tai J, Roessner C A, Scott A I. Bioorg Med Chem Lett. 1995;5:2203–2206. [Google Scholar]
- 6.Stolowich N J, Wang J, Spencer J B, Santander P J, Roessner C A, Scott A I. J Am Chem Soc. 1996;118:1657–1662. [Google Scholar]
- 7.Warren M J, Roessner C A, Ozaki S-I, Stolowich N J, Santander P J, Scott A I. Biochemistry. 1992;31:603–609. doi: 10.1021/bi00117a043. [DOI] [PubMed] [Google Scholar]
- 8.Vorlander D. Z Anal Chem. 1929;77:242–268. [Google Scholar]
- 9.Friedle, J. (1982) Ph.D. dissertation (Univ. Stuttgart, Germany).
- 10.Stadtman E R, Burton R M. Methods Enzymol. 1955;1:518–523. [Google Scholar]
- 11.Blanche F, Cameron B, Crouzet J, Debussche L, Thibaut D, Vailhorgne M, Leeper F J, Battersby A R. Angew Chem Intl Ed Engl. 1995;34:383–411. [Google Scholar]
- 12.Min C, Atshaves B P, Roessner C A, Stolowich N J, Spencer J B, Scott A I. J Am Chem Soc. 1993;115:10380–10381. [Google Scholar]
- 13.Blanche F, Thibaut D, Debussche L, Hertle R, Müller G. Angew Chem Intl Ed Engl. 1993;32:1651–1653. [Google Scholar]
- 14.Kajiwara M, Ho K S, Klein H, Scott A I, Gossauer A, Engel J, Neumann E, Zilch H. Bioorg Chem. 1977;6:397–402. [Google Scholar]
- 15.Scott A I, Stolowich N J, Atshaves B P, Karuso P, Warren M J, Williams H J, Kajiwara M, Kuramaya K, Okazaki T. J Am Chem Soc. 1991;113:9891–9892. [Google Scholar]
- 16.Arigoni D. CIBA Found Symp. 1994;180:280–281. [Google Scholar]