

Abstract
The laboratory rat (Rattus norvegicus) is a key tool for the study of medicine and pharmacology for human health. A large database of phenotypes for integrated fields such as cardiovascular, neuroscience, and exercise physiology exists in the literature. However, the molecular characterization of the genetic loci that give rise to variation in these traits has proven to be difficult. Here we show how one obstacle to progress, the fine-mapping of quantitative trait loci (QTL), can be overcome by using an outbred population of rats. By use of a genetically heterogeneous stock of rats, we map a locus contributing to variation in a fear-related measure (two-way active avoidance in the shuttle box) to a region on chromosome 5 containing nine genes. By establishing a protocol measuring multiple phenotypes including immunology, neuroinflammation, and hematology, as well as cardiovascular, metabolic, and behavioral traits, we establish the rat HS as a new resource for the fine-mapping of QTLs contributing to variation in complex traits of biomedical relevance.
The rat has for long been a favored organism for physiological and behavioral analyses and is increasingly attracting the attention of geneticists (Jacob and Kwitek 2002). Over the last century, a wealth of disease models have been developed, which compared with the mouse have proved easier to analyze at an organ and cellular level because of the rat’s larger size. Rat models of cardiovascular disease, inflammatory diseases, and susceptibility to cancer and toxic substances have been crucial in understanding the biology of common human disorders. The rat has also been a focus of classical neuroanatomical studies and electrophysiological slice studies; rat experiments have been critical for understanding many neurobiological processes, including learning and memory, and for providing models for the neuropsychology of human behavioral disorders (Weiss and Feldon 2001).
Genetic analyses of experimental crosses have led to the localization of hundreds of rat quantitative trait loci (QTLs) containing genes that confer susceptibility to the disease models (Flint et al. 2005). However, as with the study of complex traits in other model organisms, few complex trait genes per se have been identified, relative to the number of QTLs. One obstacle to progress has been the difficulties besetting attempts to refine QTL intervals, initially identified in inbred strain crosses, to gene-level resolution. Here we apply a strategy developed in the mouse that combines the identification and fine-mapping of QTLs in one population (Mott et al. 2000; Valdar et al. 2006b). The method exploits recombinants that have accumulated over many generations of outbreeding in genetically heterogeneous stocks (HS) that are derived from inbred strains.
Theoretically, an HS makes genome-wide genetic association studies possible, as has been shown in the mouse, where 843 QTLs with an average 95% confidence interval of 2.8 Mb were identified for 97 phenotypes (Valdar et al. 2006b). However, the suitability of an HS for the analysis of complex traits depends not only on its genetic constitution, but also on the quality and nature of the phenotypes that it can deliver (Solberg et al. 2006). For an HS to be widely exploited by the rat genetics community, it needs to be shown that HS phenotypes are commensurate with those found in standard laboratory strains and, so that the large genotyping costs incurred in whole genome association are minimized, that multiple phenotypes can be measured on individual animals. Obtaining multiple phenotypes from each animal raises problems of interaction between phenotypes (particularly for behavioral assays) and potentially limits the amount of information that can be obtained.
Here we explore the potential of a rat HS for high-resolution mapping of multiple quantitative traits. The rat HS (hereafter NIH-HS) was established in the 1980s in NIH and is descended from eight inbred progenitors: BN/SsN, MR/N, BUF/N, M520/N, WN/N, ACI/N, WKY/N, and F344/N (Hansen and Spuhler 1984). Three strains (MR/N, WN/N, and WKY/N) trace their ancestry to Wistar stock. Since its creation, the stock has been bred using a standard outbreeding regime through its fiftieth generation.
Our experiment had two aims: to determine the mapping resolution obtainable for an average QTL expected to be segregating in the stock, and to determine the stock’s suitability for whole genome association. To address the first issue, we focused our analysis on chromosome 5, since we have mapped a locus controlling fear-related behaviors to this chromosome (FernándezTeruel et al. 2002), in an F2 intercross between the Roman high avoidance (RHA) and low avoidance (RLA) strains (derived from the Wistar stock) (Driscoll and Battig 1982). This locus explained 5.7% of the variance in two-way active avoidance in a shuttle box, an effect size typical of those identified in many crosses between inbred strains (Flint et al. 2005). To address the second issue, we developed protocols for mapping multiple phenotypes in the same animal where laboratories with the necessary expertise for each phenotype were contributing.
Results
We assessed the genetic constitution of the stock using 786 NIH-HS rats. After phenotyping, we selected and genotyped 83 markers on chromosome 5 and 27 markers on chromosome 15 using the rat single nucleotide polymorphism (SNP) database (Saar et al. 2008). We thus obtained an average intermarker interval of ∼350 kilobases. Four markers were monomorphic, consistent with an expected rate of fixation of about 5% (Valdar et al. 2006b).
Figure 1 shows the decay in linkage disequilibrium (LD, measured as r2), with distance (Mb). We calculated LD taking into account family relationships (Abecasis et al. 2002). The results indicate that LD decays to nonsignificant levels within 3 Mb, consistent with expectations that the rat HS can deliver high-resolution mapping and in agreement with the data from our mouse experiment (Valdar et al. 2006b). We found no significant correlation between markers on different chromosomes.
Figure 1.

Linkage disequilibrium decay in the NIH-HS rats. The figure plots the decay in mean r2 (R2) on the vertical axis with distance (megabases) on the horizontal axis. The data are from 110 SNP markers on chromosomes 5 and 15.
We mapped variation in the number of shuttle-box avoidances (obtained over 40 trials) using the genotypes on chromosome 5. The NIH-HS is a structured population, containing animals of different degrees of relatedness, so that the significance threshold cannot be obtained by permuting the relationship between genotypes and phenotypes (Valdar et al. 2006b). In our analysis of the HS mice, our solution was to average over multiple QTL models (Valdar et al. 2006b). In this method, the apparent association between phenotype and genotype at any one locus is corrected by the pattern of associations over the rest of the genome. However, in the rat, HS genotypes were available for only two chromosomes, so we could not use the remainder of the genome as we had done previously. Instead, we applied a conservative threshold determined by simulating phenotypes that incorporate sibship correlation structure (see Methods). After applying this strict threshold, we detected a single QTL estimated to explain 7.1% of the phenotypic variance (Fig. 2).
Figure 2.

Genetic mapping of shuttle box avoidances on chromosome 5. The solid line shows the strength of association (y-axis) between the phenotype and the estimated haplotypes at each locus. The x-axis gives the location on chromosome 5 in megabases. The y-axis units are the P-values, expressed as negative logarithms to the base 10 (i.e., logPs), from the likelihood ratio test of association. A conservative 5% chromosome-wide significance threshold was calculated by simulation (dashed line) to help interpret these values. The 95% confidence interval of the QTL (93.37–106.16 Mb), determined by positional bootstrapping (see Methods), is shown as a gray bar.
We mapped the most likely position of the QTL by nonparametric bootstrapping (Manichaikul et al. 2006). The 95% confidence interval spans from 95.39–106.16 Mb (Fig. 2). Since at this distance genotype correlation between the flanking markers is less than 0.1, the effect may arise from more than one genetic effect, as suggested by the distribution of the bootstrapped peak positions (Fig. 3).
Figure 3.

Positional bootstraps of the QTL on chromosome 5. The figure shows the number of times a QTL peak occurred at locations over a 30-Mb region on chromosome 5. A total of 500 bootstrap samples were analyzed. The vertical axis shows the percentage of frequency of the peak positions; the horizontal axis gives the distance in megabases (Mb).
We next set out to develop protocols for mapping multiple phenotypes. In collaboration with six European laboratories, with expertise in different areas of biomedical research, we developed a continuous battery of assays that started when rats were 5 wk old with the implantation of a microchip, a blood sample for hormonal measures, and an ear punch for wound healing measurements. Thereafter, the rats were subjected to behavioral tests, immune cell phenotyping, blood pressure measurements, and a glucose tolerance test and immunized for experimental autoimmune encephalomyelitis (EAE). The protocol and the participating laboratories are summarized in Table 1.
Table 1.
Overview of the high-throughput phenotyping protocol

For each test, we recorded environmental factors such as experimenter, time of day, and humidity to enable the study of gene by environment interactions (Valdar et al. 2006c). Table 1 gives the order of tests and the age at which they were carried out. We did not explore the effects of altering the order of tests. We collected phenotype data for 220 rats and present the results in Tables 2 and 3. We compared our results with those reported for the parental strains (hematology, blood pressure, organ and body weights) (Mashimo et al. 2005) and show in Table 2 that our findings are compatible with those reported in the literature. We also measured the immunology phenotypes in three of the parental strains that we have available (Table 2).
Table 2.
Means and standard deviations of phenotypes in NIH-HS animals and their eight progenitor strains

ND, not determined.
Table 3.
Means and standard deviations of behavioral phenotypes in NIH-HS animals and RHA and RLA inbred strains

One particular concern was whether the HS rats would be useful for dissecting organ specific immune-mediated disease because of the large effect of genes in the MHC region. EAE and MS are controlled by both multiple sclerosis histocompatibility complex (MHC) and non-MHC genes. The non-MHC influence is likely to be controlled by many genes, each with small effects (Olsson et al. 2006), while major genetic effects are dependent on haplotypes of the MHC. This effect is well established in multiple sclerosis (Olerup and Hillert 1991) and in myelin oligodendrocye glcoprotein (MOG)-induced EAE (Weissert et al. 1998), and most previous F2 crosses and experiments in advanced intercross lines have therefore used strain combinations with differing non-MHC genes, but identical MHC haplotypes, to increase the power for detecting non-MHC QTLs (Becanovic et al. 2006).
Non-MHC QTLs are the primary target for the HS studies since the MHC complex, and in particular the class II genes, are well characterized and studied by other means. The currently used HS stock has a variety of different MHC haplotypes that potentially could lessen the power for detecting non-MHC QTLs. It was therefore important to perform a pilot study in the HS stock with the intended EAE model to establish if there was enough variation in disease outcome depending on non-MHC genes. If so, the setup could offer advantages in that gene–gene interactions with a variety of MHC haplotypes potentially can be unraveled in a large scale experiment.
Table 4 shows our findings and those reported in literature regarding EAE susceptibility in the NIH-HS founder strains. To ensure that influence from non-MHC genes could be mapped in this population, we performed a pilot study of 25 rats to determine the variance of phenotypes between and within MHC types. Two homozygous MHC groups could be identified (AV1 and L), while the other group contains MHC heterozygotes. The large phenotype variation within the various MHC haplotype groups strongly suggest influence of non-MHC genes that can be mapped in the NIH-HS, which is why we went ahead and phenotyped EAE also in the bigger NIH-HS experiment. The results are presented in Table 5.
Table 4.
Experimental autoimmune encephalomyelitis response in NIH-HS progenitor strains

MHC indicates major histocompatibility complex; EAE, experimental autoimmune encephalomyelitis; MOG, myelin oligodendrocyte glycoprotein; SC, spinal cord; MBP, myelin basic protein; and ND, not determined.
Table 5.
Range of experimental autoimmune encephalomyelitis phenotypes within MHC types (in pilot experiment) and in the NIH-HS rats

Incidence of EAE (INC) is defined as 0 = absent and 1 = present; onset of EAE (ONS) is defined as the first day that clinical signs are observed; duration of EAE (DUR) is the number of days with signs of disease; maximum EAE score (MAX) is the highest score obtained during the experiment; and cumulative EAE score (CUM) is the sum of all scores received during the experiment.
Discussion
We have performed the first genetic study in HS rats, proving the potential of the NIH-HS to identify and to fine-map QTLs. Previously, using an F2 intercross, we mapped a QTL to a 40-cM interval on chromosome 5 that influences behavior in multiple models of anxiety, including the acquisition of two-way active avoidance (Fernández-Teruel et al. 2002). Here we report the analysis of chromosome 5 in 786 NIH-HS rats for the avoidance phenotype and the identification of at least one QTL located between 93.37 and 106.16 Mb, a region containing nine genes, none of which has been previously shown to influence anxiety-related behavior.
Our genetic analysis of the rat HS was confined to the analysis of two chromosomes. However, they are likely to be representative of the genetic structure of other chromosomes for the following reasons. Analysis of the available genotypes from the progenitors of the rat HS structure indicates that there are no large (>2 Mb) regions without SNPs and no unusually large regions of LD (Saar et al. 2008). It should be noted that compared with the mouse, there is less variation in the extent and amount of large LD and haplotype blocks in the rat genome (Saar et al. 2008). We have no reason to expect large differences between the progenitor chromosomes.
However, observations made of inbred strains do not necessarily reflect what will be found in crosses derived from them. Stochastic allele fluctuations may give rise to large regions of LD, and this cannot be definitely excluded without whole genome analysis. Nevertheless, the data presented here, together with data from the mouse HS (Valdar et al. 2006b) and simulations of the HS breeding scheme (Valdar et al. 2006a), indicate that this is unlikely. Therefore, we believe that our results are typical of the rest of the genome and that the NIH-HS can be used for whole genome association studies.
It should be stressed that our advance depends on the deployment of novel analytical methods. The NIH-HS is a genetically structured population in which individuals are genetically related to different degrees. The need to take into account family structure while fitting multiple QTLs, as well as environmental and other covariates means that methods developed for whole-genome analysis of a classical intercross (Ball 2001; Broman and Speed 2002; Morris et al. 2002; Sillanpaa and Corander 2002) are not directly applicable: In an HS, more loci are tested than individuals, so it is not possible to use methods that fit all markers simultaneously.
A necessary next step for the widespread use of the NIH-HS is the demonstration that phenotypes in the stock are comparable with those obtained using classical laboratory rats. We have developed a phenotyping protocol capable of capturing many aspects of the rat’s behavior and physiology, and we have shown that the measures we obtain are indeed consistent with those reported for inbred strains.
In contrast to standard QTL mapping experiments, where each laboratory maintains, breeds, phenotypes, and genotypes its own animals, we brought scientists with different phenotyping skills from different laboratories to a single site where the animals are bred. This model brings considerable savings, since each animal provides a considerable amount of information. The breadth of phenotypic and genotypic information can later be combined with expression data to provide the basis for a systems biology approach to complex phenotypes in general.
A current major challenge in biology is to understand how phenotypes arise from the resulting complex web (Hartwell et al. 1999; Barabasi and Oltvai 2004). Critical to this endeavor is the collection of sufficiently dense data sets, integrating data from genetic variants, gene transcription, and cellular components, and from whole body experiments, to identify networks. The combined approach to using the NIH-HS rats will provide a significant international resource for systems biology applications.
Methods
Animals
The rat NIH-HS used originate from the colony established in the 1980s in NIH (Hansen and Spuhler 1984). Since its creation, the stock has been bred using a rotational outbreeding regime in order to minimize the extent of inbreeding, drift, and fixation (Boucher and Cotterman 1990). The scheme is implemented as follows: Given a population of 2m individuals, split equally between the sexes, couples are put into cages labeled 1–m. Cages labeled are assigned 1′–m′ for the next generation. The female from cage 1 is mated with the male from cage 2, and their offspring are placed in cage 1′. Similarly, the female from cage k is mated with the male from cage k + 1 (or the male from cage 1 if k = m) and offspring placed in cage k′, and so on for all remaining k. The procedure is repeated for each generation.
Forty breeding pairs of NIH-HS rats were sent from Northwestern University (Chicago) and established at the Autonomous University of Barcelona to generate animals for phenotyping. Subjects used for the chromosome 5 QTL study were 786 N/NIH-HS rats, approximately half of each sex, which were 3 mo old at the beginning of the experiments (weight 250–350 g). Two hundred twenty NIH-HS rats were used to establish the high-throughput phenotyping protocol (125 males and 95 females). Animals were housed in pairs (males) or three and three (females) in macrolon cages (50 × 25 × 14 cm) and maintained with food and tap water available ad libitum, under conditions of controlled temperature (22°C ± 2°C) and a 12-h light-dark cycle (lights on at 0800 h). Experiments were performed during the light cycle, between 0900 and 1900 h in accordance with the Spanish legislation on “Protection of Animals Used for Experimental and Other Scientific Purposes” and the European Communities Council Directive (86/609/EEC) on this subject.
Genotyping
Information on polymorphic SNPs was provided through the effort of the STAR consortium to identify SNPs and haplotypes in the rat to assist complex trait analysis as part of the EURATools consortium. SNPs were selected with an algorithm taking into account the strain distribution pattern to maximize the information available for mapping in an HS. DNA was extracted from tails using a standard proteinase K protocol. Genotyping was performed using the Sequenom MassArray platform according to the manufacturer’s instructions (www.sequenom.com).
Chromosome 5 scan
We tested the association of the number of avoidances in 40 trials to variation in estimated haplotype composition along chromosome 5 using a combination of hierarchical binomial logistic regression and the multipoint haplotype reconstruction method HAPPY (Mott et al. 2000). HAPPY estimates the expected proportion of each founder haplotype between each consecutive pair of markers (marker interval) in each animal. The effect of haplotype variation at marker interval m on the number of shuttle-box avoidances yi of animal i was modeled as an overdispersed binomial such that
 |
where pi is the probability of avoidance in any one trial, logit(pi) is the log odds, xi is the sex of the animal, wi is its weight, gi(m) is the vector of haplotype proportions estimated by HAPPY for marker interval m, εi is a normally distributed random variate that accounts for overdispersion (e.g., Gelman and Hill 2007), and μ, βs, βw, βm, and σ2 are parameters estimated by the model. The statistical significance of association is judged by a likelihood ratio test (LRT) performed at each marker interval in turn that compares the fit of the above model against that of a model excluding the genetic term βmTgi(m). The LRT gives a P-value, which we report as a logP (i.e., the log to base 10 of the P-value).
Establishing genome-wide significance thresholds parametric bootstrap from a multilevel model incorporating sibship covariance
In an F2 cross, the sibship of an animal is statistically independent of any polygenic structure because the F1 generation parents are identical across sibships. In an HS intercross line, this is not the case. Rather, since parents are genetically distinct, the allocation of alleles to individuals varies by sibship, with the result that genetic structure and family structure, which incorporate influential locus-generic environmental and biological factors, are confounded. A conservative approach to establish significance thresholds is then to allow the null model of no QTLs to nonetheless incorporate the sibship correlation structure. We first fit the hierarchical model
 |
where sk[i] is the effect of the sibship k with animal i as its member and is a normally distributed random intercept with variance σs2. We then generate null phenotypes that mimic the observed covariate and sibship covariance structure by parametric bootstrap (Davison and Hinkley 2005) from the fitted model, first drawing new sibship effects from Sk∼N(0,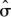 s2), and individual dispersion effects from εi∼N(0,2), and then drawing conditional phenotypes for each animal i from Yi | Sk, εi ∼ Binomial(40, logit−1
s2), and individual dispersion effects from εi∼N(0,2), and then drawing conditional phenotypes for each animal i from Yi | Sk, εi ∼ Binomial(40, logit−1 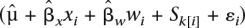 ), where carats denote parameter estimates from fitting the model in Equation 2 to the data. The single locus model in Equation 1 is then fit to the simulated phenotypes and the logP from the most significant marker interval recorded. We repeated this 200 times to generate the (null) distribution of maximum logPs we would expect from genome scans of a phenotype affected by the observed covariate effects and sibship covariance structure. A generalized extreme value distribution (GEV) was fit to these maxima (Valdar et al. 2006b), and from that fitted distribution, the α = 0.05 upper-tail quantile, our estimate for the 5% genome-wide significance threshold, was estimated at logP = 3.6. All models were fit using the statistical language R (R-Development-Core-Team 2004) and the add-on packages lme4 and evd.
), where carats denote parameter estimates from fitting the model in Equation 2 to the data. The single locus model in Equation 1 is then fit to the simulated phenotypes and the logP from the most significant marker interval recorded. We repeated this 200 times to generate the (null) distribution of maximum logPs we would expect from genome scans of a phenotype affected by the observed covariate effects and sibship covariance structure. A generalized extreme value distribution (GEV) was fit to these maxima (Valdar et al. 2006b), and from that fitted distribution, the α = 0.05 upper-tail quantile, our estimate for the 5% genome-wide significance threshold, was estimated at logP = 3.6. All models were fit using the statistical language R (R-Development-Core-Team 2004) and the add-on packages lme4 and evd.
Determining positional uncertainty of the mapped QTL
To determine the uncertainty in the location of the mapped QTL, we used positional bootstrapping (Visscher et al. 1996). Specifically, we sampled the observed animals with replacement to generate a nonparametric bootstrap resample of the data set. To each of 500 of such resampled data sets, we performed chromosome scans using Equation 1 and recorded the marker interval showing the strongest association to the phenotype. We then used the distribution of those top marker intervals to characterize the uncertainty in QTL location, reporting the Mb positions covered by the central q% of the distribution as the q% confidence interval.
Two-way active, shuttle-box avoidance acquisition
Active avoidance acquisition sessions were performed in three identical shuttle boxes (Letica Instruments), each one placed in independent sound-attenuating boxes consisting of two equal-sized compartments (25 × 25 × 28 cm) connected by an opening (8 × 10 cm). Rats were allowed a 4-min period of familiarization to the box. Immediately after that period, a 40-trial session/rat was administered, each trial consisting of a 10-sec CS (conditioned stimulus; 2400 Hz, 63-dB tone plus a 7-W small light) followed after termination by a 20-sec US (unconditioned stimulus; scrambled 0.7-mA foot shock) delivered through the grid floor. Crossings to the other compartment during the CS (avoidances) or US (escapes) switched off the stimuli and were followed by a 60-sec intertrial interval.
Automated novel-cage activity
The apparatus (Panlab) consisted of a horizontal surface (50 × 50 cm) provided with photobeams that detect movement and measure it automatically, loading the data in a computer. The subjects were placed in transparent Plexiglas cages (40 × 40 × 40 cm). They were situated in a white fluorescent (60 W) illuminated chamber. Spontaneous horizontal activity was measured for the first 5 min (ACT-DIST5; measure of novelty-induced—open filed-like—activity) and for the last 5 min (ACT-DIS30; as a measure of habituated activity) of a 30-min session.
Elevated zero maze
The maze comprised an annular platform (105 cm diameter; 10 cm in width) made of black plywood and elevated to 65 cm above the ground level. It had two open sections (quadrants) and two enclosed ones (with walls 40 cm in height). The subject was placed in an enclosed section facing the wall. The apparatus was situated in a black testing room, dimly illuminated with red fluorescent light, and the behavior was videotaped and measured outside the testing room. Latency to enter into an open section, time spent in the open sections, number of entries in the open sections, number of stretched attend postures, and number of defecation boluses were measured for 5 min.
Intraperitoneal glucose tolerance test (IPGTT)
Conscious rats in the post-absorptive state were injected intraperitoneally with a solution of glucose (2 g/kg body weight). Blood samples were collected for glucose reading before glucose injection and 30, 60, and 120 min afterward by tail tipping. Blood glucose concentration was determined using a glucose meter (Accucheck, Roche Diagnostics). Cumulative glycemia (AUC-G) was calculated as the increment of the values of plasma glucose during the IPGTT. Incremental plasma glucose values above baseline integrated over 120 min after an injection of glucose were used to calculate the index of glucose tolerance (DeltaG).
Blood pressure
Systolic blood pressure was measured by tail-cuff plethysmography in conscious, restrained animals as previously described (Evans et al. 1994; Brosnan et al. 2002; Graham et al. 2004). Briefly, rats were heated to 34°C for 20 min and wrapped in a cloth, and an inflatable cuff was placed on their tail along with a piezoceramic transducer (Hartmann and Braun type 2) for pulse direction. An average of six to eight pressure readings were taken for each rat per sitting.
Full blood count
Red blood cells, Hb, hematocrit, white blood cells, and platelets were counted by an automatic hemocytometer (ADVIA 120 Hematology analyzer from Bayer, Siemens Diagnostics) from a 350-μL tail vein blood sample collected in a tube pretreated with the anticoagulant EDTA-2K (Sangüesa).
Immunology
Blood samples were collected from the lateral tail vein into heparinized (Heparin LEO, Lövens, 10 units/mL blood) tubes (Eppendorf Safe-Lock microcentrifuge tubes) and further stained in duplicates with FITC-, PE, PerCP, or biotinylated antibodies to αβTCR (R73), CD4 (OX35), CD8a (OX8), CD25 (OX39), CD28 (JJ319), RT1.B (rat MHC class II, OX6), RT1.A (rat MHC class I, OX18), CD45R/A (B cell–specific isoform, OX33), CD45 (leukocyte common antigen, OX1), and neutrophilic granulocytes (His48) using 20 μLwhole blood per sample (all antibodies were purchased from Pharmingen, except OX33, which was purchased from BioLegend). Streptavidin-APC (Pharmingen) was used as a secondary reagent. All incubations were performed at 4°C for 20 min in the dark). After antibody incubation, red blood cells were lyzed using 0.84% NH4Cl (0.01M Tris-HCl, 0.01% NaN3 at pH 7.4) and leukocytes were fixed overnight at 4°C on an orbital rotator at 300 rpm using 2% paraformaldehyde (PFA, Sigma-Aldrich) in PBS-D (Dulbecco’s phosphate buffered saline; GIBCO) supplemented with 0.5% bovine serum albumin (Sigma Aldrich). One hundred thousand total cells were acquired on a FACScan (Becton Dickinson), with gates set to include all viable cells to be further analyzed using Flowjo 2.1 software (Tree Star Inc.)
Induction and clinical evaluation of experimental autoimmune encephalomyelitis (EAE)
Recombinant rat MOG (rMOG), amino acids 1–125 from the N terminus, was expressed in Escherichia coli and purified to homogeneity by metal chelate affinity chromatography (Amor et al. 1994) and ion exchange chromatography. Rats were anesthetized with isoflurane (Servicios Genéticos Porcinos) and immunized subcutaneously in the dorsal tail base with 200 μL inoculum containing rMOG (females 50 μg and males 120 μg) in phosphate buffered-saline (PBS; Life Technologies) emulsified 1:1 with Freund’s adjuvant (Sigma-Aldrich) containing 200 μg Mycobacterium tuberculosis (H37 RA, Sigma).
Signs of EAE and body weight were monitored daily from day 8 until day 28 post-immunization (p.i.), after which the animals were euthanized by sanguination under anesthesia. The scale for EAE scoring was as follows: 0 = healthy; 1 = tail weakness or tail paralysis; 2 = hind leg paresis or hemiparesis; 3 = hind leg paralysis or hemiparalysis; 4 = tetraplegy, urinary, and/or fecal incontinence; and 5 = death. If severe disease (score 4) was observed for two consecutive days, the rats were sacrificed due to ethical reasons.
The following clinical parameters were assessed: Incidence of EAE (INC) was defined as 0 = absent and 1 = present; onset of EAE (ONS) was defined as the first day that clinical signs were observed; duration of EAE (DUR) was the number of days with signs of disease; maximum EAE score (MAX) was the highest score obtained during the experiment; and cumulative EAE score (CUM) was the sum of all scores received during the experiment.
Tissue dissection of HS rats
Twenty-eight days after immunization and of clinical scoring for EAE symptoms, the rats were euthanized by sanguination under isofluorane anesthesia. The thyroid gland was carefully dissected out and the heart. Thereafter the ears, brain, and spinal cord were dissected in parallel with spleen, liver, adrenal glands, kidneys, and bones. Tissue was either snap frozen in liquid nitrogen or kept in RNAlater. Blood was incubated for 6 h at room temperature and kept at 4°C overnight and spun at 2000 rpm for 20 min, and sera was aliquoted and kept at −80°C until use. Tissues were sent to each laboratory responsible for investigating the phenotype of interest.
Acknowledgments
This work was funded by the Wellcome Trust and EURATools (European Commission Contract no. LSHG-CT-2005-019015), SAF2003-03480 (A.F.-T.), and 2005SGR-00885 (A.F.-T.). M.J. was funded by a Marie Curie Intra-European fellowship within the Sixth European Framework Programme. M.D. was funded by The Swedish Brain Foundation. D.G. holds a Wellcome Trust Senior Fellowship in Basic Biomedical Science (057733). A.D. was supported by British Heart Foundation Chair and Programme Grant (BHFRG/07/005/23633), P.S.G. was supported by the National Institutes of Arthritis and Musculoskeletal and Skin Disease (NIAMS/NIH) grants AR046213 and AR052439. Wellcome Trust Cardiovascular Functional Genomics Initiative (066780/2/012) (D.G., A.D.). W.V. was funded by BioSapiens (European Union Framework 6 Programme, contract no LHSG-CT-2003-503265).
Footnotes
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.081497.108.
References
- Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Amor S., Groome N., Linington C., Morris M.M., Dornmair K., Gardinier M.V., Matthieu J.M., Baker D. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J. Immunol. 1994;153:4349–4356. [PubMed] [Google Scholar]
- Ball R.D. Bayesian methods for quantitative trait loci mapping based on model selection: Approximate analysis using the Bayesian information criterion. Genetics. 2001;159:1351–1364. doi: 10.1093/genetics/159.3.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi A.L., Oltvai Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Becanovic K., Jagodic M., Sheng J.R., Dahlman I., Aboul-Enein F., Wallstrom E., Olofsson P., Holmdahl R., Lassmann H., Olsson T. Advanced intercross line mapping of Eae5 reveals Ncf-1 and CLDN4 as candidate genes for experimental autoimmune encephalomyelitis. J. Immunol. 2006;176:6055–6064. doi: 10.4049/jimmunol.176.10.6055. [DOI] [PubMed] [Google Scholar]
- Boucher W., Cotterman C.W. On the classification of regular systems of inbreedin. J. Math. Biol. 1990;28:293–305. doi: 10.1007/BF00178778. [DOI] [PubMed] [Google Scholar]
- Broman K.W., Speed T.P. A model selection approach for the identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. Ser. B Stat. Methodol. 2002;64:641–656. doi: 10.1534/genetics.108.094565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosnan M.J., Hamilton C.A., Graham D., Lygate C.A., Jardine E., Dominiczak A.F. Irbesartan lowers superoxide levels and increases nitric oxide bioavailability in blood vessels from spontaneously hypertensive stroke-prone rats. J. Hypertens. 2002;20:281–286. doi: 10.1097/00004872-200202000-00018. [DOI] [PubMed] [Google Scholar]
- Dahlman I., Wallstrom E., Weissert R., Storch M., Kornek B., Jacobsson L., Linington C., Luthman H., Lassmann H., Olsson T. Linkage analysis of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in the rat identifies a locus controlling demyelination on chromosome 18. Hum. Mol. Genet. 1999;8:2183–2190. doi: 10.1093/hmg/8.12.2183. [DOI] [PubMed] [Google Scholar]
- Davison A.C., Hinkley D.V. Bootstrap methods and their application. Cambridge University Press; Cambridge, UK: 2005. [Google Scholar]
- Driscoll P., Battig K. Behavioral, emotional and neurochemical profiles of rats selected for extreme differences in active two way avoidance. In: Lieblich I., editor. Genetics of the brain. Elsevier; Amsterdam: 1982. pp. 95–123. [Google Scholar]
- Evans A.L., Brown W., Kenyon C.J., Maxted K.J., Smith D.C. Improved system for measuring systolic blood pressure in the conscious rat. Med. Biol. Eng. Comput. 1994;32:101–102. doi: 10.1007/BF02512487. [DOI] [PubMed] [Google Scholar]
- Fernández-Teruel A., Escorihuela R.M., Gray J.A., Aguilar R., Gil L., Giménez-Llort L., Tobeña A., Bhomra A., Nicod A., Mott R., et al. A quantitative trait locus influencing anxiety in the laboratory rat. Genome Res. 2002;12:618–626. doi: 10.1101/gr.203402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J., Valdar W., Shifman S., Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat. Rev. Genet. 2005;6:271–286. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- Gelman A., Hill J. Data analysis using regression and multilevel/hierarchical models. Cambridge University Press; Cambridge, UK: 2007. [Google Scholar]
- Goldmuntz E.A., Wilder R.L., Goldfarb Y., Cash J.M., Zha H., Crofford L.J., Mathern P., Hansen C.T., Remmers E.F. The origin of the autoimmune disease-resistant LER rat: An outcross between the buffalo and autoimmune disease-prone Lewis inbred rat strains. J. Neuroimmunol. 1993;44:215–219. doi: 10.1016/0165-5728(93)90046-2. [DOI] [PubMed] [Google Scholar]
- Graham D., Hamilton C., Beattie E., Spiers A., Dominiczak A.F. Comparison of the effects of omapatrilat and irbesartan/hydrochlorothiazide on endothelial function and cardiac hypertrophy in the stroke-prone spontaneously hypertensive rat: Sex differences. J. Hypertens. 2004;22:329–337. doi: 10.1097/00004872-200402000-00017. [DOI] [PubMed] [Google Scholar]
- Hansen C., Spuhler K. Development of the National Institues of Health genetically heterogeneous rat stock. Alcohol. Clin. Exp. Res. 1984;8:477–479. doi: 10.1111/j.1530-0277.1984.tb05706.x. [DOI] [PubMed] [Google Scholar]
- Hartwell L.H., Hopfield J.J., Leibler S., Murray A.W. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- Jacob H.J., Kwitek A.E. Rat genetics: Attaching physiology and pharmacology to the genome. Nat. Rev. Genet. 2002;3:33–42. doi: 10.1038/nrg702. [DOI] [PubMed] [Google Scholar]
- Levine S., Wenk E.J. Induction of experimental allergic encephalomyelitis in rats without the aid of adjuvant. Ann. N. Y. Acad. Sci. 1965;122:209–226. doi: 10.1111/j.1749-6632.1965.tb20204.x. [DOI] [PubMed] [Google Scholar]
- Manichaikul A., Dupuis J., Sen S., Broman K.W. Poor performance of bootstrap confidence intervals for the location of a quantitative trait locus. Genetics. 2006;174:481–489. doi: 10.1534/genetics.106.061549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T., Voigt B., Kuramoto T., Serikawa T. Rat phenome project: The untapped potential of existing rat strains. J. Appl. Physiol. 2005;98:371–379. doi: 10.1152/japplphysiol.01006.2004. [DOI] [PubMed] [Google Scholar]
- Morris A.P., Whittaker J.C., Balding D.J. Fine-scale mapping of disease loci via shattered coalescent modelling of genealogies. Am. J. Hum. Genet. 2002;70:686–707. doi: 10.1086/339271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott R., Talbot C.J., Turri M.G., Collins A.C., Flint J. A method for fine mapping quantitative trait loci in outbred animal stocks. Proc. Natl. Acad. Sci. 2000;97:12649–12654. doi: 10.1073/pnas.230304397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olerup O., Hillert J. HLA class II-associated genetic susceptibility in multiple sclerosis: A critical evaluation. Tissue Antigens. 1991;38:1–15. doi: 10.1111/j.1399-0039.1991.tb02029.x. [DOI] [PubMed] [Google Scholar]
- Olsson T., Jagodic M., Piehl F., Wallstrom E. Genetics of autoimmune neuroinflammation. Curr. Opin. Immunol. 2006;18:643–649. doi: 10.1016/j.coi.2006.08.001. [DOI] [PubMed] [Google Scholar]
- R-Development-Core-Team . A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna: 2004. [Google Scholar]
- Saar K., Beck A., Bihoreau M.T., Birney E., Brocklebank D., Chen Y., Cuppen E., Demonchy S., Dopazo J., Flicek P., et al. SNP and haplotype mapping for genetic analysis in the rat. Nat. Genet. 2008;40:560–566. doi: 10.1038/ng.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpaa M.J., Corander J. Model choice in gene mapping: What and why? Trends Genet. 2002;18:301–307. doi: 10.1016/S0168-9525(02)02688-4. [DOI] [PubMed] [Google Scholar]
- Solberg L.C., Valdar W., Gauguier D., Nunez G., Taylor A., Burnett S., Arboledas-Hita C., Hernandez-Pliego P., Davidson S., Burns P., et al. A protocol for high-throughput phenotyping, suitable for quantitative trait analysis in mice. Mamm. Genome. 2006;17:129–146. doi: 10.1007/s00335-005-0112-1. [DOI] [PubMed] [Google Scholar]
- Stefferl A., Brehm U., Storch M., Lambracht-Washington D., Bourquin C., Wonigeit K., Lassmann H., Linington C. Myelin oligodendrocyte glycoprotein induces experimental autoimmune encephalomyelitis in the “resistant” Brown Norway rat: Disease susceptibility is determined by MHC and MHC-linked effects on the B cell response. J. Immunol. 1999;163:40–49. [PubMed] [Google Scholar]
- Stevens D.B., Gold D.P., Sercarz E.E., Moudgil K.D. The Wistar Kyoto (RT1l) rat is resistant to myelin basic protein-induced experimental autoimmune encephalomyelitis: Comparison with the susceptible Lewis (RT1l) strain with regard to the MBP-directed CD4+ T cell repertoire and its regulation. J. Neuroimmunol. 2002;126:25–36. doi: 10.1016/s0165-5728(02)00045-0. [DOI] [PubMed] [Google Scholar]
- Storch M.K., Stefferl A., Brehm U., Weissert R., Wallstrom E., Kerschensteiner M., Olsson T., Linington C., Lassmann H. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 1998;8:681–694. doi: 10.1111/j.1750-3639.1998.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D., Whitaker J.N., Wilson D.B. Regulatory T cells in experimental allergic encephalomyelitis. III. Comparison of disease resistance in Lewis and Fischer 344 rats. Eur. J. Immunol. 1999;29:1101–1106. doi: 10.1002/(SICI)1521-4141(199904)29:04<1101::AID-IMMU1101>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Valdar W., Flint J., Mott R. Simulating the collaborative cross: Power of quantitative trait loci detection and mapping resolution in large sets of recombinant inbred strains of mice. Genetics. 2006a;172:1783–1797. doi: 10.1534/genetics.104.039313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar W., Solberg L.C., Gauguier D., Burnett S., Klenerman P., Cookson W.O., Taylor M.S., Rawlins J.N., Mott R., Flint J. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat. Genet. 2006b;38:879–887. doi: 10.1038/ng1840. [DOI] [PubMed] [Google Scholar]
- Valdar W., Solberg L.C., Gauguier D., Cookson W.O., Rawlins J.N., Mott R., Flint J. Genetic and environmental effects on complex traits in mice. Genetics. 2006c;174:959–984. doi: 10.1534/genetics.106.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher P.M., Thompson R., Haley C.S. Confidence intervals in QTL mapping by bootstrapping. Genetics. 1996;143:1013–1020. doi: 10.1093/genetics/143.2.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss I.C., Feldon J. Environmental animal models for sensorimotor gating deficiencies in schizophrenia: A review. Psychopharmacology. 2001;156:305–326. doi: 10.1007/s002130100800. [DOI] [PubMed] [Google Scholar]
- Weissert R., Wallstrom E., Storch M.K., Stefferl A., Lorentzen J., Lassmann H., Linington C., Olsson T. MHC haplotype-dependent regulation of MOG-induced EAE in rats. J. Clin. Invest. 1998;102:1265–1273. doi: 10.1172/JCI3022. [DOI] [PMC free article] [PubMed] [Google Scholar]