

Abstract
Natural antisense transcripts (NATs) have been shown to play important roles in post-transcriptional regulation through the RNA interference pathway. We have combined pyrophosphate-based high-throughput sequencing and computational analysis to identify and analyze, in genome scale, cis-NAT and trans-NAT small RNAs that are derived under normal conditions and in response to drought and salt stresses in the staple plant Oryza sativa. Computationally, we identified 344 cis-NATs and 7142 trans-NATs that are formed by protein-coding genes. From the deep sequencing data, we found 108 cis-NATs and 7141 trans-NATs that gave rise to small RNAs from their overlapping regions. Consistent with early findings, the majority of these 108 cis-NATs seem to be associated with specific conditions or developmental stages. Our analyses also revealed several interesting results. The overlapping regions of the cis-NATs and trans-NATs appear to be more enriched with small RNA loci than non-overlapping regions. The small RNAs generated from cis-NATs and trans-NATs have a length bias of 21 nt, even though their lengths spread over a large range. Furthermore, >40% of the small RNAs from cis-NATs and trans-NATs carry an A as their 5′-terminal nucleotides. A substantial portion of the transcripts are involved in both cis-NATs and trans-NATs, and many trans-NATs can form many-to-many relationships, indicating that NATs may form complex regulatory networks in O. sativa. This study is the first genome-wide investigation of NAT-derived small RNAs in O. sativa. It reveals the importance of NATs in biogenesis of small RNAs and broadens our understanding of the roles of NAT-derived small RNAs in gene regulation, particularly in response to environmental stimuli.
Post-transcriptional gene regulation at the RNA level has been recently shown to be more widespread and important than previously assumed (Behm-Ansmant and Izaurralde 2006; Brodersen and Voinnet 2006). While various regulatory RNA molecules have been reported in animals and plants, two prominent types of regulatory small RNAs are microRNAs (miRNAs) and endogenous short interfering RNAs (siRNAs). miRNAs can be encoded in their own genes, which are transcribed by RNA polymerase II or exist in introns of protein-coding genes (Bartel 2004; Jones-Rhoades et al. 2006). Primary transcripts of miRNAs are processed to give rise to short 20- to 24-nt-long mature miRNAs. SiRNA species, on the other hand, seem to be diverse. In plants, up to date, four major groups of endogenous siRNAs—natural antisense siRNAs (nat-siRNAs) (Borsani et al. 2005; Katiyar-Agarwal et al. 2006; Jin et al. 2008), trans-acting short interfering RNAs (tasiRNAs) (Allen et al. 2005; Williams et al. 2005; Axtell et al. 2006), heterochromatic siRNAs (Volpe et al. 2002; Moazed et al. 2006; Buhler et al. 2007), and long siRNAs (lsiRNAs) (Katiyar-Agarwal et al. 2007)—have been reported. In animals, the Piwi-interacting RNAs (O’Donnell and Boeke 2007) and nat-siRNAs (Czech et al. 2008; Ghildiyal et al. 2008; Kawamura et al. 2008; Okamura et al. 2008a, b) have been investigated. Both miRNAs and siRNAs are associated with Argonaute (AGO) proteins to form effector complexes, RNA-induced silencing complexes (RISCs). RISCs are guided by small RNAs within them to their RNA or chromatin targets based on sequence complementarity and trigger translational repression or cleavage of target mRNAs, or modification of target chromatin (Hannon 2002). Sorting specific small RNAs into distinct AGOs is very important for post-transcription gene regulation and chromatin modification. In Arabidopsis, the 5′-terminal nucleotide of small RNAs is the most important factor in sorting them into different Argonaute complexes (Mi et al. 2008; Montgomery et al. 2008).
Endogenous siRNAs can be derived from long, double-stranded RNAs (dsRNAs), which are formed by transcribed repeat sequences, products of RNA-dependent RNA polymerase (RdRP) activities, and overlapped reverse complementary transcripts (Rajagopalan et al. 2006). Natural antisense transcripts (NATs) form double-stranded RNAs in their overlapping regions and have been reported in many model species. There are two classes of NATs, cis-NATs (Shendure and Church 2002; Osato et al. 2003; Yelin et al. 2003; Jen et al. 2005; Wang et al. 2005; Zhang et al. 2006; Henz et al. 2007; Jin et al. 2008) and trans-NATs (Rosok and Sioud 2004; Steigele and Nieselt 2005; Wang et al. 2006). cis-NATs are formed by convergent or divergent antisense transcripts at the same genomic loci, while sense and antisense transcripts of trans-NATs are derived from different loci. Endogenous siRNAs derived from NATs are referred to as “natural antisense siRNAs” (nat-siRNAs). nat-siRNAs direct post-transcriptional gene silencing. In the founding example of post-transcriptional gene silencing directed by nat-siRNA, a pair of cis-NATs, SRO5 and P5CDH, was shown to have antagonistic functions in the regulation of salt tolerance in Arabidopsis (Borsani et al. 2005). P5CDH is expressed constitutively, while SRO5 is induced in response to salt stress. Once SRO5 is expressed, a 24-nt-long siRNA is produced from the overlapped region of these two genes through the action of DCL2, NRPD1A, RDR6, and SGS3. This siRNA directs the cleavage of P5CDH transcripts, and the consequent terminus is used by RdRP to produce dsRNA. Subsequently, the secondary siRNAs are processed from the dsRNAs by DCL1, and these secondary siRNAs can also target P5CDH messages (Borsani et al. 2005). Later, another endogenous nat-siRNA, nat-siRNA ATGB2, was found to be specifically induced in Arabidopsis by a bacterial pathogen (Katiyar-Agarwal et al. 2006). Moreover, endogenous siRNAs derived from NATs have recently been reported in Drosophila melanogaster (Czech et al. 2008; Ghildiyal et al. 2008; Kawamura et al. 2008; Okamura et al. 2008a, b). siRNAs typically arise from both genomic strands and are usually enriched in overlapping portions of cis-NATs (Czech et al. 2008; Okamura et al. 2008a). These recent discoveries revealed the unexpected complexity of the regulatory small RNAs and may open a window onto understanding the functions of nat-siRNAs in both plant and animal species.
Since the discovery of the founder example of cis-NATs, SRO5 and P5CDH, NATs have been believed to be an important biogenesis mechanism of endogenous siRNAs (Borsani et al. 2005). Large-scale analyses in several model species have indicated the widespread existence of cis-NATs (Farrell and Lukens 1995; Lehner et al. 2002; Shendure and Church 2002; Osato et al. 2003; Yelin et al. 2003; Wang et al. 2005; Zhang et al. 2006). The reported frequencies of cis-NATs in different species vary because the different search criteria were used and the available samples of various sizes were considered. Generally, 5%–10% of all neighboring gene pairs in a genome of question seem to be cis-NATs. In the human genome, 4%–9% of all transcript pairs overlap, while in the murine genome, 1.7%–14% have been reported to be overlapping (Lehner et al. 2002; Shendure and Church 2002; Yelin et al. 2003). Much effort has been devoted to discovering NATs in Arabidopsis. A transcriptome analysis with whole-genome tiling arrays has detected antisense expression of 7600 transcripts, which are roughly 25% of all annotated genes (Yamada et al. 2003). An in silicon analysis using the Arabidopsis UniGene (Build 45) data set and genome annotation data (tair5 version) predicted 1340 potential cis-NAT pairs in Arabidopsis, and further analysis of full-length cDNAs and massively parallel signature sequencing (MPSS) data confirmed sense and antisense transcripts of 957 cis-NAT pairs (Wang et al. 2005). Based on some qualitative criteria, these studies concluded that the majority of cis-NATs showed highly anticorrelated expression (Yamada et al. 2003; Wang et al. 2005). However, there are counterexamples or exceptions; some independent studies did not find any evidence of anticorrelated expression, in general, to be greater than expected by chance (Jen et al. 2005; Henz et al. 2007). Henz et al. (2007) found anticorrelation of cis-NAT expression in only a small number of cis-NAT pairs. Thus, the general biological functions and regulatory mechanisms of cis-NATs remain elusive. In addition to cis-NATs, 1320 putative trans-NATs in Arabidopsis have been recently reported (Wang et al. 2006). Interestingly, a substantial number of transcripts were predicted to form both cis-NATs and trans-NATs, suggesting that antisense transcripts may form a complex regulatory network (Wang et al. 2006), which idea, nevertheless, needs to be further investigated.
If a pair of cis-NATs or trans-NATs is coexpressed in the same cells under the same conditions, they may form a double-stranded RNA duplex, which would presumably be necessary for them to spawn endogenous siRNAs. However, there are currently few data to support the coexpression of cis-NAT or trans-NAT pairs in individual cells. The advent of high-throughput sequencing techniques made it efficient to profile, in genome scale, all small RNAs species including nat-siRNAs. Rajagopalan et al. (2006) recently applied high-throughput pyrosequencing to analyze small RNAs in Arabidopsis. More than 340,000 unique small RNA sequences were obtained from libraries from whole seedlings, rosette leaves, whole flowers, and siliques (Rajagopalan et al. 2006). They also noticed that some protein-coding genes in Arabidopsis had a particularly high propensity for spawning small RNAs. Eleven of the top 20 hotspot genes of siRNAs in Arabidopsis were found to be convergently transcribed with neighboring genes. These include an antisense gene pair, At2g16580/At2g16575. Both genes have open reading frames (ORFs) with unknown functions, with one ORF falling largely within an intron of the other (Rajagopalan et al. 2006). With the same high-throughput sequencing technique, Kasschau et al. (2007) profiled and analyzed small RNAs for silencing pathway mutants with defects in three RNA-dependent RNA polymerase (RDR) and four Dicer-like (DCL) genes. Our recent study of cis-NATs in Arabidopsis identified 1008 cis-NAT pairs of protein-coding genes (Jin et al. 2008). A further analysis of small RNA data sets obtained by high-throughput sequencing techniques found that at least one gene in 646 pairs out of these 1008 cis-NATs generates small RNAs under certain conditions or in certain development stages (Jin et al. 2008). Moreover, high-throughput sequencing techniques have also been applied to study endogenous siRNAs in Drosophila melanogaster (Czech et al. 2008; Ghildiyal et al. 2008; Kawamura et al. 2008; Okamura et al. 2008a, b). These sequencing-based profiling techniques have successfully discovered miRNAs that have escaped earlier detection because they are not well-conserved in related genomes or they are expressed with low abundance (Berezikov et al. 2006; Lu et al. 2006; Ruby et al. 2006, 2007; Fahlgren et al. 2007; Sunkar et al. 2008).
In this study, we take advantage of two complementary approaches, computational analysis and high-throughput sequencing, to study cis-NATs and trans-NATs in the model plant species Oryza sativa. First, we perform a genome-wide computational analysis to identify cis-NATs and trans-NATs in O. sativa. Then, we deep-clone small RNAs in seedlings of O. sativa grown under normal and stress conditions and apply a high-throughput pyrosequencing technique to sequence and profile small RNA species (Margulies et al. 2005). Finally, we identify and analyze the small RNAs derived from the identified NATs. Different from recent studies of nat-siRNAs in Arabidopsis, which only focus on cis-NATs, we consider endogenous siRNAs derived from trans-NATs as well as cis-NATs in O. sativa.
Results and Discussion
Antisense transcript pairs in O. sativa
In order to analyze small RNAs spored from NATs, we first identified cis-NATs, which are located at the same or adjacent loci. The O. sativa genome contains 67,450 transcription units based on the version 5 annotation from TIGR (http://www.tigr.org). To identify potential NAT pairs, we compared the genomic loci of all annotated transcription units to search for gene pairs that overlap in an antiparallel manner. As shown in Table 1, 530 pairs of neighboring transcription units are reverse-complementarily overlapped. According to the directions of the involved transcription units, cis-NATs can be categorized in three groups, “convergent” (with 3′-ends overlapped), “divergent” (with 5′-ends overlapped), and “enclosed” (with one transcription unit being entirely overlapped by the other) cis-NATs. Among the 530 cis-NAT pairs, 514 (97%) are arranged in the convergent orientation, and 16 (3%) are enclosed cis-NATs, but none of them is divergent (see Table 1). Plant genomes contain many transposable elements. In O. sativa, more than 24,000 transcription units annotated by TIGR are transposons. In some of the cis-NATs (44, 8%), both transcription units are transposons, and in some other cis-NATs (142, 27%), one transcript is a transposon. As we expected, the majority of cis-NATs (344, 65%) are from protein-coding genes that are not found in any transposons. Since many transposons also code for protein-coding genes, in the rest of this paper, “protein-coding genes” refer to the subset of protein-coding genes not found in any transposons.
Table 1.
Statistics of candidate cis-NATs in O. sativa

aConvergent cis-NAT (with 3′-ends overlapped).
bOne transcription unit being completely reverse-complementarily overlapped by the other.
ccis-NAT pairs of transposons and protein-coding genes.
dcis-NAT pairs of transposons.
ecis-NAT pairs of protein-coding genes.
The number of cis-NATs that we identified in O. sativa is lower than what was previously reported for Arabidopsis. A previous study of adjacent genes in Saccharomyces cerevisiae suggested that there may be evolutionary pressure to select against convergent genes (Dujon 1996). Since O. sativa has a much larger genome than Arabidopsis and both genomes encode a similar amount of transcription units, we expect rice to have fewer cis-NATs. Moreover, we identified 514 convergent cis-NATs but no diverged cis-NATs. A plausible explanation for this is that transcriptional exclusion mechanisms are preferentially inhibitory to transcriptional initiation. We also note that our analysis may underestimate the number of cis-NATs in O. sativa, because the current rice genome annotation may still lack the extreme 5′-ends and 3′-ends for many transcripts and even miss some transcription units.
Besides cis-NATs, which are derived from the same genomic loci, there are many trans-NATs, which are produced by transcription units from distinct genomic regions. As a major group of antisense transcripts, trans-NATs also widely exist and may have important functions. We performed a genome-wide screen of trans-encoded NATs in O. sativa. Specifically, we searched for transcript pairs sharing sequence complementarities using NCBI BLAST. Two transcripts were considered as a trans-NAT pair if their overlapping region was longer than 100 nt and could form RNA–RNA duplexes. We classified the resulting trans-NATs into three categories based on their origins. The first category contains 7142 trans-NATs originated all from protein-coding genes, the second has 25,677 trans-NATs from the combinations of transcripts of protein-coding genes and transposons, and the third category includes 504,935 trans-NATs composed purely of transposons.
Small RNAs derived from NATs
The founder example of cis-NATs involved in post-transcriptional gene regulation was discovered to be related to salt stress (Borsani et al. 2005). We expected more cis-NATs to be underlying various abiotic stress and also anticipated identifying more NAT-associated siRNAs in O. sativa that are related to drought and salt stresses. We thus performed a pyrophosphate-based, high-throughput sequencing experiment to profile small RNAs in O. sativa under drought and salt stresses and the normal condition and obtained a total of 714,202 raw sequence reads. Specifically, 58,781, 43,003, and 80,990 unique small RNAs from the control, drought, and salt libraries, respectively, match perfectly to the rice genome (see below for details).
We searched in our three libraries for small RNAs that match 100% to the putative cis-NATs and trans-NATs discussed above. We found 49 cis-NAT pairs and 4769 trans-NATs whose overlapping regions spawned small RNAs in at least one of our three libraries. In addition to our three small RNA libraries, we also searched two public data sets, MPSS (Nobuta et al. 2007) and CSRDB (Cereal Small RNA Database) (Johnson et al. 2007), for small RNAs with a 100% match to O. sativa cis-NATs and trans-NATs. Specifically, we identified 84 cis-NATs and 5190 trans-NATs whose overlapping regions match small RNAs in the MPSS data set, and two cis-NATs and 2338 trans-NATs that match small RNAs in the CSRDB data set. In total, for 108 cis-NATs and 7141 trans-NATS, we found small RNAs in at least one of the three data sets with a 100% match to their overlapping regions. Supplemental Table S1 shows the number of unique small RNAs derived from cis-NATs and trans-NATs in different stages or under different conditions in all three data sets. In summary, 2592 unique small RNAs match 323 pairs of cis-NAT genes, and 56,790 unique small RNAs match 7141 pairs of trans-NAT genes.
Among these 49 cis-NATs matching small RNAs in our libraries, 40 (81.6%) are specific to one of the three conditions, that is, 11, 13, and 16 of these 49 cis-NATs exclusively match small RNAs in the control, drought, and salt libraries, respectively. In addition, three of the 49 cis-NATs appear in the control and drought libraries, four match small RNAs in the control and salt libraries, and two match those in the drought and salt libraries; but none of them appears in all three libraries. Supplemental Table S2 lists these 49 cis-NATs, the conditions under which they spawn siRNAs, and the annotation of the genes involved. Similar observations of cis-NATs were also obtained in the MPSS data set. As shown in Table 2, most cis-NATs produce small RNAs in specific developmental stages/tissues. All these observations suggest that small RNAs derived from cis-NATs are associated with specific conditions or developmental stages. However, as shown in Supplemental Figure 1 and Supplemental Table S3, the trans-NATs related to specific conditions are relatively few when compared with the trans-NATs that generate small RNAs under all conditions.
Table 2.
Numbers of cis-NATs in O. sativa that spawn small RNAs under both conditions/stages listed in the second row and the first column

aSeedlings treated with abscisic acid (ABA).
bNipponbare immature panicles; 90-d-old plants.
cGerminating seedlings infected with Magnaporthe grisea.
dGerminating seedlings.
eStem.
fSeedling control for ABA treatment. The small RNAs were downloaded from the MPSS small RNA database (Nobuta et al. 2007).
To further explore whether NATs exhibit a higher likelihood to give rise to small RNAs than other protein-coding genes, we computed the density of small RNA loci in the overlapping regions, along the whole regions of the genes involved in NATs and along the whole regions of all protein-coding genes in the O. sativa genome, respectively, in all three data sets. Here, the density is the number of small RNA loci within 1 kb. As shown in Table 3, small RNAs do not seem to be enriched in cis-NAT genes. The densities of small RNA loci in the whole regions of cis-NAT genes and in whole regions of all protein-coding genes are similar. Nevertheless, small RNAs appear to be enriched in the overlapping regions of cis-NATs. The density of small RNA loci in overlapping regions is at least three times greater than that in the whole regions of cis-NATs. This is consistent with earlier reports about derivation of small RNAs from cis-NATs (Henz et al. 2007). We further assessed the statistical significance of the enrichment of small RNA loci in the cis-NAT overlapping regions with P-values obtained by a randomization procedure (see below). The small P-values in Table 3 suggest that the enrichment of small RNA loci in the overlapping regions is statistically significant. The case for trans-NATs is more complicated. In all the data sets, small RNAs are enriched in overlapping regions of trans-NATs. The density of small RNA loci in the overlapping regions is fivefold and sixfold greater than that in the whole regions of trans-NATs and all protein-coding genes, respectively. The enrichment of small RNA loci in trans-NAT overlapping regions is statistically significant, with their P-values being <0.0001 (Table 3). However, the density of small RNA loci spans a broad range across different data sets. For example, in the small RNA library that we profiled under the normal condition as control, the density in the whole regions of trans-NAT genes is about twofold that in the whole regions of all protein-coding genes. But, in the other two libraries that we sequenced, the MPSS data set and the CSRDB data set, there is little difference in density between the whole regions of trans-NAT genes and all protein-coding genes.
Table 3.
Density of small RNA loci in different genome regions in O. sativa

The density is the number of small RNA loci per kilobase.
aWhole regions of all protein-coding genes.
bOverlapping regions that generate small RNAs.
cWhole regions of all genes involved, respectively, in cis-NATs and trans-NATs whose overlapping regions generate small RNAs.
dEnrichment score, the ratio of small RNA locus densities in overlapping regions and whole regions (Ao/Ag).
eP-value for the enrichment score. The development stages and treatment conditions of the MPSS data set are the same as in Table 2.
Characteristics of small RNAs derived from NATs
In D. melanogaster, two genes on the opposite strands of cis-NATs contribute approximately the same number of small RNAs (Czech et al. 2008; Ghildiyal et al. 2008; Kawamura et al. 2008; Okamura et al. 2008a, b). However, in our study, we found that the majority of cis-NATs and trans-NATs spawn small RNAs only from one gene in a NAT pair. Specifically, among the 58 pairs of cis-NATs that produce small RNAs in our three libraries, 52 generate small RNAs only from one transcript of the pair, two cis-NATs generate at least twofold more small RNA reads from one transcript than the other in the pair, while only four spawn small RNA reads roughly equally from both strands. Here, these numbers of cis-NATs are different from the numbers described above since several cis-NATs give rise to small RNAs in more than one library. As shown in Table 4, in the MPSS data set, cis-NATs also produce small RNAs with a strand bias. Moreover, a similar observation on trans-NATs was also obtained. In all the data sets, the majority of trans-NATs prefer one strand in the pair to spawn small RNAs.
Table 4.
Strand bias of cis-NATs and trans-NATs that give rise to small RNAs in O. sativa

aOnly one strand of the NATs gives rise to small RNAs.
bOne strand of the NATs spawns at least twofold more small RNAs than the other.
cBoth strands of the NATs contribute roughly an equal number of small RNAs. The development stages and treatment conditions of the MPSS data set are the same as Table 2.
Endogenous small RNAs from cis-NATs in D. melanogaster have been shown to be included in the AGO2 complex (Kawamura et al. 2008). However, no direct evidence has been collected for which Argonaute complexes are associated with plant nat-siRNAs. In Arabidopsis, sorting of small RNAs in Argonaute complexes is likely to be directed by the 5′-terminal nucleotides (Mi et al. 2008; Montgomery et al. 2008). AGO2 and AGO4 preferentially recruit small RNAs with a 5′-terminal adenosine (Mi et al. 2008). However, no similar study on O. sativa has been reported so far. To gain further insight into endogenous small RNAs in O. sativa, we analyzed the lengths and 5′-terminal nucleotides of small RNAs derived from cis-NATs and trans-NATs in the three libraries that we sequenced. As shown in Figure 1, >40% of the small RNAs from NATs have an “A” as their 5′-terminals. We further computed the natural logarithm odds ratios (Agresti 1996) of cis-NAT and trans-NAT small RNAs to all small RNAs that are not mapped to any NATs. Specifically, the log odds ratios of cis-NAT and trans-NAT small RNAs are 0.48 and 0.44, respectively. We also analyzed the first nucleotide frequencies in the total reads of NAT-derived small RNAs and obtained very similar results. These NAT-derived small RNAs also have a detectable 21-nt length bias, even though their lengths spread over a wide range from 17 to 31 nt (Fig. 2). It has been reported that different biogenesis mechanisms of small RNAs in Arabidopsis will generate small RNAs with different sizes (Xie et al. 2004; Pontes et al. 2006). However, no such study on O. sativa has been reported.
Figure 1.

Distribution of the first nucleotide of small RNAs generated from cis-NATs and trans-NATs.
Figure 2.

Distribution of the lengths of small RNAs generated from cis-NATs and trans-NATs.
Networks formed by NATs
It has been reported that several Arabidopsis genes are involved in two cis-NATs. One is a convergent cis-NAT with overlapped 3′-ends, while the other is a divergent cis-NAT with overlapped 5′-ends (Wang et al. 2005; Jin et al. 2008). In general, one transcript in a cis-NAT pair has only one antisense partner, and we did not find any gene with more than one cis-NAT in O. sativa. However, 162 of the 688 genes involved in the 344 cis-NATs also formed trans-NATs with other genes. Moreover, one transcript may have more than one antisense transcription partner at a different location to form more than one trans-NAT. As discussed above, we identified 7142 trans-NATs with overlapping regions >100 nt in O. sativa. Of these, 7141 spawn small RNAs in at least one of the data sets that we studied. These trans-NATs consist of 4753 genes, among which 271 (11%) genes have at least 10 antisense transcription partners, 2028 (37%) have more than one but fewer than 10 antisense partners, and the remaining 2454 (51.6%) have single antisense partners. These observations indicate that antisense transcripts might form complex regulatory networks in O. sativa.
Theoretically, multiple paralogous genes may form RNA duplexes with the same antisense transcripts. We further analyzed the structures of possible networks formed by these 7141 trans-NATs. The first is a star structure (see Fig. 3 for an example). LOC_Os03g46690, encoding an F-box-domain-containing protein, forms trans-NATs with 12 other transcripts. The Gene Ontology (GO) annotations of these 12 genes are listed in Figure 3B. Eleven of these 12 antisense transcripts are reverse-complementary to the same region of LOC_Os03g46690, while the last one base-pairs with a different region of this gene. This example shows that a transcript may pair with more than one antisense transcript at different locations of its sequence. Another type of structure can be represented by a bipartite graph as shown in Figure 4. Trans-NATs involved in this kind of network have many-to-many relationships. The two networks shown in Figure 4 may function in regulation of the photosynthesis process. We did not find any clique structure formed by three genes in the trans-NAT network.
Figure 3.

One transcript may form trans-NATs with multiple antisense transcripts. The network formed by these NATs shows a star structure. (A) The positions where the NATs are formed; (B) the annotations of genes that are involved in the NATs.
Figure 4.

Many-to-many relationship in trans-NAT networks with bipartite structures. Two examples of trans-NAT networks with bipartite structure and the GO annotations of the genes involved in these two networks are shown.
Networks formed by NATs reflect the complexity of their post-transcriptional regulation. However, under certain conditions, some genes may not be transcribed. Thus, many NAT pairs may not appear under specific conditions. As discussed, we applied the 454 high-throughput sequencing technique to identify small RNAs induced by salt and drought stresses. Eight-hundred-two and 866 trans-NAT pairs specifically spawn small RNAs under drought and salt stress conditions, respectively. We used an enrichment analysis of Gene Ontology (GO) terms (The Gene Ontology Consortium 2004) to functionally characterize these two sets of condition-specific trans-NATs. The 802 drought-specific trans-NAT pairs are formed by 1056 genes. Only 429 of these 1056 genes have function annotation. The 866 salt-specific trans-NAT pairs consist of 987 genes. Similarly, only 442 of these 987 genes have been annotated. As shown in Supplemental Figure 2, among GO terms related to biological processes, “cellular process,” “metabolic process,” and “response to stimulus” are the three most enriched terms in both sets of genes that form drought- and salt-specific trans-NATs. “Binding,” “catalytic,” “transcription regulator,” and “molecule transducer” are the four most enriched GO terms that are related to molecular functions. All these enriched GO terms have P-values < 0.001, which were obtained by a hypergeometric distribution test. When Bonferroni correction was applied, the enrichment of these GO terms is not statistically significant. The main reason is that the function annotation of O. sativa genome is very preliminary, so the GO terms that we obtained are very general, and most analyzed genes have not been annotated yet.
UDP-glucosyl transferase family proteins are important enzymes for catalyzing transportation of sugars. In Arabidopsis, it has been reported that 44 of 115 UDP-glucosyl transferase family members have one or more pairing trans-NATs, and five of these 44 genes also have putative cis-NATs (Wang et al. 2006). Another 13 UDP glucosyl transferase genes have pairing cis-NATs only (Wang et al. 2006). In O. sativa, 28 genes have been annotated as UDP-glucosyl transferase proteins. Our analysis showed that 10 of these 28 genes form trans-NATs with their antisense partners, and two of them have putative cis-NATs. This implies that regulations of some biological processes through the mechanism of NATs may be conserved to a certain degree.
Methods
Data sets
Genomic sequences and annotation information of version 5 of the O. sativa genome were downloaded from the TIGR Plant Genomics Database (http://www.tigr.org).
We included three sets of genome-matched small RNAs. The first set of small RNAs was cloned and sequenced in this study with the 454 high-throughput sequencing technique. Our small-RNA sequence data have been deposited into the NCBI GEO database with access number GSE12317. The second set of 11,809 sequences was downloaded from the CSRDB (Cereal Small RNA Database, http://sundarlab.ucdavis.edu/smrnas/) (Johnson et al. 2007). The third set of MPSS 320,531 17-bp signature sequences was retrieved from the University of Delaware (http://mpss.udel.edu/rice/?) (Nobuta et al. 2007). This data set contains six libraries constructed in different development stages or with certain stress treatments.
Search for cis-NATs and trans-NATs
Putative cis-NATs were identified on the basis of O. sativa genome annotation. If a pair of overlapping genes was located on opposite strands at the same locus of the genome and the overlapped region was longer than 30 nt, they were considered as a cis-NAT pair.
Trans-NATs were identified by pairwise alignment of transcripts to search for transcript pairs with high sequence complementarity to one another. In this study, a pair of transcripts from different genomic loci was considered as a trans-NAT if they satisfied the following two criteria: they have a continuous perfect pairing region longer than 100 nt, and their overlapping region can form an RNA–RNA duplex. We applied a computational tool, DINAMelt (Dimitrov and Zuker 2004; Markham and Zuker 2005), to inspect whether the overlapping regions of a trans-NAT pair can melt into an RNA–RNA duplex in silico. Some protein-coding genes contain transposons in their introns. In order to exclude the effect of intronic transposons on the identification of trans-NATs, for these genes, we performed a trans-NAT search on the coding sequences, which do not include any introns and untranslated regions.
Small RNA sequence profiling and analysis
Four-week-old rice seedlings were dehydrated for 12 h (drought stress), treated with 150 mM NaCl for 24 h (salt stress), or grown under normal conditions (control). Three corresponding small RNA libraries were constructed from these three samples as described previously (Sunkar et al. 2008). Briefly, total RNA was isolated from the frozen seedlings by using TRIzol (Invitrogen) according to the manufacturer’s instructions. Low-molecular-weight RNA was enriched by NaCl and PEG precipitation. About 100 μg of low-molecular-weight RNA was separated on a denaturing 15% polyacrylamide gel. Labeled RNA oligonucleotides with 18 nt and 26 nt were used as size standards. The RNAs from 18 to 26 nt were excised, purified, and ligated with adaptors. Reverse transcription was performed after ligation with adapters, followed by PCR amplification (Sunkar et al. 2008). These three libraries were sequenced with the 454 high-throughput pyro-sequencing platform (Margulies et al. 2005) at 454 Life Sciences. A total of 714,202 raw sequence reads from three independent libraries were parsed to remove the 5′- and 3′-adaptors. In total, 54,016, 174,530, and 102,876 sequence reads that match TIGR version 5.0 rice genome sequences (http://www.tigr.org) were obtained from drought, salt, and control libraries, respectively. After removing sequences that could map to rRNA, tRNA, and sn/snoRNA, we obtained 58,781, 43,003, and 80,990 unique small RNAs that match perfectly to the rice genome from the control, salt, and drought libraries, respectively. The estimated breakdown products were varied in the range of 5%–8% among the three libraries. Note that the 454 sequencing techniques may result in small insertions and a small number of mismatches. If we allowed one unmatched nucleotide, we obtained 3912, 3334, and 6382 additional mapped reads in the control, drought, and salt libraries, respectively. The remaining sequences that could not be mapped to the O. sativa genome were discarded. Possible reasons for the unmapped sequences are as follows: these sequences may contain too many sequence errors, they may be affected by RNA editing, they may be derived from the unsequenced genomic regions or splicing junction sites of cDNAs that have not been characterized, or they are simply contaminants.
To provide candidate data sets of small RNAs that match protein-coding genes, we also removed small RNAs that match transposons, retrotransposons, or repeats in the TIGR rice genome annotation. We then extracted five sets of small RNAs from the candidate data sets for each of the three libraries: small RNAs that match any protein-coding genes; small RNAs mapped to the full-length sequences of any genes involved in cis-NATs and trans-NATs, respectively; and small RNAs matched to the overlapping regions of cis-NATs and trans-NATs, respectively. The last set of small RNAs was considered as cis-NAT- or trans-NAT-derived small RNAs.
Enrichment of endogenous small RNAs in the overlapping regions of NATs
For each NAT (i.e., cis-NATs or trans-NATs), we computed the densities of small RNA loci in the overlapping region and along the whole regions of the two NAT genes as follows: We first counted the number of unique small RNAs, No, mapping to the overlapping region and the total number of unique small RNAs, Ng, matching the two genes. Then we measured the length of the overlapping region, Lo, and the sum of the length of the two genes, Lg. Finally, the ratios No/Lo and Ng/Lg were considered as small-RNA locus densities in the overlapping regions and the whole regions of the NAT genes, respectively. For each data set described and each library constructed in this study, we computed the average densities in the overlapping regions (Ao) and along whole regions of the NAT genes (Ag) that spawn small RNAs. The ratio Ao/Ag was used as the enrichment score.
The significance of the enrichment of small RNAs in the overlapping regions of NATs was quantified by the probability that the enrichment is more than that in arbitrary regions (with the same length of the NAT overlap regions) of a set of randomly chosen gene pairs; the smaller the probability, the more statistically significant the enrichment. This probability was taken as the P-value of the enrichment. To estimate this P-value, we adopted a randomization procedure. Briefly, we first computed the enrichment score (ratio of Ao/Ag) and the average length of the overlapping regions, l, of a given set of n pairs of NATs. Secondly, we arbitrarily chose n pairs of protein-coding genes in the genome. For each pair of genes in the arbitrary set, we randomly chose a region of length l from each gene in the pair and treated it as the “overlapping region.” Then, following the steps described above, we computed the enrichment score of the arbitrary set, which constituted one sample of the randomization procedure. We collected a large number of such samples, specifically, 10,000 in our study. We then estimated the P-value by the frequency that a sample has a bigger enrichment score than that of the given set of NATs.
Functional gene analysis
The statistical significance of enrichment of a GO term t was measured by a cumulative hypergeometric test (Altman 1991). Given M genes in a genome (e.g., O. sativa), assume that N of these M genes have a particular property (e.g., involved in salt-specific trans-NATs), m of the M genes in the genome contain term t in their annotations, and n of the N genes (which are involved in salt- or drought-specific trans-NATs) have the term t in their annotations. We calculated a P-value for the statistical significance of the GO term t as the probability under which we would expect at least n genes to have t if we randomly selected m genes from the given M genes in the genome. Specifically, the P-value was computed as follows:
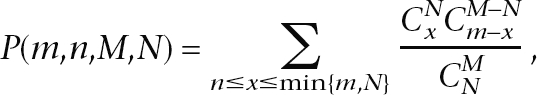 |
where
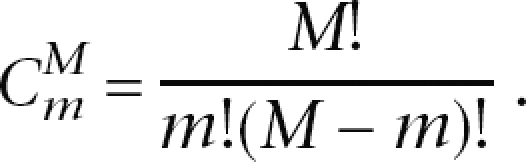 |
Acknowledgments
The research was supported in part by NSF grants IIS-0535257 and DBI-0743797, a grant from the Alzheimer’s Association, a grant from Monsanto Corporation to W.Z., and NIH grant R01GM059138 and USDA grant 2008-35100-04518 to J.-K.Z.
Footnotes
[Supplemental material is available online at www.genome.org. The small-RNA sequence data from this study have been submitted to NCBI Gene Expression Omnibus (http:www.ncbi.nlm.nih.gov/geo/) under accession no. GSE12317.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.084806.108.
References
- Agresti A. An introduction to categorical data analysis. Wiley & Sons; New York: 1996. [Google Scholar]
- Allen E., Xie Z., Gustafson A.M., Carrington J.C. microRNA-directed phasing during transacting siRNA biogenesis in plants. Cell. 2005;121:207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Altman D.G. Practical statistics for medical research. Chapman & Hall/CRC; Boca Raton: 1991. [Google Scholar]
- Axtell M.J., Jan C., Rajagopalan R., Bartel D.P. A two-hit trigger for siRNA biogenesis in plants. Cell. 2006;127:565–577. doi: 10.1016/j.cell.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I., Izaurralde E. Quality control of gene expression: A stepwise assembly pathway for the surveillance complex that triggers nonsense-mediated mRNA decay. Genes & Dev. 2006;20:391–398. doi: 10.1101/gad.1407606. [DOI] [PubMed] [Google Scholar]
- Berezikov E., Thuemmler F., van Laake L.W., Kondova I., Bontrop R., Cuppen E., Plasterk R.H. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006;38:1375–1377. doi: 10.1038/ng1914. [DOI] [PubMed] [Google Scholar]
- Borsani O., Zhu J., Verslues P.E., Sunkar R., Zhu J.K. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell. 2005;123:1279–1291. doi: 10.1016/j.cell.2005.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen P., Voinnet O. The diversity of RNA silencing pathways in plants. Trends Genet. 2006;22:268–280. doi: 10.1016/j.tig.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Buhler M., Haas W., Gygi S.P., Moazed D. RNAi-dependent and -independent RNA turnover mechanisms contribute to heterochromatic gene silencing. Cell. 2007;129:707–721. doi: 10.1016/j.cell.2007.03.038. [DOI] [PubMed] [Google Scholar]
- Czech B., Malone C.D., Zhou R., Stark A., Schlingeheyde C., Dus M., Perrimon N., Kellis M., Wohlschlegel J.A., Sachidanandam R., et al. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov R.A., Zuker M. Prediction of hybridization and melting for double-stranded nucleic acids. Biophys. J. 2004;87:215–226. doi: 10.1529/biophysj.103.020743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. The yeast genome project: What did we learn? Trends Genet. 1996;12:263–270. doi: 10.1016/0168-9525(96)10027-5. [DOI] [PubMed] [Google Scholar]
- Fahlgren N., Howell M.D., Kasschau K.D., Chapman E.J., Sullivan C.M., Cumbie J.S., Givan S.A., Law T.F., Grant S.R., Dangl J.L., et al. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;2:e219. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell C.M., Lukens L.N. Naturally occurring antisense transcripts are present in chick embryo chondrocytes simultaneously with the down-regulation of the alpha 1 (I) collagen gene. J. Biol. Chem. 1995;270:3400–3408. doi: 10.1074/jbc.270.7.3400. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M., Seitz H., Horwich M.D., Li C., Du T., Lee S., Xu J., Kittler E.L., Zapp M.L., Weng Z., et al. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science. 2008;320:1077–1081. doi: 10.1126/science.1157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon G.J. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- Henz S.R., Cumbie J.S., Kasschau K.D., Lohmann J.U., Carrington J.C., Weigel D., Schmid M. Distinct expression patterns of natural antisense transcripts in Arabidopsis. Plant Physiol. 2007;144:1247–1255. doi: 10.1104/pp.107.100396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen C.H., Michalopoulos I., Westhead D.R., Meyer P. Natural antisense transcripts with coding capacity in Arabidopsis may have a regulatory role that is not linked to double-stranded RNA degradation. Genome Biol. 2005;6:R51. doi: 10.1186/gb-2005-6-6-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., Vacic V., Girke T., Lonardi S., Zhu J.K. Small RNAs and the regulation of cis-natural antisense transcripts in Arabidopsis. BMC Mol. Biol. 2008;9:6. doi: 10.1186/1471-2199-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C., Bowman L., Adai A.T., Vance V., Sundaresan V. CSRDB: A small RNA integrated database and browser resource for cereals. Nucleic Acids Res. 2007;35:D829–D833. doi: 10.1093/nar/gkl991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Rhoades M.W., Bartel D.P., Bartel B. MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- Kasschau K.D., Fahlgren N., Chapman E.J., Sullivan C.M., Cumbie J.S., Givan S.A., Carrington J.C. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;5:e57. doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar-Agarwal S., Morgan R., Dahlbeck D., Borsani O., Villegas A., Zhu J.K., Staskawicz B.J., Jin H. A pathogen-inducible endogenous sirna in plant immunity. Proc. Natl. Acad. Sci. 2006;103:18002–18007. doi: 10.1073/pnas.0608258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar-Agarwal S., Gao S., Vivian-Smith A., Jin H. A novel class of bacteria-induced small RNAs in Arabidopsis. Genes & Dev. 2007;21:3123–3134. doi: 10.1101/gad.1595107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y., Saito K., Kin T., Ono Y., Asai K., Sunohara T., Okada T.N., Siomi M.C., Siomi H. Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature. 2008;453:793–797. doi: 10.1038/nature06938. [DOI] [PubMed] [Google Scholar]
- Lehner B., Williams G., Campbell R.D., Sanderson C.M. Antisense transcripts in the human genome. Trends Genet. 2002;18:63–65. doi: 10.1016/s0168-9525(02)02598-2. [DOI] [PubMed] [Google Scholar]
- Lu C., Kulkarni K., Souret F.F., MuthuValliappan R., Tej S.S., Poethig R.S., Henderson I.R., Jacobsen S.E., Wang W., Green P.J., et al. MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res. 2006;16:1276–1288. doi: 10.1101/gr.5530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham N.R., Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S., Cai T., Hu Y., Chen Y., Hodges E., Ni F., Wu L., Li S., Zhou H., Long C., et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell. 2008;133:116–127. doi: 10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D., Buhler M., Buker S.M., Colmenares S.U., Gerace E.L., Gerber S.A., Hong E.J., Motamedi M.R., Verdel A., Villen J., et al. Studies on the mechanism of RNAi-dependent heterochromatin assembly. Cold Spring Harb. Symp. Quant. Biol. 2006;71:461–471. doi: 10.1101/sqb.2006.71.044. [DOI] [PubMed] [Google Scholar]
- Montgomery T.A., Howell M.D., Cuperus J.T., Li D., Hansen J.E., Alexander A.L., Chapman E.J., Fahlgren N., Allen E., Carrington J.C. Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell. 2008;133:128–141. doi: 10.1016/j.cell.2008.02.033. [DOI] [PubMed] [Google Scholar]
- Nobuta K., Venu R.C., Lu C., Belo A., Vemaraju K., Kulkarni K., Wang W., Pillay M., Green P.J., Wang G.L., et al. An expression atlas of rice mRNAs and small RNAs. Nat. Biotechnol. 2007;25:473–477. doi: 10.1038/nbt1291. [DOI] [PubMed] [Google Scholar]
- O’Donnell K.A., Boeke J.D. Mighty Piwis defend the germline against genome intruders. Cell. 2007;129:37–44. doi: 10.1016/j.cell.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K., Balla S., Martin R., Liu N., Lai E.C. Two distinct mechanisms generate endogenous siRNAs from bidirectional transcription in Drosophila melanogaster. Nat. Struct. Mol. Biol. 2008a;15:581–590. doi: 10.1038/nsmb.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K., Chung W.J., Ruby J.G., Guo H., Bartel D.P., Lai E.C. The Drosophila hairpin RNA pathway generates endogenous short interfering RNAs. Nature. 2008b;453:803–806. doi: 10.1038/nature07015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osato N., Yamada H., Satoh K., Ooka H., Yamamoto M., Suzuki K., Kawai J., Carninci P., Ohtomo Y., Murakami K., et al. 2003Antisense transcripts with rice full-length cDNAs Genome Biol. 5R5 . http://genomebiology.com/2003/5/1/R5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes O., Li C.F., Nunes P.C., Haag J., Ream T., Vitins A., Jacobsen S.E., Pikaard C.S. The Arabidopsis chromatin-modifying nuclear siRNA pathway involves a nucleolar RNA processing center. Cell. 2006;126:79–92. doi: 10.1016/j.cell.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Rajagopalan R., Vaucheret H., Trejo J., Bartel D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes & Dev. 2006;20:3407–3425. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosok O., Sioud M. Systematic identification of sense-antisense transcripts in mammalian cells. Nat. Biotechnol. 2004;22:104–108. doi: 10.1038/nbt925. [DOI] [PubMed] [Google Scholar]
- Ruby J.G., Jan C., Player C., Axtell M.J., Lee W., Nusbaum C., Ge H., Bartel D.P. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Ruby J.G., Stark A., Johnston W.K., Kellis M., Bartel D.P., Lai E.C. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–1864. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J., Church G.M. Computational discovery of sense-antisense transcription in the human and mouse genomes. Genome Biol. 2002;3:RESEARCH0044. doi: 10.1186/gb-2002-3-9-research0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigele S., Nieselt K. Open reading frames provide a rich pool of potential natural antisense transcripts in fungal genomes. Nucleic Acids Res. 2005;33:5034–5044. doi: 10.1093/nar/gki804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R., Zhou X., Zheng Y., Zhang W., Zhu J.K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008;8:25. doi: 10.1186/1471-2229-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe T.A., Kidner C., Hall I.M., Teng G., Grewal S.I., Martienssen R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- Wang X.J., Gaasterland T., Chua N.H. Genome-wide prediction and identification of cis-natural antisense transcripts in Arabidopsis thaliana. Genome Biol. 2005;6:R30. doi: 10.1186/gb-2005-6-4-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Chua N.H., Wang X.J. Prediction of trans-antisense transcripts in Arabidopsis thaliana. Genome Biol. 2006;7:R92. doi: 10.1186/gb-2006-7-10-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L., Carles C.C., Osmont K.S., Fletcher J.C. A database analysis method identifies an endogenous trans-acting short-interfering RNA that targets the Arabidopsis ARF2, ARF3, and ARF4 genes. Proc. Natl. Acad. Sci. 2005;102:9703–9708. doi: 10.1073/pnas.0504029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Johansen L.K., Gustafson A.M., Kasschau K.D., Lellis A.D., Zilberman D., Jacobsen S.E., Carrington J.C. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004;2:e104. doi: 10.1371/journal.pbio.0020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K., Lim J., Dale J.M., Chen H., Shinn P., Palm C.J., Southwick A.M., Wu H.C., Kim C., Nguyen M., et al. Empirical analysis of transcriptional activity in the Arabidopsis genome. Science. 2003;302:842–846. doi: 10.1126/science.1088305. [DOI] [PubMed] [Google Scholar]
- Yelin R., Dahary D., Sorek R., Levanon E.Y., Goldstein O., Shoshan A., Diber A., Biton S., Tamir Y., Khosravi R., et al. Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol. 2003;21:379–386. doi: 10.1038/nbt808. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu X.S., Liu Q.R., Wei L. Genome-wide in silico identification and analysis of cis natural antisense transcripts (cis-NATs) in ten species. Nucleic Acids Res. 2006;34:3465–3475. doi: 10.1093/nar/gkl473. [DOI] [PMC free article] [PubMed] [Google Scholar]