

Abstract
Organisms must adapt to make optimal use of the metabolic system in response to environmental changes. In the long-term, this involves evolution of the genomic repertoire of enzymes; in the short-term, transcriptional control ensures that appropriate enzymes are expressed in response to transitory extracellular conditions. Unicellular organisms are particularly susceptible to environmental changes; however, genome-scale impact of these modulatory effects has not been explored so far in bacteria. Here, we integrate genome-scale data to investigate the evolutionary trends and transcriptional control of metabolism in Escherichia coli K12. Globally, the regulatory system is organized in a clear hierarchy of general and specific transcription factors (TFs) that control differing ranges of metabolic functions. Further, catabolic, anabolic, and central metabolic pathways are targeted by distinct combinations of these TFs. Locally, enzymes catalyzing sequential reactions in a metabolic pathway are co-regulated by the same TFs. Regulation is more complex at junctions: General TFs control the overall activity of all connecting reactions, whereas specific TFs control individual enzymes. Divergent junctions play a special role in delineating metabolic pathways and decouple the regulation of incoming and outgoing reactions. We find little evidence for differential usage of isozymes, which are generally co-expressed in similar conditions, and thus are likely to reinforce the metabolic system through redundancy. Finally, we show that enzymes controlled by the same TFs have a strong tendency to co-evolve, suggesting a significant constraint to maintain similar regulatory regimes during evolution. Catabolic, anabolic, and central energy pathways evolve differently, emphasizing the role of the environment in shaping the metabolic system. Many of the observations also occur in yeast, and our findings may apply across large evolutionary distances.
Small-molecule metabolism is the set of all chemical reactions that allow a cell to assimilate environmental nutrients, generate energy, and synthesize precursors necessary for macromolecular synthesis. In order to survive in a habitat, organisms must use the nutrients that exist in the environment efficiently and adapt to changes in their availability. In the long term, prolonged exposure to particular habitats leads to the evolution of the metabolic enzymes encoded in the organism’s genome (Herring et al. 2006). In bacteria—which are generally highly streamlined and efficient organisms—small-molecule metabolism assumes special importance as typically a quarter of gene content is devoted to metabolism. In fact, the number of enzymatic genes is a key determinant of bacterial genome size (Ranea et al. 2005). Systematic analyses of metabolic evolution are now possible owing to the availability of several hundred bacterial genome sequences accompanied by information on organism habitat and phenotype (Shlomi et al. 2007b; Kreimer et al. 2008). More recently, the dependence between bacterial gene content and the environment has been highlighted by metagenomic studies, which have suggested that specific metabolic functions act as signatures for particular types of habitats (Gill et al. 2006; Turnbaugh et al. 2006; Dinsdale et al. 2008).
The enzymatic gene content of an organism represents just one dimension of the metabolic system, as many bacteria live in variable environments and not all enzymes are required at all times. In the short term, an adaptive response to changing nutrient conditions can be achieved through transcriptional regulation of intracellular enzyme concentrations. A powerful approach to study metabolic activity has been through the use of network representations, in which enzymatic reactions are depicted as directed edges and small molecules as nodes. The availability of genome sequences, coupled with the biochemical characterization of enzymes, has led to high-quality computational reconstruction of metabolic networks for a wide variety of organisms. Graph-theoretical analyses and simulations such as flux-balance analysis have been applied to these networks to study their structural and functional properties (Jeong et al. 2000; Ravasz et al. 2002; Ibarra et al. 2003; Almaas et al. 2004, 2005).
Control of metabolic activity can be studied by overlaying a transcriptional regulatory network in which edges represent regulatory interactions from transcription factors (TFs) to target genes. Data for the best-studied bacterium Escherichia coli are available in RegulonDB, which is a compilation of more than 2000 regulatory interactions derived largely from literature describing small-scale experiments (Salgado et al. 2006). This substantial data set has been used to improve phenotypic predictions by integrating regulatory information into metabolic flux balance simulations (Covert and Palsson 2002; Herrgard et al. 2003; Covert et al. 2004; Barrett et al. 2005; Shlomi et al. 2007a). It has also been used to characterize recurring patterns of TF–target gene interactions termed “network motifs” that confer different kinetic properties to metabolic circuits (Shen-Orr et al. 2002; Mangan et al. 2003, 2006; Zaslaver et al. 2004; Alon 2007).
In parallel, three genome-scale investigations have made important contributions to our understanding of general principles that underlie transcriptional regulation of the metabolic system (Ihmels et al. 2004; Kharchenko et al. 2005; Notebaart et al. 2008). In Saccharomyces cerevisiae, Ihmels et al. (2004) combined large-scale gene expression data with the metabolic network to demonstrate that transcriptional regulation ensures coherent metabolite flow between sequential enzymes. Kharchenko et al. (2005) reported, again in yeast, that enzymes show more similar expression profiles if they are closer together in the metabolic network. Very recently in both S. cerevisiae and E. coli, Notebaart et al. (2008) argued that strong correlations in metabolite flow calculated from flux balance simulations are better predictors of coexpression than simple distance separation in the metabolic network.
The above studies are largely based on the metabolic and regulatory apparatus of S. cerevisiae. Although both S. cerevisiae and E. coli are unicellular organisms, their regulatory machineries are vastly different (Fink 1987). For example, whereas more than half of all E. coli metabolic regulators are activated by small-molecule binding (Anantharaman et al. 2001; Madan Babu and Teichmann 2003), most eukaryotic TFs respond to complex signaling cascades (Reece et al. 2006). It is unclear if the findings are applicable to prokaryotes, and it is important to perform an independent study focused on a bacterial system.
Here we study, on a genomic scale, how the transcriptional regulatory system controls small-molecule metabolism in E. coli. First, we investigate the regulation of different types of metabolic pathways on a global scale, and also introduce a functional hierarchy of TFs. Next, we examine regulatory patterns at a local scale, by assessing how gene expression is mediated for neighboring metabolic reactions, with special attention on the control of pathway junctions. Finally, we study the evolution of metabolic pathways and evaluate whether there is a relationship between the coordinated regulation and the conservation of enzymes. In doing so, we establish rules of transcriptional regulatory system that are generally applicable to bacterial metabolic systems.
Results and Discussion
Our study uses five distinct data sets:
A metabolic network comprising 788 reactions mapped onto 781 enzyme genes and 628 small molecules from the EcoCyc database (Keseler et al. 2005);
a transcriptional regulatory network involving 111 TFs regulating 388 enzyme genes (49.7% of all enzymes) via 913 regulatory interactions, sourced from RegulonDB (Salgado et al. 2006);
43 binding interactions between 40 TFs and 39 small molecules from EcoCyc representing post-translational regulation of TF activity;
Affymetrix microarray data covering 221 mRNA hybridizations across diverse cellular conditions from the M3D database (Faith et al. 2007); and
protein sequences for E. coli K12 MG1655 and 380 other prokaryotic organisms with completely sequenced genomes obtained from the KEGG database (Kanehisa et al. 2006).
The first four data sets contain information for E. coli K12 only.
Global regulation of metabolic enzymes
Hierarchy of general and specific TFs
The E. coli transcriptional regulatory network was previously shown to have a pyramid-shaped hierarchical topology, with a few master TFs at the top level regulating lower-level TFs (Ma et al. 2004; Balazsi et al. 2005; Yu and Gerstein 2006). Recent studies have further shown that metabolic pathways are regulated by shorter cascades of TFs than functions such as motility and biofilm formation (Shen-Orr et al. 2002; Martinez-Antonio et al. 2008). Here we show that the regulation of the metabolic system is also hierarchical in terms of the functionality of the targeted enzymes (Fig. 1). All the results presented in this paper are robust against perturbations in the underlying data set (i.e., introduction of errors and deletion of data points), indicating that our findings are likely to remain valid as data sets are updated in the future (Methods).
Figure 1.

Regulatory targets of general and specific TFs. Schematic representation of transcriptional regulation of E. coli small-molecule metabolism displaying general TFs (red circles), specific TFs (blue), and target enzyme genes (black). TFs are labeled with gene names, and enzymes are grouped according to their COG annotations. Regulatory interactions are shown as lines directed from general TF to functionally enriched target (red), general TF to non-enriched function (gray), and specific TF to target (blue).
At the top of the hierarchy, general TFs regulate genes belonging to multiple functional categories as described by the COG classification system (Tatusov et al. 2003). Ten of the 111 TFs in our data set belong to this group, and it includes six of the seven global TFs described in an earlier publication (Martínez-Antonio and Collado-Vides 2003). Despite the breadth of their regulation, all these TFs display a statistical enrichment for a single functional category that reflects the nature of the input signal sensed by the TF concerned. For example, Lrp binds leucine and preferentially targets amino acid metabolism. Even Fis and IHF—which do not explicitly contain signal-sensing domains, but whose activities are growth-phase-dependent (Ali Azam et al. 1999)—favor regulation of energy metabolism (Blot et al. 2006).
On the other hand, specific TFs restrict their target genes to those in the same EcoCyc pathway or the same functional category: 54 TFs are pathway specific, and 18 others are function specific (i.e., regulate more than one pathway but only those of single functional category). Again, the activities of many specific TFs are post-translationally controlled through small-molecule binding (Anantharaman et al. 2001; Madan Babu and Teichmann 2003); however, here, the regulatory metabolite tends to originate from the pathway that is targeted by the relevant TF, effectively forming a local feedback loop. A classic example is the LacI repressor of the lactose utilization operon: On binding allo-lactose, this TF immediately affects the corresponding catabolic pathway. We could not classify the remaining 29 TFs, as there were too few targets with metabolic COG or pathway annotations in the current data set.
In direct relation to the diversity of target gene functions, we find that general TFs regulate more genes than specific TFs; thus the topological and functional hierarchies of the regulatory network are closely linked. The two classes of TFs also differ in several other ways (Supplemental Fig. 1):
General TFs display higher mRNA expression levels than specific TFs when we examine data from Affymetrix GeneChips (PMann-Whitney = 5.6 × 10−5). This reflects the fact that cells require larger absolute quantities of general TFs since they have to bind many more targets (Lozada-Chavez et al. 2008). For example, it is well known that CRP, which targets the entire carbohydrate metabolism system, is present in 1400–6600 copies per cell (Ishizuka et al. 1993), compared with LacI, which controls only lactose degradation and is present in only 10–20 copies per cell (Gilbert and Muller-Hill 1966).
In line with previous observations (Menchaca-Mendez et al. 2005), we also observe that the genes of specific TFs tend to be encoded much closer to their target binding sites, in contrast to general TFs (PMann-Whitney < 2.2 × 10−16). In prokaryotes, transcription and translation are tightly coupled, ensuring that the protein product is generally produced close to the encoding gene on the chromosome. As specific TFs are expressed in small quantities and have comparatively few targets, they would locate binding sites more efficiently if their genes were proximal on the chromosome (Janga et al. 2007; Kolesov et al. 2007).
Finally, small molecules that regulate the activity of specific TFs tend to be closer in the metabolic network to the target enzymes of these TFs, compared with those that bind general TFs (see Local regulation of metabolic enzymes and Methods). This indicates that the feedback loop involving specific TFs is more local in nature.
Catabolism, anabolism, and central energy metabolism are regulated differently
Next, we investigated whether distinct regulatory principles operate in different metabolic subsystems (Table 1): (1) the catabolic subsystem assimilates diverse nutrients from the environment, and feeds these products into energy-generating pathways; (2) the anabolic subsystem synthesizes a wide range of small-molecule products from a limited set of precursor molecules; and (3) the central/energy subsystem is situated between catabolic and anabolic pathways, generating ATP and biosynthetic precursors. We studied this by testing for differences in the numbers of TFs that regulate genes from these metabolic functions and also for differences in the TF classes involved.
Table 1.
Metabolic subfunctions

As shown in Figure 2A, each subsystem is regulated differently. Anabolic pathways are controlled by few TFs, with 67% of enzymes targeted by a single regulator each. In contrast, central energy metabolism is heavily regulated: >75% of enzymes are controlled by at least three TFs each. Catabolic enzymes lie between the two extremes.
Figure 2.

Numbers of TFs controlling metabolic enzymes. (A) Histogram of numbers of TFs regulating all, catabolic, anabolic, and central energy enzymes. (*) Overrepresented groups; (§) underrepresented groups. (B) Box plots of numbers of general and specific TFs targeting different classes of enzymes. (C) Histogram of numbers of enzymes regulated by general TFs only, specific TFs only, and combinations of general and specific TFs.
The subsystems also differ in the balance of general and specific TFs (Fig. 2B,C). Catabolic enzymes tend to be regulated by a combination of both TF types (e.g., CRP and LacI), and the need for multiple regulators is illustrated by the control of carbohydrate-processing pathways. Under normal circumstances, E. coli favors glucose as the main carbon source. However, in the absence of glucose and the presence of an alternative sugar (e.g., arabinose), CRP, a general regulator for many catabolic systems, and a specific TF (e.g., AraC) jointly activate the appropriate pathways. This ensures that alternative carbon sources are not used when glucose is available in the medium.
In contrast, anabolic enzymes are usually targeted by a lone regulator, with no preference for general or specific TF type. As discussed above, many of these TFs bind small molecules that are related to the pathways they control (e.g., substrate or product of the regulated pathway), thus creating extensive feedback between enzyme expression and cellular demands for the anabolic product. A general regulator that demonstrates this principle is Lrp, which binds leucine and preferentially targets a large group of amino acid biosynthetic pathways. An example of a specific regulator is BirA, which binds biotin-5-AMP and controls biotin biosynthesis. By varying a given TFs binding affinity to different target promoters, a single input regulation facilitates a program of “just-in-time” transcription in which enzymes at the beginning of pathways are expressed earlier than those at the end (Zaslaver et al. 2004). This form of control is most likely to benefit anabolic pathways as they tend to involve more reactions than catabolic pathways (Supplemental Fig. 2), and metabolites take longer to process.
Finally, central energy metabolism is almost exclusively controlled by combinations of general TFs. This subsystem is a hub to which nutrients assimilate and from which anabolic pathways radiate out. This means that these enzymes need to respond to numerous environmental conditions, which is best achieved through control by general TFs expressed under multiple conditions (Martínez-Antonio and Collado-Vides 2003).
Local regulation of metabolic enzymes
Connectivity of enzyme pairs in the metabolic network
Having studied how the metabolic network and its subsystems are controlled on a global scale, we now examine the regulatory properties of individual enzymes at a local level. For this we classified pairs of neighboring enzymes by their relative positioning in the metabolic network (Fig. 3).
Figure 3.

Configurations for neighboring enzymatic reactions. Example reactions are given for each configuration.
“Flow” reactions describe enzyme pairs arranged in a sequential manner so that metabolites proceed from one reaction to the next. These can occur in linear sections of the metabolic system, or at junctions; for the latter, enzyme pairs can be in divergent or convergent configurations. “Non-flow” reactions occur only at junctions, and represent enzyme pairs that are positioned nonsequentially. These can once again be convergent (both reactions feed into a common product) or divergent (both enzymes emerge from a common reactant). In total, we identify 9798 enzyme pairs in these configurations, of which nearly a third have known regulators for both reactions (Table 2). Note that these arrangements are not exclusive, and enzymes can belong to more than one configuration.
Table 2.
Transcriptional co-regulation in local metabolic network patterns

aThe number of enzymes with overlapping TFs does not include those with identical TFs.
Linear-flow reactions are strongly co-regulated
We assess the extent to which enzyme pairs are co-regulated (Fig. 4A; Table 2); that is, targeted by identical sets of TFs. Simple linear stretches of the metabolic network are tightly co-regulated, as 75% of linear-flow reactions are targeted by the same TFs. An example of such regulation is that of purB and purC in purine biosynthesis by the TF PurR. We note that a substantial proportion (58%) of co-regulated enzyme pairs reside in the same operon; thus genomic organization of enzyme genes is a major driving force for coordinated regulation. However, nearly half of co-regulated enzymes belong to different operons, and it is clear that transcriptional control extends well beyond the confines of operon structure.
Figure 4.

Co-regulation and coexpression of metabolic enzyme pairs. (A) Histogram of numbers of pairs of enzymes that are co-regulated by identical sets of TFs in the regulatory network. (B) Box plot of distributions of Pearson correlation coefficients for gene expression profiles of enzyme pairs. The horizontal dashed line displays the median correlation for all pairs of enzymes in the metabolic network.
We confirm the observations made from the transcriptional network by testing the coexpression of enzyme pairs (Fig. 4B). Here, we measure coexpression of gene pairs by calculating the Pearson correlation coefficient of their expression profiles across 221 Affymetrix hybridizations (Faith et al. 2007). Although this calculation is independent of information from the transcriptional regulatory network, enzyme pairs in linear-flow configurations display higher levels of coexpression than other pairs (PMann-Whitney < 2.2 × 10−16).
Pathway junctions are intricately regulated
At first glance, the amount of co-regulation appears much lower at pathway junctions (8% for flow and 10% for non-flow) (Fig. 4A). This is partly because most junctions have numerous incoming and outgoing reactions resulting in a large number of pairwise comparisons. In fact, more than half of junctions (52%) contain at least one pair of co-regulated flow reactions (i.e., one incoming and one outgoing reaction), but fewer than a third (30%) contain a pair of co-regulated non-flow reactions (i.e., both incoming or outgoing). This is also reflected by the microarray data in which co-regulated reactions display significantly higher levels of coexpression compared with other enzymes (Supplemental Fig. 3). In addition, many reactions connected at junctions have at least one TF in common (51% of all flow pairs, 36% of non-flow pairs) (Supplemental Fig. 4); in 95% of these cases, a general TF acts as the overlapping regulator.
An example of such co-regulation occurs at a junction centered on the L-ribulose-5-phosphate. This metabolite is produced by AraB and consumed by AraD of the arabinose pathway, and by UlaE and UlaF of the ascorbate utilization pathway. The first pair is targeted by the TFs CRP and AraC, and the second pair by CRP, IHF, and UlaR. Thus, distinct but overlapping sets of regulators ensure a coherent flow of metabolites through two complementary pathways in the junction.
These observations demonstrate that there is an intricate system of control at pathway junctions. General TFs determine the overall activity of junctions by targeting all connecting reactions. Specific TFs are then used to fine-tune the expression of individual reactions. In many cases, one or more pairs of reactions—commonly an incoming and outgoing pair—are controlled by identical sets of TFs to provide a major thoroughfare for the flow of metabolites. Alternative or additional reactions are then activated to divert metabolic flow depending on the cellular conditions.
Regulation is decoupled at divergent junctions
We can examine the control of junctions by dividing them into those that are convergent and divergent (Figs. 3 and 4; Table 2). Here, we find that flow reaction pairs are more likely to be co-regulated if they traverse convergent junctions (17% with identical TFs, 85% with overlapping TFs) than divergent junctions (5% and 47%, respectively).
The difference in level of control is also reflected in the expression data. As previously described by Kharchenko et al. (2005), coexpression tends to fall with increasing distance between enzyme pairs in the metabolic network (measured as the number of separating metabolites). However, the pattern of coexpression depends on the nature of separation between the enzymes (Fig. 5). Reactions uninterrupted by any junctions retain relatively high levels of coexpression (Fig. 5A). Introduction of a convergent junction causes a slight drop in coexpression, and the signal is not affected substantially beyond this point (Fig. 5B). In contrast, a single divergent junction is sufficient to abolish all coexpression (Fig. 5C).
Figure 5.

Coexpression of enzyme pairs at different levels of separation in the metabolic network. Box plots of Pearson correlation coefficients for gene expression profiles between enzyme pairs separated by different numbers of (A) linear metabolites, (B) convergent junctions, and (C) divergent junctions. There is an upper limit of three intermediate convergent junctions in the metabolic network. The horizontal dashed line displays the median correlation for all pairs of enzymes.
Notebaart et al. (2008) recently reported that enzymes with coupled metabolic fluxes—where a nonzero metabolic flux for one enzyme implies a nonzero flux for the other and vice versa—tend to show similar expression profiles (Supplemental Fig. 5). In fact, there are fewer divergent junctions between flux-coupled enzymes than expected by chance. Moreover, these junctions appear to provide natural boundaries for the definition of pathways: 62% of reaction pairs connected at convergent junctions share the same EcoCyc pathway, but only 11% of pairs linked at divergent junctions do so (PFisher-test < 2.2 × 10−16) (Supplemental Fig. 6). Thus, we suggest that the topology of the metabolic network is an important determinant of flux coupling, as well as co-regulation.
A possible underlying reason for the unique behavior of divergent junctions is that they are decision points in the network; that is, metabolic flow is dependent on the choice of one reaction over another. In comparison, convergent arrangements are not decision-making as flux can flow only in one direction out of the junctions. Therefore, by decoupling the regulation of connecting reactions at divergent junctions, incoming metabolic flux can be directed toward the required product according to independent cellular signals.
Isozymes are partially co-regulated
Isozymes are two or more enzymes that differ in amino acid sequence but catalyze the same reaction. There are several possible ways in which they could be beneficial to the organism. First, through selective utilization under different conditions, isozymes could permit fine-tuning of a metabolic pathway, as they often display differing kinetics. Next, the use of dedicated isozymes in distinct pathways containing a common reaction could help reduce cross-talk. Finally, isozymes could imply increased metabolic flow through a reaction and also provide redundancy to compensate against mutations.
The E. coli network contains 97 reactions that are mediated by 196 isozymes. Although only 7% of isozyme pairs are co-regulated by identical TFs, >65% have overlapping TFs, indicating substantial regulatory coordination (Supplemental Fig. 7). For example, the E. coli genome encodes for two acetylornithine transaminases, ArgD and AstC; the general TF ArgR controls both, but another TF, GlnG, targets only argD.
In the yeast metabolic network, Ihmels et al. (2004) described a special type of coordinated regulation termed a “linear switch” that operates at divergent junctions, in which different incoming isozyme reactions are coexpressed with alternative outgoing branches. Such an arrangement could modulate the direction of metabolic flow in a condition-specific manner. We searched for similar patterns of regulation in E. coli using the expression data (Supplemental Fig. 8A). However, out of 68 isozyme pairs occurring at junctions (i.e., including both convergent and divergent), we could find only eight cases of linear switches. Instead, 47 isozyme pairs are coexpressed with the same upstream or downstream reaction in the junction.
Both the regulatory network and gene expression data indicate a high level of co-regulation of isozymes, thus ruling out the first two possibilities above. Instead, the primary effect of isozymes appears to be that of redundancy against mutations (Supplemental Fig. 8B). Isozyme reactions are not preferentially present in particular COG functional categories. However, in general, they tend to be connected with larger numbers of substrate and product metabolites compared with other enzymes (PMann-Whitney = 3.6 × 10−5). In addition, a higher proportion of isozyme reactions (28 out of 97 reactions; 29%) are involved in multiple metabolic pathways compared with other reactions (13%; PFisher-test = 7.8 × 10−6). Therefore, isozymes appear to be enriched in highly connected reactions that would have a great impact on the organism should they break down.
Global and local patterns of enzyme evolution
Having examined the patterns of transcriptional regulation of enzymes, we now study how these regulatory properties relate to the evolution of the metabolic system. For this, we identified the ortholog of the E. coli metabolic enzymes in our data set across 380 bacterial and archaeal genome sequences. Every enzyme was then assigned a phylogenetic profile represented by a series of binary values indicating the presence or absence of orthologs in each genome.
Conservation of catabolic, anabolic, and central metabolic pathways
First, we assessed the conservation of different types of metabolic pathways by comparing the percentage of genomes in which an enzyme has a detectable ortholog (Fig. 6A). Catabolic enzymes are the least conserved, whereas anabolic enzymes are the most conserved. Members of the central metabolic pathways lie between the two.
Figure 6.

Conservation and coexpression of metabolic enzymes across 380 bacterial genomes. (A) Box plot of proportion of genomes containing orthologs of catabolic, anabolic, and central energy enzymes. (B) Scatterplot between the Pearson correlation coefficients measuring coexpression and Phi correlation measuring coevolution of enzyme pairs. Data points are shaded according to a normalized proportion of observations in the data set, with darker shades representing higher proportions. (Inset) The density distribution displays the mutual information between the two sets of correlations for the actual data and random expectation from 1000 simulations.
In general, pathways that are exposed to long-term extracellular changes tend to be less conserved than internal pathways; this underlines the importance of the environment in driving bacterial evolution (Borenstein et al. 2008; Kreimer et al. 2008). Catabolism is poorly conserved because the presence or absence of specific pathways is governed by the organisms’ habitat and the access to different sources of nutrients. It should be noted that the reference organism, E. coli, is metabolically versatile and therefore contains a large complement of catabolic enzymes compared with most other organisms. In contrast, anabolism is highly conserved because similar metabolic products—including amino acids, nucleotides, and lipids—are required for macromolecular synthesis regardless of the organisms’ lifestyle. The only exceptions occur in species that are auxotrophic for some molecules. This difference in conservation of catabolism and anabolism has also been observed in eukaryotes (Lopez-Bigas et al. 2008).
Central energy metabolism displays intermediate conservation because organisms encode for either the TCA cycle or fermentative pathways, or both, depending on their lifestyles. E. coli contains both sets of enzymes, reflecting its capacity to perform aerobic and anaerobic respiration. In general, organisms classified as being aerobic or facultative (i.e., both aerobic and anaerobic, like E. coli) encode for aerobic respiration genes more often than anaerobic species (P < 3.6 × 10−5) (Supplemental Fig. 9). However, we do not observe a similar trend for fermentative genes and anaerobic species; this could be because different substrates can be used for these pathways.
Coexpressed genes also coevolve
Next, we examined how enzyme pairs in different types of flow and non-flow configurations evolve. For this, we calculate the correlation between the phylogenetic profiles of each enzyme pair. All the trends observed for co-regulation are reproduced here (Supplemental Fig. 8). We find that (1) enzyme pairs coevolve most frequently when they occur at linear flow reactions; (2) of pairs at junctions, those in nonconvergent reactions display greater coevolution compared with divergent forks; (3) enzymes in continuous sections of nondivergent flow reactions show high levels of coevolution, but this trend is decoupled by divergent junctions; and (4) enzyme pairs with coupled fluxes tend to coevolve.
These observations can be explained by the clear agreement between the correlations in expression and phylogenetic profiles of enzyme pairs (Fig. 6B, mutual information = 0.09; P < 0.001). Coevolving enzymes tend to be coexpressed (Fig. 6B, first quadrant), and to a lesser extent inversely coexpressed also (second quadrant). However, there is little evidence for enzyme pairs to coevolve negatively (third and fourth quadrants).
These observations indicate a strong evolutionary pressure to preserve the co-regulation of enzymes that coevolve. The transcriptional regulatory network is known to evolve rapidly in bacteria (Lozada-Chavez et al. 2006; Madan Babu et al. 2006; Price et al. 2007). Therefore, the repertoire of TFs and the details of regulatory interactions probably differ substantially between species but the gene expression program dictated by the regulatory system is maintained. An important contribution to this trend is undoubtedly the strong evolutionary pressure to maintain similar chromosomal organization of genes, especially in cases in which entire pathways are encoded within the same operon (Supplemental Fig. 11).
Conclusions
Summary of results
In this study, we presented a genome-scale study of the transcriptional regulation and evolution of the metabolic system of a facultative model bacterium that is capable of assimilating a large variety of nutrients.
At the global level, regulators operate in a two-tier hierarchy of general and specific TFs that control differing ranges of metabolic functions. The two types of regulators also differ in other characteristics such as expression level and chromosomal distance between genes encoding the TF and target gene. We also show that catabolic, anabolic, and central metabolic pathways operate under different regulatory regimes. Catabolic pathways tend to be regulated by a combination of general and specific TFs. Such an arrangement allows the system to respond to a combination of several environmental inputs, one of which is more dominant than others. This is exemplified by the role of the general TF CRP in carbohydrate metabolism, which represses all alternative sugar assimilation pathways in the presence of glucose. In fact, the expression of even a single additional pathway can adversely affect the organism’s fitness (Dekel and Alon 2005). Anabolic pathways are mostly targeted by a single TF, which permits a “just-in-time” regulatory output as presented by Alon and colleagues (Zaslaver et al. 2004). As anabolism tends to involve the longest pathways in the network, this type of organization is advantageous in allowing enzymes at the end of pathways to be expressed later. Finally, central metabolism is a hub whose activity should respond to diverse conditions, and accordingly these are regulated by multiple TFs, among which, the general TFs are known to be expressed under many conditions (Martínez-Antonio and Collado-Vides 2003).
At the local level, we examined the principles of co-regulation of neighboring reactions. Here, in common with S. cerevisiae, there is a strong tendency to coexpress enzymes that are arranged sequentially. Interestingly, divergent junctions have gained a special status in the metabolic network, as they play an important role in decoupling the regulation of connecting pathways. Through the introduction of this modularity, divergent junctions allow pathways and their regulation to evolve independently of each other.
It is important to note that although we do find a strong general relationship between the regulation and expression of enzymes (i.e., genes with identical TFs display similar expression profiles), a recent high-resolution study shows that the precise kinetics of target gene expression can differ substantially even when identical regulatory architectures are involved (Kaplan et al. 2008). Therefore, detailed understanding of how specific TF combinations will produce particular outputs will only be possible once we consider the structure of the promoter and the nature of upstream regulatory circuitry (such as the presence of feedback and feed-forward loops).
Lastly, we show that enzymes controlled by the same TFs display a strong tendency to coevolve, suggesting a significant constraint to maintain similar regulatory regimes during evolution. In particular, differences in the evolution of catabolic, anabolic, and central metabolic genes across the prokaryotic kingdom clearly illustrate the role of the environment in dictating bacterial evolution. By incorporating information about this dependency, it may be possible to fine-tune predictions of an organism’s habitat given its enzyme gene content, or more ambitiously vice versa (Borenstein et al. 2008; Kreimer et al. 2008).
Comparison to other genome-scale studies of metabolic regulation
This study complements and expands on earlier genome-scale studies of metabolic regulation, most notably that of Ihmels et al. (2004). By using a large compendium of yeast gene expression data, the investigators established that the transcriptional regulation of the metabolic system in S. cerevisiae drives flux toward linearity. Even at junctions, there is a tendency for a single pair of incoming and outgoing reactions to be coexpressed, thus prioritizing the flow of metabolites through selected pathways.
The major findings for S. cerevisiae also apply to E. coli: There is very high co-regulation of linear-flow reactions, and junctions preferentially target a single pair of flow-connected enzymes. However, in contrast to the above study, we do not see differential regulation of isozymes at pathway junctions. Much of this difference may be due to the contrasting accuracy in assigning isozymes to pathways (see Methods).
A study by Kharchenko et al. (2005) showed that coexpression decreases with increased separation between enzyme pairs in the metabolic network. Most recently, Notebaart et al. (2008) reasoned that the level of enzyme coexpression is best predicted by looking at the correlation of their metabolic fluxes. In fact, we demonstrate that both observations can be explained by considering the nature of the separation between enzymes; we suggest that the presence of a divergent junction serves to decouple both the transcriptional regulation and metabolic flux of enzymes.
Of the above studies (Ihmels et al. 2004; Kharchenko et al. 2005; Notebaart et al. 2008), only Notebaart et al. made use of available transcriptional regulatory data, whereas the other two inferred patterns of co-regulation from gene expression data. Here, we incorporated regulatory interaction data in addition to gene expression information, and both data sets support all our observations, confirming the robustness of the results. More importantly, the use of direct interaction data allows us to gain insights that are inaccessible from gene expression alone, including the hierarchical classification of general and specific TFs; differences in regulation of catabolic, anabolic, and central metabolic pathways; and combined use of overlapping and distinct TFs at pathway junctions.
Impact of perturbations on metabolic regulation
Given the observed precision of the metabolic regulatory apparatus, it is surprising that the few TF-knockout experiments that have been performed—even those involving major regulators—generally do not cause lethality in E. coli (Covert et al. 2004; Perrenoud and Sauer 2005; Blot et al. 2006; Bradley et al. 2007). Mutant strains lacking FNR, Fis, and ArcA display the expected gene expression changes (i.e., the pathways that are under direct regulation), but show only slight differences in growth rate. Moreover, a recent study has shown that E. coli tolerates substantial artificial rewiring of the regulatory network through the introduction of new binding sites to promoters (Isalan et al. 2008). We anticipate that the modularity imposed by divergent junctions is a major underlying reason for this robustness, as they ensure that detrimental regulatory perturbations do not spill over into neighboring pathways. Indeed, the metabolic system appears to maintain stable small-molecule concentrations by recruiting alternative pathways, even when central metabolic enzymes are deleted (Ishii et al. 2007). These responses are likely to impose a cost on the organism: Most knockouts are tested in isolation, but significant growth defects may become apparent if they are grown in competition with the wild-type strain in the appropriate condition. Although deletions of general regulators are not lethal to the cell, it has been shown, using systematic genome-scale gene deletion data sets in yeast, that these TFs tend to have a greater impact on cell growth than specific TFs (Yu et al. 2004). It would be interesting to test if this is true in E. coli using similar genome-scale experimental studies and computational analysis.
These observations suggest that transcriptional regulators of metabolic processes are probably not good targets for future antibiotic design. Instead, it may be more fruitful to target the enzymes themselves. We propose that isozymes make good candidates, as they are generally coexpressed at highly connected junctions. By targeting isozyme pairs at strategic locations, it should be possible to choke an organism’s metabolic system.
Our findings also have implications for the synthetic introduction of new metabolic enzymes into a bacterium. A major challenge when synthetic pathways overlap with the indigenous cellular metabolic network is the prevention of potentially disruptive interference between pathways. This could be minimized by ensuring that enzymes are incorporated close to divergent junctions in the existing network so that there is no cross-talk.
Future work to complete the metabolic regulatory network
Our work here has benefited from the availability of large, genome-scale descriptions of the metabolic and regulatory systems, and the results are robust to gaps and errors in the underlying data. However, it is clear that we still do not have a complete picture of metabolic regulation. There is an uneven distribution of information, as more is known about regulators of catabolic pathways than anabolic ones. And, as highlighted by Figure 1, certain functions such as energy production, sugar, and amino acid metabolism are better represented. In fact, we lack regulatory data for more than half the enzymes in the metabolic network; in particular, little is known about the control of lipid metabolism, secondary metabolite metabolism, and cell wall biosynthesis, which are critical for cell survival. An important area of future experimental work in microbiology will be to build a complete network of bacterial transcriptional regulation.
Finally, many of the results presented here have also been observed for S. cerevisiae (Ihmels et al. 2004). This is remarkable because of the enormous divergence between the regulatory machineries of E. coli and yeast. Given this, we propose that the patterns of transcriptional control that we report might apply to many prokaryotic systems and, perhaps, even to some eukaryotic organisms.
Methods
Data sources
Metabolic network
Metabolite, enzyme, reaction, and pathway information were obtained from EcoCyc 9.0 (Keseler et al. 2005). We used EcoCyc pathway annotations as they are manually curated by human experts and have been shown to be functionally coherent (Green and Karp 2006).
We removed transport (reaction type TR* or ABC* in EcoCyc) and tRNA charging reactions. We also excluded interactions mediated by the following compounds: ATP, ADP, AMP, Pi, NAD, NADH, NADP, NADPH, FAD, FADH2, NH3, NH4+, CO2, H2O2, O2, H2, CoA, H2O, and any other metabolite labeled as “non-metabolic-compounds,” “anions,” “cations,” “coenzymes,” or “coenzyme-groups.”
This gives a data set of 628 metabolites, 781 enzymes, 788 reactions, and 158 pathways.
Transcriptional regulatory network
Transcription factor to target gene regulatory interactions were obtained from RegulonDB 5.0 (Salgado et al. 2006). Additional regulatory interactions for CRP were included from ChIP-chip data (Grainger et al. 2005). From this we excluded target genes that did not belong to the metabolic network above. This resulted in a data set of 111 TFs, 388 targets, and 913 regulatory interactions. Data regarding 43 TF-metabolite interactions were obtained from EcoCyc.
Gene expression data
Raw .CEL files for 221 transcriptomic hybridizations to Affymetrix E. coli Antisense v2 GeneChips were downloaded from the M3D database (Faith et al. 2007).
Other data
Functional classifications for E. coli genes were obtained from the Clusters of Orthologous Groups database (Tatusov et al. 2003);
information about the operon organization of E. coli genes was obtained from RegulonDB 5.0;
the set for 2777 enzyme pairs with coupled fluxes was taken from Notebaart et al. (2008);
protein sequences for ortholog identification were downloaded from the Kyoto Encyclopedia of Genes and Genomes database (Kanehisa et al. 2006); and
organism phenotype data (aerobic, anaerobic, facultative) were obtained from the NCBI microbial genomes database (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi).
Classification of catabolic, anabolic, and central energy metabolic enzymes
Enzymes were classified as catabolic, anabolic, or central energy metabolism according to EcoCyc annotations. All enzymes belonging to the glycolysis, TCA cycle, glyoxylate shunt, and pentose phosphate pathways were classed as central energy metabolism. Enzymes belonging to two or more categories (e.g., catabolism and anabolism) were excluded from the relevant sections of analysis.
Classification of general and specific TFs
TFs were classified according to the functional annotations of target genes: specific if all targets belonged to the same EcoCyc pathway or COG functional category; general if targets belonged to more than one COG functional category. For general TFs, the enrichment for functional categories was assessed using the Fisher-test followed by False Discovery Rate correction for multiple testing (Benjamini and Hochberg 1995).
TF was classified as pathway (or COG) specific if all its targets shared a common EcoCyc pathway (or COG function) membership. General TFs were identified as follows: For each TF, enrichment of a given COG function among its targets was assessed using Fisher-test followed by FDR multiple testing. This procedure has been applied to identification of enriched functional categories in a given set of genes (Al-Shahrour et al. 2004). A TF was flagged as general if at least one function was statistically enriched (FDR ≤ 0.01) and there were at least 10 targets belonging to this function.
Derivation of enzyme pair configurations
We first defined a metabolic reaction network as a bipartite graph with two types of edges: (1) leading from a metabolite to a reaction that consumes the metabolite and (2) from a reaction to its product. Directionality of reactions was derived based on pathway annotations.
The above network was then used to identify enzyme pairs connected in different configurations as given below:
Flow configurations
These are networks that represent metabolite flow from one reaction to the next.
Linear flow: E1 (enzyme 1) is connected to E2 (enzyme 2) if the product of E1 is product of no other enzyme and is reactant to only E2;
Junction flow: E1 is connected to E2 if a product of E1 is substrate to E2 and this edge is not part of the linear-flow network. Here, any metabolite that is consumed by more than one reaction is a divergent metabolite; all other metabolites are convergent metabolites.
Non-flow configurations
These networks do not represent the direction of metabolite flow.
3. Convergent: E1 is connected to E2 if they share a common product.
4. Divergent: E1 is connected to E2 if they share a common reactant.
In the first stage, the networks were represented as pairs of reactions represented by EcoCyc reaction IDs. Connectivity properties of reactions (not individual enzyme genes) were used to derive the four network types. In the second step, reaction pairs were converted to corresponding enzyme pairs; any reaction pair might be represented by more than one enzyme pair because of the involvement of several genes in catalyzing a single reaction.
In the above analysis, all isozymes are mapped to the same reactions without any discrimination. A separate analysis was done for isozymes in order to achieve a more complete comparison with the work of Ihmels et al. (2004). In EcoCyc, every reaction ID is mapped to an enzymatic conversion. Each enzymatic conversion is represented by an enzyme that might be composed of one or more genes. Any reaction that is mapped to more than one enzymatic conversion was taken to be regulated by isozymes. Every pair of enzymatic conversions assigned to the same reaction ID was then analyzed at the level of individual enzyme genes.
In order to calculate the nature of regulation of isozymes at junctions, we made a list of junctions involving reactions using isozymes. If the reaction involving isozymes fed into the junction, then there should exist more than one outgoing reaction. Similarly, if the reaction involving isozymes led out of the junction, then there should be more than one incoming reaction. The nature of regulation was calculated for every quadruplet of enzymes: two isozymes for the same reaction and two enzymes that operate immediately upstream or downstream of the isozymes-involving reaction. Any pair of reactions was flagged as being co-regulated if the coexpression correlation across 221 arrays was ≥0.30.
Path lengths in metabolic networks
The distance between any two reactions was calculated using the nonheuristic Breadth First Search algorithm. For these calculations, the union of the linear-flow and the junction-flow networks was used. The distance between a metabolite and a reaction (used to measure the separation between a metabolite that binds a TF and an enzyme regulated by the TF) was defined as either (1) the number of reactions upstream of the metabolite and downstream from the target reaction or (2) the number of reactions downstream from the metabolite and upstream of the target reaction along the shortest path separating one of the reactions directly involving the metabolite and the target reaction. The shortest of the above two distances was used.
Enzymes involved in the shortest paths between any two enzymes were identified using the iGraph package implemented in R.
Microarray data processing
Raw CEL files were processed using the RMA procedure (Irizarry et al. 2003) implemented in Bioconductor (Reimers and Carey 2006), as it was previously shown to be the best procedure for this data set (Faith et al. 2007). This results in a matrix of log-normalized expression measures in which each row represents a gene and each column an array. Pearson correlation coefficients between gene expression profiles of every pair of enzymes across all arrays were calculated from this expression matrix.
Identification of orthologs
Orthologs were identified using the standard approach of bidirectional best-hit FASTA (Pearson and Lipman 1988). For this, FASTA version 34 and an upper expectation cutoff of 10−4 were used.
Statistical tests
Standard tests
Five statistical tests were used in this study: (1) a one-tailed Mann-Whitney test, a nonparametric test, for comparing two distributions; (2) a one-tailed Fisher exact test, followed by application of FDR in which more than one P-value was computed, for categorical data; (3) a Pearson correlation coefficient to assess coexpression; (4) a Phi correlation coefficient for nominal variables to measure coevolution; and (5) a Mutual information score for testing association between coexpression and coevolution correlations. Details of where each of these tests was used are presented in context in the Results section.
In our calculations of Pearson correlations, we found that 75% of all gene pairs with correlation P-value < 0.001 have correlation coefficients greater than 0.30. This cut-off was used to define co-regulation primarily in the analysis of isozymes because of a lack of relevant information in the transcriptional regulatory network.
Random simulations for significance testing of the highest of n coexpression correlations
For any given set of m metabolites, with any metabolite mediating connections between ni (1 ≤ i ≤ m) pairs of enzymes, a distribution (size = m) of highest expression correlations among all ni pairs of enzymes was obtained. In addition, 1000 random distributions, each of size m, and each value being the highest coexpression correlation among ni randomly chosen pairs of enzyme genes, were obtained. The actual distribution was compared against each of the 1000 random distributions using the Mann-Whitney test, and P-values (under the null hypothesis that the actual distribution is less than or equal to the random distribution) were derived. This was done for the junction-flow (where each P-value was <2.2 × 10−16) and for the non-flow (P-values ranging from 0.004 and 0.2) networks.
Random simulation for significance testing of the mutual information between coexpression and coevolution
We obtained 1000 sets of 9798 random pairs of correlation coefficients, one value representing coexpression and the other coevolution correlation, while maintaining the distributions of each of the two coefficients. For each of the 1000 random sets, mutual information between the two variables was calculated. A Z-score for the actual mutual information value was derived using the formula:
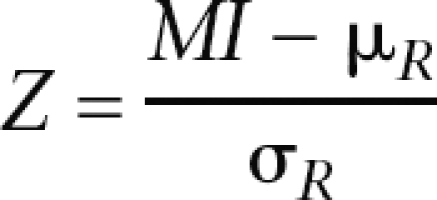 |
where MI is the actual mutual information score, μR is the mean, and σR is the standard deviation of mutual information scores calculated for the 1000 random data sets.
All these calculations were done using a combination of Perl (http://www.perl.com) and the R environment (http://www.r-project.org). Mutual information was calculated using the R package supplied by Daub et al. (2004).
Normalization of coexpression versus coevolution correlations
In Figure 6B, we used the following normalization procedure to obtain a measure of the overlap between coexpression and coevolution correlations corresponding to each bin.
If p is the number of bins for coexpression correlations and q is that for coevolution correlations, and Ni,j represents the number of entries belonging to the intersection of the ith coexpression bin and the jth coevolution bin, then
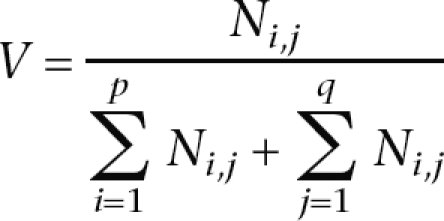 |
Robustness of results
In order to test the robustness of results obtained, we generated transcriptional regulatory networks with deletion of or errors in 5%, 10%, 15%, 20%, 25%, and 30% of all edges. We then compared the medians of the degree distributions obtained for catabolic, anabolic, and central metabolic genes across these networks against the real network. We also used the percentage of enzyme pairs with matching sets of regulators in the flow and the non-flow metabolic configurations (local regulation) as a parameter to test the robustness of results. The effect on local regulation of similar deletions and alterations in the various forms of the metabolic network was also tested. In general, the results from these calculations are qualitatively similar, suggesting that our findings are unlikely to be affected by any incompleteness or inaccuracies in the data set (Supplemental Fig. 12A,B).
Acknowledgments
A.S.N.S. thanks the Inlaks Foundation, Cambridge Commonwealth Trusts, and St. John’s College, Cambridge for funding. M.M.B. acknowledges the MRC Laboratory of Molecular Biology, Schlumberger Ltd., and Darwin College for support. G.M.F. and N.M.L. acknowledge funding from the BBSRC Grant “Genomic Analysis of Regulatory Networks for Bacterial Differentiation and Multicellular Behaviour.”
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.079715.108.
References
- Ali Azam T., Iwata A., Nishimura A., Ueda S., Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J. Bacteriol. 1999;181:6361–6370. doi: 10.1128/jb.181.20.6361-6370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaas E., Kovacs B., Vicsek T., Oltvai Z.N., Barabasi A.L. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature. 2004;427:839–843. doi: 10.1038/nature02289. [DOI] [PubMed] [Google Scholar]
- Almaas E., Oltvai Z.N., Barabasi A.L. The activity reaction core and plasticity of metabolic networks. PLoS Comput. Biol. 2005;1:e68. doi: 10.1371/journal.pcbi.0010068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U. Network motifs: Theory and experimental approaches. Nat. Rev. Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- Al-Shahrour F., Diaz-Uriarte R., Dopazo J. FatiGO: A web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- Anantharaman V., Koonin E.V., Aravind L. Regulatory potential, phyletic distribution and evolution of ancient, intracellular small-molecule-binding domains. J. Mol. Biol. 2001;307:1271–1292. doi: 10.1006/jmbi.2001.4508. [DOI] [PubMed] [Google Scholar]
- Balazsi G., Barabasi A.L., Oltvai Z.N. Topological units of environmental signal processing in the transcriptional regulatory network of Escherichia coli. Proc. Natl. Acad. Sci. 2005;102:7841–7846. doi: 10.1073/pnas.0500365102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett C.L., Herring C.D., Reed J.L., Palsson B.O. The global transcriptional regulatory network for metabolism in Escherichia coli exhibits few dominant functional states. Proc. Natl. Acad. Sci. 2005;102:19103–19108. doi: 10.1073/pnas.0505231102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J.R. Stat. Soc. Ser. B Methodol. 1995;57:125–133. [Google Scholar]
- Blot N., Mavathur R., Geertz M., Travers A., Muskhelishvili G. Homeostatic regulation of supercoiling sensitivity coordinates transcription of the bacterial genome. EMBO Rep. 2006;7:710–715. doi: 10.1038/sj.embor.7400729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borenstein E., Kupiec M., Feldman M.W., Ruppin E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc. Natl. Acad. Sci. 2008;105:14482–14487. doi: 10.1073/pnas.0806162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley M.D., Beach M.B., de Koning A.P., Pratt T.S., Osuna R. Effects of Fis on Escherichia coli gene expression during different growth stages. Microbiology. 2007;153:2922–2940. doi: 10.1099/mic.0.2007/008565-0. [DOI] [PubMed] [Google Scholar]
- Covert M.W., Palsson B.O. Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J. Biol. Chem. 2002;277:28058–28064. doi: 10.1074/jbc.M201691200. [DOI] [PubMed] [Google Scholar]
- Covert M.W., Knight E.M., Reed J.L., Herrgard M.J., Palsson B.O. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- Daub C.O., Steuer R., Selbig J., Kloska S. Estimating mutual information using B-spline functions—an improved similarity measure for analysing gene expression data. BMC Bioinformatics. 2004;5:118. doi: 10.1186/1471-2105-5-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekel E., Alon U. Optimality and evolutionary tuning of the expression level of a protein. Nature. 2005;436:588–592. doi: 10.1038/nature03842. [DOI] [PubMed] [Google Scholar]
- Dinsdale E.A., Edwards R.A., Hall D., Angly F., Breitbart M., Brulc J.M., Furlan M., Desnues C., Haynes M., Li L., et al. Functional metagenomic profiling of nine biomes. Nature. 2008;452:629–632. doi: 10.1038/nature06810. [DOI] [PubMed] [Google Scholar]
- Faith J.J., Hayete B., Thaden J.T., Mogno I., Wierzbowski J., Cottarel G., Kasif S., Collins J.J., Gardner T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink G.R. Global regulation in fungi. Antonie Van Leeuwenhoek. 1987;53:353–356. doi: 10.1007/BF00400560. [DOI] [PubMed] [Google Scholar]
- Gilbert W., Muller-Hill B. Isolation of the Lac repressor. Proc. Natl. Acad. Sci. 1966;56:1891–1898. doi: 10.1073/pnas.56.6.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S.R., Pop M., Deboy R.T., Eckburg P.B., Turnbaugh P.J., Samuel B.S., Gordon J.I., Relman D.A., Fraser-Liggett C.M., Nelson K.E. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger D.C., Hurd D., Harrison M., Holdstock J., Busby S.J. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc. Natl. Acad. Sci. 2005;102:17693–17698. doi: 10.1073/pnas.0506687102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M.L., Karp P.D. The outcomes of pathway database computations depend on pathway ontology. Nucleic Acids Res. 2006;34:3687–3697. doi: 10.1093/nar/gkl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrgard M.J., Covert M.W., Palsson B.O. Reconciling gene expression data with known genome-scale regulatory network structures. Genome Res. 2003;13:2423–2434. doi: 10.1101/gr.1330003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring C.D., Raghunathan A., Honisch C., Patel T., Applebee M.K., Joyce A.R., Albert T.J., Blattner F.R., van den Boom D., Cantor C.R., et al. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat. Genet. 2006;38:1406–1412. doi: 10.1038/ng1906. [DOI] [PubMed] [Google Scholar]
- Ibarra R.U., Fu P., Palsson B.O., DiTonno J.R., Edwards J.S. Quantitative analysis of Escherichia coli metabolic phenotypes within the context of phenotypic phase planes. J. Mol. Microbiol. Biotechnol. 2003;6:101–108. doi: 10.1159/000076740. [DOI] [PubMed] [Google Scholar]
- Ihmels J., Levy R., Barkai N. Principles of transcriptional control in the metabolic network of Saccharomyces cerevisiae. Nat. Biotechnol. 2004;22:86–92. doi: 10.1038/nbt918. [DOI] [PubMed] [Google Scholar]
- Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isalan M., Lemerle C., Michalodimitrakis K., Horn C., Beltrao P., Raineri E., Garriga-Canut M., Serrano L. Evolvability and hierarchy in rewired bacterial gene networks. Nature. 2008;452:840–845. doi: 10.1038/nature06847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N., Nakahigashi K., Baba T., Robert M., Soga T., Kanai A., Hirasawa T., Naba M., Hirai K., Hoque A., et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science. 2007;316:593–597. doi: 10.1126/science.1132067. [DOI] [PubMed] [Google Scholar]
- Ishizuka H., Hanamura A., Kunimura T., Aiba H. A lowered concentration of cAMP receptor protein caused by glucose is an important determinant for catabolite repression in Escherichia coli. Mol. Microbiol. 1993;10:341–350. doi: 10.1111/j.1365-2958.1993.tb01960.x. [DOI] [PubMed] [Google Scholar]
- Janga S.C., Salgado H., Collado-Vides J., Martinez-Antonio A. Internal versus external effector and transcription factor gene pairs differ in their relative chromosomal position in Escherichia coli. J. Mol. Biol. 2007;368:263–272. doi: 10.1016/j.jmb.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Jeong H., Tombor B., Albert R., Oltvai Z.N., Barabasi A.L. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Hattori M., Aoki-Kinoshita K.F., Itoh M., Kawashima S., Katayama T., Araki M., Hirakawa M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan S., Bren A., Zaslaver A., Dekel E., Alon U. Diverse two-dimensional input functions control bacterial sugar genes. Mol. Cell. 2008;29:786–792. doi: 10.1016/j.molcel.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseler I.M., Collado-Vides J., Gama-Castro S., Ingraham J., Paley S., Paulsen I.T., Peralta-Gil M., Karp P.D. EcoCyc: A comprehensive database resource for Escherichia coli. Nucleic Acids Res. 2005;33:D334–D337. doi: 10.1093/nar/gki108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko P., Church G.M., Vitkup D. Expression dynamics of a cellular metabolic network. Mol. Syst. Biol. 2005;1:0016. doi: 10.1038/msb4100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesov G., Wunderlich Z., Laikova O.N., Gelfand M.S., Mirny L.A. How gene order is influenced by the biophysics of transcription regulation. Proc. Natl. Acad. Sci. 2007;104:13948–13953. doi: 10.1073/pnas.0700672104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreimer A., Borenstein E., Gophna U., Ruppin E. The evolution of modularity in bacterial metabolic networks. Proc. Natl. Acad. Sci. 2008;105:6976–6981. doi: 10.1073/pnas.0712149105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bigas N., De S., Teichmann S.A. Functional protein divergence in the evolution of Homo sapiens. Genome Biol. 2008;9:R33. doi: 10.1186/gb-2008-9-2-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada-Chavez I., Janga S.C., Collado-Vides J. Bacterial regulatory networks are extremely flexible in evolution. Nucleic Acids Res. 2006;34:3434–3445. doi: 10.1093/nar/gkl423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada-Chavez I., Angarica V.E., Collado-Vides J., Contreras-Moreira B. The role of DNA-binding specificity in the evolution of bacterial regulatory networks. J. Mol. Biol. 2008;379:627–643. doi: 10.1016/j.jmb.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H.W., Buer J., Zeng A.P. Hierarchical structure and modules in the Escherichia coli transcriptional regulatory network revealed by a new top-down approach. BMC Bioinformatics. 2004;5:199. doi: 10.1186/1471-2105-5-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan Babu M., Teichmann S.A. Evolution of transcription factors and the gene regulatory network in Escherichia coli. Nucleic Acids Res. 2003;31:1234–1244. doi: 10.1093/nar/gkg210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan Babu M., Teichmann S.A., Aravind L. Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J. Mol. Biol. 2006;358:614–633. doi: 10.1016/j.jmb.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Mangan S., Zaslaver A., Alon U. The coherent feedforward loop serves as a sign-sensitive delay element in transcription networks. J. Mol. Biol. 2003;334:197–204. doi: 10.1016/j.jmb.2003.09.049. [DOI] [PubMed] [Google Scholar]
- Mangan S., Itzkovitz S., Zaslaver A., Alon U. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J. Mol. Biol. 2006;356:1073–1081. doi: 10.1016/j.jmb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Martínez-Antonio A., Collado-Vides J. Identifying global regulators in transcriptional regulatory networks in bacteria. Curr. Opin. Microbiol. 2003;6:482–489. doi: 10.1016/j.mib.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Martínez-Antonio A., Janga S.C., Thieffry D. Functional organisation of Escherichia coli transcriptional regulatory network. J. Mol. Biol. 2008;381:238–247. doi: 10.1016/j.jmb.2008.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menchaca-Mendez R., Janga S.C., Collado-Vides J. The network of transcriptional interactions imposes linear constrains in the genome. OMICS. 2005;9:139–145. doi: 10.1089/omi.2005.9.139. [DOI] [PubMed] [Google Scholar]
- Notebaart R.A., Teusink B., Siezen R.J., Papp B. Co-regulation of metabolic genes is better explained by flux coupling than by network distance. PLoS Comput. Biol. 2008;4:e26. doi: 10.1371/journal.pcbi.0040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson W.R., Lipman D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrenoud A., Sauer U. Impact of global transcriptional regulation by ArcA, ArcB, Cra, Crp, Cya, Fnr, and Mlc on glucose catabolism in Escherichia coli. J. Bacteriol. 2005;187:3171–3179. doi: 10.1128/JB.187.9.3171-3179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P. Orthologous transcription factors in bacteria have different functions and regulate different genes. PLoS Comput. Biol. 2007;3:1739–1750. doi: 10.1371/journal.pcbi.0030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranea J.A., Grant A., Thornton J.M., Orengo C.A. Microeconomic principles explain an optimal genome size in bacteria. Trends Genet. 2005;21:21–25. doi: 10.1016/j.tig.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Ravasz E., Somera A.L., Mongru D.A., Oltvai Z.N., Barabasi A.L. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- Reece R.J., Beynon L., Holden S., Hughes A.D., Rebora K., Sellick C.A. Nutrient-regulated gene expression in eukaryotes. Biochem. Soc. Symp. 2006;2006:85–96. doi: 10.1042/bss0730085. [DOI] [PubMed] [Google Scholar]
- Reimers M., Carey V.J. Bioconductor: An open source framework for bioinformatics and computational biology. Methods Enzymol. 2006;411:119–134. doi: 10.1016/S0076-6879(06)11008-3. [DOI] [PubMed] [Google Scholar]
- Salgado H., Gama-Castro S., Peralta-Gil M., Diaz-Peredo E., Sanchez-Solano F., Santos-Zavaleta A., Martinez-Flores I., Jimenez-Jacinto V., Bonavides-Martinez C., Segura-Salazar J., et al. RegulonDB (version 5.0): Escherichia coli K-12 transcriptional regulatory network, operon organization, and growth conditions. Nucleic Acids Res. 2006;34:D394–D397. doi: 10.1093/nar/gkj156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr S.S., Milo R., Mangan S., Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- Shlomi T., Eisenberg Y., Sharan R., Ruppin E. A genome-scale computational study of the interplay between transcriptional regulation and metabolism. Mol. Syst. Biol. 2007a;3:101. doi: 10.1038/msb4100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T., Herrgard M., Portnoy V., Naim E., Palsson B.O., Sharan R., Ruppin E. Systematic condition-dependent annotation of metabolic genes. Genome Res. 2007b;17:1626–1633. doi: 10.1101/gr.6678707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R.L., Fedorova N.D., Jackson J.D., Jacobs A.R., Kiryutin B., Koonin E.V., Krylov D.M., Mazumder R., Mekhedov S.L., Nikolskaya A.N., et al. The COG database: An updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J., Ley R.E., Mahowald M.A., Magrini V., Mardis E.R., Gordon J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Yu H., Gerstein M. Genomic analysis of the hierarchical structure of regulatory networks. Proc. Natl. Acad. Sci. 2006;103:14724–14731. doi: 10.1073/pnas.0508637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Greenbaum D., Xin Lu H., Zhu X., Gerstein M. Genomic analysis of essentiality within protein networks. Trends Genet. 2004;20:227–231. doi: 10.1016/j.tig.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Zaslaver A., Mayo A.E., Rosenberg R., Bashkin P., Sberro H., Tsalyuk M., Surette M.G., Alon U. Just-in-time transcription program in metabolic pathways. Nat. Genet. 2004;36:486–491. doi: 10.1038/ng1348. [DOI] [PubMed] [Google Scholar]