

Abstract
The Daphniphyllum alkaloids are a group of highly
complex polycyclic alkaloids. Examination of the structures if several
members of this family of natural products led to a hypothesis about
their mode of biosynthesis (depicted in Scheme SI). Based on this
hypothetical biosynthetic pathway, a laboratory synthesis was designed
that incorporated as a key transformation the novel one-pot
transformation of dialdehyde 24 to pentacyclic unsaturated amine 25.
This process turned out to be an exceptionally efficient way to
construct the pentacyclic nucleus of the Daphniphyllum
alkaloids. However, a purely fortuitous discovery, resulting from
accidental use of methylamine rather than ammonia, led to a great
improvement in the synthesis and suggests an even more attractive
possible biosynthesis.
Organic chemists who like to design and execute multistep syntheses or complex molecules have the goal of eventually putting themselves out of business. We hope to do this by becoming so proficient at what we do that synthesis becomes a routine task that can be relegated to a well-trained technician, or even a machine. We can fantasize that some 25th century physician, encountering a new disease that requires a certain specific organic molecule, may simply draw the structure of that molecule, complete with stereochemical information, and receive in return a detailed recipe for its synthesis. Better still, the computer might program a robot to actually perform the synthesis and deliver an actual sample of the desired molecule.
The foregoing fanciful scenario is obviously the stuff that popular
television shows are made of and bears little resemblance to modern
reality. It is true that organic chemists have become quite good at
executing multistep synthesis, and some truly impressive molecules have
been prepared in the laboratory. Classic examples are the syntheses of
cobyric acid, which was conquered in the 1970s by Woodward,
Eschenmoser, and coworkers (1–3), and palytoxin, which yielded to
Kishi and coworkers in the 1980s (4). However, these classic syntheses,
although important because they demonstrate that we can
synthesize virtually any molecule for which we can write a structure,
were both pitifully inefficient for the purpose of providing
significant quantities of material in a timely manner. Indeed, each of
these landmark accomplishments required large teams of experimentalists
who painstakingly assembled a few milligrams of the target molecule
using more than a hundred separate reactions over a decade or
more.
Indeed, when confronted with the need for significant amounts of a
complex organic compound, modern-day chemists are almost helpless. A
notable example, which actually represents something of an
embarrassment to the whole field, is taxol. This diterpenoid,
originally isolated from the Pacific yew, an endangered species that
occurs mostly in old-growth Redwood forests, has useful properties for
the treatment of certain cancers. Although a relatively abundant source
of a related compound was eventually discovered, there was a period of
more than a decade when total synthesis appeared to be the only
solution to the supply problem. During this time a great many research
teams worked on the taxol problem The first syntheses were only
achieved in 1994 (5–7) and none of the taxol syntheses that have been
reported to date are sufficiently efficient to provide
pharmaceutically-relevant quantities of the
drug.
Yet there seems to be a widely-held view that organic synthesis is such a “mature” subject that there is no longer a need for basic research in the field. In fact, nothing could be farther from the truth. Now that we have provided abundant proof that, with large teams of trained experimentalists and given years to achieve the goal, we can synthesize such molecules as cobyric acid, palytoxin, and taxol, we are obliged to take the next step and discover how to perform such tasks efficiently. This goal will only be met if organic chemists continue to explore the margins of synthetic practicality by attempting to solve synthetic problems of ever-increasing complexity, and if we continue to revisit the old problems and try to solve them in new, more efficient ways. In this way the art of organic synthesis will continue to become more and more sophisticated.
One of the strategies we have been used to look for efficient synthetic routes to complex natural products is to try and figure out how nature has solved the problem. The basic assumption of this approach is that nature is the quintessential process development chemist. We think that the molecular frameworks of most natural products arise by intrinsically favorable chemical pathways—favorable enough that the skeleton could have arisen by a nonenzymic reaction in the primitive organism. If a molecule produced in this purely chemical manner was beneficial to the organism, enzymes would eventually have evolved to facilitate the production of this useful material. Further optimization of the biological activity might then have been accomplished by cytochrome P450-mediated oxidations. Once again, those oxidation products that conferred an evolutionary advantage to the organism would have promoted selection of oxidase variants with appropriate binding selectivity.
A corollary of the foregoing hypothesis is that the coexistence of two structurally related molecules in an organism implies some reasonable chemical pathway from one to the other. Sometimes the chemical relationship is trivial and the pathway from one structure to the other is obvious. However, in other cases one is forced to speculate a chemical conversion that is unknown in the laboratory. A “biomimetic” synthesis is a laboratory synthesis that is based on such reasoning.
To illustrate this approach to discovering efficient organic syntheses,
I would like to briefly account our synthetic work on the
Daphniphyllum alkaloids, a structurally diverse group of
alkaloids that are elaborated by trees of the species
Daphniphyllum macropodum. A full account of this extensive
project has appeared elsewhere (8–15). Although there are now more
than 30 members of the Daphniphyllum alkaloid family,
comprising about seven different skeletal types, for the purpose of the
present discussion we shall consider only the six compounds
1–6, which illustrate four different skeletal
classes. Daphniphylline (1) and secodaphniphylline
(3) represent two of the three basic classes of C-30
Daphniphyllum alkaloids. They are accompanied in nature by
their C-22 counterparts, methyl homodaphniphyllate (4) and
methyl homosecodaphniphyllate (6). Of these two basic
skeletal types, daphniphylline is more common than secodaphniphylline.
For example 1000 kg of D. macropodum leaves yielded 100
g of compound 1 and only 1.1 g of compound 3
(16). Co-occurring with these alkaloids are the highly oxygenated C-22
compound yuzurimine (2) and the C-23 compound daphnilactone
A (5).
Can we deduce anything from the structures of these six alkaloids about
their biosynthesis? In the skeleton of secodaphniphylline
(3) we see that the unbroken squalene molecule may be traced
through the pentacyclic domain. To convert squalene into
secodaphniphylline, four C—C bonds must be formed: C-10 to C-14; C-6
to C-15; C-3 to the C-15 methyl group; and C-7 to the C-10 methyl
group. In addition, the nitrogen is inserted between C-7 and the C-15
methyl group. For daphniphylline, however, the nitrogen seems to have
been inserted between C-10 and its methyl group, which has also become
bonded to C-7. Thus, it is likely that secodaphniphylline precedes
daphniphylline biosynthetically, and that an unsaturated amine such as
compound 7 provides a plausible biosynthetic link between
the two skeletons.
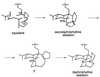
The hypothetical unsaturated amine 7 also contains the
bicyclo[4.4.1]undecane feature that is seen in yuzurimine
(2) and could account for the “extra” carbon that is
found in daphnilactone A (5), if one postulates an
intramolecular Mannich-type cyclization:
![]()
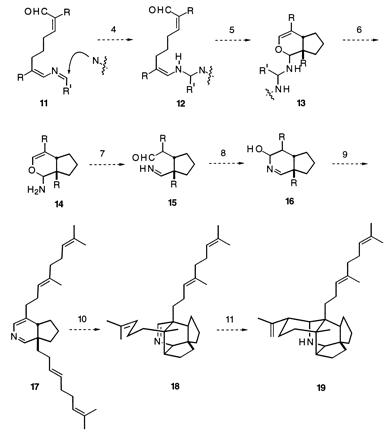
This hypothesis led us to postulate various scenarios whereby squalene might acquire a nitrogen atom and be transformed into the pentacyclic secodaphniphylline skeleton. Eventually, the possible path set forth in Scheme SI emerged. The rough outlines of this proposal are as follows. Step 1 is an oxidative transformation of squalene (8) into a dialdehyde, 9. [Squalene derivatives have been described in which two methyl groups are in the aldehyde oxidation state. One example is petrodial (17).] In step 2 it is proposed that some primary amine, perhaps pyridoxamine or an amino acid, condenses with one of the carbonyl groups of compound 9, giving imine 10. Step 3 is the prototopic rearrangement of a 1-azadiene to a 2-azadiene, a process that is well-precedented for the imines formed from α,β-unsaturated carbonyl compounds and benzylamine (18). Although potassium tert-butoxide was used for the prototopic rearrangement of benzylimines, one can imagine that an imine derived from pyridoxamine or an amino acid would rearrange under much milder conditions. The 2-azadiene that would result from the foregoing prototopic rearrangement is an enimine, and its double bond is not especially nucleophilic. However, if some nucleophilic species adds to the imine double bond, as in step 4, the product 11 is a nucleophilic enamine. The subsequent cyclization to give compound 12 has an exact in vitro precedent in the work of Schreiber et al. (19). In steps 6–9 the resulting bicyclic dihydropyran derivative 12 is transformed into a dihydropyridine derivative (17) by straightforward proton-mediated addition and elimination processes. According to our biosynthetic supposition, 17 would then be converted into compound 18 by a catalyzed Diels–Alder process and the final ring would result from an ene-like cyclization, giving compound 19, the putative primordial Daphniphyllum alkaloid. Because of the likelihood that 19 is the first pentacyclic substance to occur in the biosynthesis of the Daphniphyllum alkaloids, we call it proto-daphniphylline.
Scheme I.
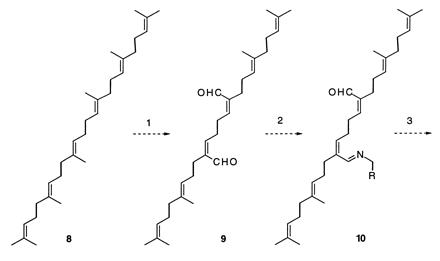
These considerations stimulated us to embark on a program to find
laboratory ways to accomplish the proposed transformations. We focused
our attention first on the final stages of proposed polycyclization
reaction leading to the secodaphniphylline skeleton (17
→ 19). Three simple building blocks, amide 20,
unsaturated ester 21, and unsaturated iodide 22,
were combined in a highly convergent conjugate addition/enolate
alkylation process to obtain ester amide 23 in high yield.
Straightforward methods were then employed to convert this substance
into dialdehyde 24. Compound 24 was treated with
ammonia and then buffered acetic acid to obtain unsaturated amine
25 in excellent yield (64% from 23 to
25).
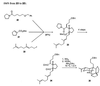
The transformation of compound 24 to 25
involves a cascade of reactions; the two intermediates can be isolated,
as shown below. Treatment of compound 24 with ammonia causes
almost instantaneous transformation of the nonpolar dialdehyde to a
complex mixture of polar materials, from which the dihydropyridine
26 can be isolated in about 45% yield. This compound reacts
rather rapidly upon being treated with ammonium acetate in acetic acid
at room temperature to give compound 27, the result of a
formal intramolecular Diels–Alder reaction. Continued treatment with
warm acetic acid converts compound 27 into the final
product, compound 25.
![]()
Mechanistically, the transformations can be depicted as shown in
the following diagram. Prins-like cyclization of the protonated
dihydropyridine 26-H+ would provide an
intermediate (28) that would cyclize to give compound
27-H+. A second Prins-like attack of the pendant
trisubstituted alkene on the immonium ion would provide a tertiary
carbocation (29), which would undergo 1,5-proton transfer to
provide compound 25-H+. Alternatively, one might
consider the first two bond-forming events to be somewhat synchronous,
with compound 28 representing a transition structure, rather
than an intermediate.
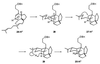
Encouraged by the success of the cyclization process, we sought to
intervene at an earlier stage in the hypothetical biosynthetic pathway
depicted in Scheme SI. To this end, we prepared the
dihydrosqualene dialdehyde 30 and treated it sequentially
with ammonia and warm acetic acid. It was gratifying to find
proto-daphniphylline (19) in the product of this
reaction. Although the isolated yield of compound 19 is only
modest (15%), a great deal has been accomplished by the use of such
simple reaction conditions.
![]()
Dialdehyde 30 presumably reacts with ammonia to give an
intermediate enamine (31) that is analogous to compound
12 in Scheme SI. The first crucial C—C
bond-forming step is an intramolecular Michael addition, in which
compound 32, containing the five-membered ring, is
produced.
![]()
The low yield observed in conversion of compound 30 to 19 must be largely due to inefficiency of this first C—C bond is formed, in light of the fact that dialdehyde 24, which already contains this bond, undergoes the cyclization reaction in 85–90% yield. This is not really surprising, in light of the fact that enamines are such highly reactive compounds. In fact, primary enamines are virtually unknown species in the literature. Much more common are secondary and tertiary enamines. Therefore, one might expect that cyclization of compound 31 would occur in higher yield if a primary or secondary amine is used, rather than ammonia. However, the Daphniphyllum alkaloids have no additional alkyl group attached to the nitrogen.
A solution to the foregoing dilemma was provided not by design,
but through a remarkable accident. At one point in our utilization of
the cyclization protocol for the synthesis of various
Daphniphyllum alkaloids, one of my graduate student
coworkers carried out the normal protocol that we had developed, using
dialdehyde 24 as the substrate. To our amazement, the
product of this reaction was not the normal one, compound
25, but its dihydro derivative compound 32
instead. Remarkably, compound 32 was produced in very good
yield (about 75%).
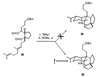
Careful examination of all of the reagents, solvents, and reaction
conditions soon revealed the cause for this unexpected result: a
mislabeled reagent. Shortly before the strange reaction was carried
out, our supply of ammonia had been exhausted and my coworker had
obtained a new lecture bottle from a friend in another research group.
The new lecture bottle, although clearly labeled “Ammonia,” was
found by mass spectral analysis to contain only methylamine. The
mysterious transformation could now be understood in terms of the
mechanism presented in Scheme II. That is, methylamine
merely substitutes for ammonia in formation of the dihydropyridinium
ion 34, which undergoes the intramolecular Diels–Alder
reaction normally to give unsaturated immonium ion 35. This
compound cyclizes as usual, providing carbocation 36. At
this point something different happens. Instead of 1,5-proton transfer,
there is a 1,5-hydride shift, leading to compound 37. Upon
aqueous workup, the immonium ion is hydrolyzed, providing compound
32.
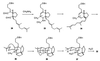
This fortuitous discovery suggested a possible solution to the problem
of low yield in the pentacyclization process with the dihydrosqualene
dialdehyde 30. Indeed, when compound 30 was
treated successively with methylamine and warm acetic acid, we were
delighted to find that dihydro-proto-daphniphylline
(38) is formed in 65% yield.
![]()
This marvelous transformation results in the formation of seven new sigma bonds and five rings. It is fully diastereoselective, and a necessary consequence of the reaction mechanism is that one of three similar carbon–carbon double bonds is regioselectively saturated! Although these are merely laboratory model studies, we think that Nature must also have discovered this easy and highly efficient method for creation of proto-daphniphylline. It is probably no coincidence that not a single one of the more than three dozen known Daphniphyllum alkaloids actually has an isopropenyl group, so the source of biosynthetic nitrogen is probably an alkylamine. Perhaps it is pyridoxamine, or maybe the nitrogen comes from an amino acid.
This project illustrates how one can take clues from the actual structures of complex natural products that can lead to the discovery of amazingly simple laboratory chemistry. We have done much more with the Daphniphyllum alkaloids, including the synthesis of the postulated unsaturated amine 7 and demonstration of its facile conversion into the daphniphylline (4) and daphnilactone (5) ring systems. One thing we have not yet done, but something that is still on our agenda, is to enter the postulated biosynthesis (Scheme SI) even earlier, by preparing squalene dialdehyde 9 and studying its reactions with likely nitrogen sources, such as pyridoxamine.
An important lesson from the project to date is the importance of serendipity. Although the idea of condensing dihydrosqualene dialdehyde 30 with ammonia came through fairly logical reasoning, the important breakthrough of using alkyl-amines instead of ammonia was completely irrational, and was made possible only because a careful student took the time to fully investigate a completely unexpected result. It is not unusual for organic reactions, even ones that have become rather routine, to go completely astray and give undesired products. It is too often the case that, confronted with a such a failed reaction, the experimentalist is concerned only with making things right again. The normal way is to repurify the organic starting material and use completely fresh materials. If my coworker had taken this path, and merely opened a fresh lecture bottle of ammonia, he would probably have been happy to find that the expected product was again formed. However, he would have missed the most exciting part of the whole project, and missed an important insight into how the Daphniphylline skeleton is really formed in nature.
References
- 1.Woodward R B. Pure Appl Chem. 1973;33:145–177. doi: 10.1351/pac197333010145. [DOI] [PubMed] [Google Scholar]
- 2.Eschenmoser A, Wintner C. Science. 1977;196:1410–1420. doi: 10.1126/science.867037. [DOI] [PubMed] [Google Scholar]
- 3.Eschenmoser A. Angew Chem Int Edn Engl. 1988;27:5–39. [Google Scholar]
- 4.Armstrong R W, Beau J-M, Cheon S H, Christ W J, Fujioka H, Ham W-H, Hawkins L D, Jin H, Kang S H, Kishi Y, Martinelli M J, McWhorter W W, Jr, Mizuno M, Nakata M, Stutz A E, Talamas F X, Taniguchi M, Tino J A, Ueda K, Uenishi J-i, White J B, Yonaga M. J Am Chem Soc. 1989;111:7530–7533. [Google Scholar]
- 5.Holton R A, Somoza C, Kim H B, Liang F, Biediger R J, Boatman P D, Shindo M, Smith C C, Kim S, Nadizadeh H, Suzuki Y, Tao C, Vu P, Tang S, Zhang P, Murthi K K, Gentile L N, Liu J H. J Am Chem Soc. 1994;116:1597–1598. [Google Scholar]
- 6.Holton R A, Kim H B, Somoza C, Liang F, Biediger R J, Boatman P D, Shindo M, Smith C C, Kim S, Nadizadeh H, Suzuki Y, Tao C, Vu P, Tang S, Zhang P, Murthi K K, Gentile L N, Liu J H. J Am Chem Soc. 1994;116:1599–1600. [Google Scholar]
- 7.Nicolaou K C, Zang Z, Liu J J, Ueno H, Nantermet P G, Guy R K, Claiborne C F, Renaud J B, Couladourus E A, Paulvannan K, Sorensen E J. Nature (London) 1994;367:630–634. doi: 10.1038/367630a0. [DOI] [PubMed] [Google Scholar]
- 8.Heathcock C H, Davidsen S K, Mills S, Sanner M A. J Org Chem. 1992;57:2531–2544. [Google Scholar]
- 9.Heathcock C H, Hansen M M, Ruggeri R B, Kath J C. J Org Chem. 1992;57:2544–2553. [Google Scholar]
- 10.Heathcock C H, Piettre S, Ruggeri R B, Ragan J A, Kath J C. J Org Chem. 1992;57:2554–2566. [Google Scholar]
- 11.Heathcock C H, Stafford J A. J Org Chem. 1992;57:2566–2575. [Google Scholar]
- 12.Heathcock C H, Stafford J A, Clark D N. J Org Chem. 1992;57:2575–2584. [Google Scholar]
- 13.Heathcock C H, Ruggeri R B, McClure K F. J Org Chem. 1992;57:2585–2594. [Google Scholar]
- 14.Heathcock C H, Kath J C, Ruggeri R B. J Org Chem. 1995;60:1120–1130. [Google Scholar]
- 15.Heathcock C H, Joe D. J Org Chem. 1995;60:1131–1142. [Google Scholar]
- 16.Toda M, Hirata Y, Yamamura S. Tetrahedron. 1972;28:1477–1484. [Google Scholar]
- 17.Isoe S, Ge Y, Yamamoto K, Katsumura S. Tetrahedron Lett. 1988;29:4591–4594. [Google Scholar]
- 18.Malhotra S K, Moakley D F, Johnson F. J Am Chem Soc. 1967;89:2794–2795. [Google Scholar]
- 19.Schreiber S L, Meyers H V, Wiberg K B. J Am Chem Soc. 1986;108:8274–8277. [Google Scholar]