

Abstract
Recent studies have demonstrated that the β2-adrenergic receptor (β2AR)-Gαi signaling pathway exerts a cardiac antiapoptotic effect. The goals of this study were to determine the intracellular signaling factors involved in β2AR-mediated protection against doxorubicin-induced apoptosis in H9c2 cardiomyocyte and explore the impact of high ambient glucose on the antiapoptotic effect. Under physiological glucose environment (100 mg/dl), β2AR stimulation prevented doxorubicin-induced apoptosis, which was attenuated by cotreatment with wortmannin, a phosphoinositide 3-kinase (PI3K) inhibitor, or transfection of a dominant-negative Akt. Inhibition of Src kinase with 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d] pyrimidine or cSrc small interfering RNA 32 also attenuated the antiapoptotic effect. Inhibition of platelet-derived growth factor receptor (PDGFR) with AG1296 reversed the β2AR-induced antiapoptotic effect. Transfection of an active Src cDNA (Y529F) alone was sufficient to render the cells resistant to apoptosis, and the resistance was blocked by wortmannin. Transfection of an active PI3K minigene (iSH2-p110) alone also induced resistance to apoptosis, and the resistance was reversed by an Akt-inhibitor but not by AG1296. High ambient glucose (450 mg/dl) caused two major effects: 1) it significantly reduced βAR-induced PDGFR phosphorylation, Src kinase activity, and activation of PI3K signaling pathway; and 2) it partially attenuated β2AR-induced antiapoptotic effect. These data provide in vitro evidence supporting a signaling cascade by which β2AR exerts a protective effect against doxorubicin-induced apoptosis via sequential involvement of Gαi, Gβγ, Src, PDGFR, PI3K, and Akt. High ambient glucose significantly attenuates β2AR-mediated cardioprotection by suppressing factors involved in this cascade including PDGFR, Src, and PI3K/Akt.
THE β-ADRENERGIC RECEPTORS (βAR) are members of the G protein-coupled receptor (GPCR) family (1). At least three βAR subtypes, β1AR, β2AR, and β3AR, have been characterized at the gene level (2,3,4). It has been shown that β1AR and β2AR modulate cardiac function differently and exert physiological responses by distinct signal transduction pathways (5). In particular, sustained β1AR stimulation promotes cardiomyocyte apoptosis, whereas sustained β2AR stimulation is known to protect cardiomyocytes from apoptotic insults (6). This finding is of great clinical interest because selective pharmacological activation of β2AR-mediated inotropy or its overexpression through gene therapy might be used as a novel therapeutic approach in the types of failing heart.
Doxorubicin (Dox), an anthracycline antibiotic, has been established as an effective agent against a wide range of cancers (7). However, the severe cardiotoxicity of Dox is a major limiting factor for its effective use in the treatment of all malignancies (8). It is believed that mitochondria play a significant role in the toxicity as isolated heart mitochondria and myocytes have been shown to give rise to increased levels of oxygen radicals after Dox treatment (9). Mitochondria play a pivotal role in regulating apoptosis by mechanisms that result in the activation of caspases and subsequent cell death (10). Apoptosis has been attracting increasing attention as a possible mechanism underlying cardiomyopathy and chronic heart failure induced by Dox (11).
Cardiomyopathy is also a common complication in patients with diabetes mellitus (12). Acute responses of cardiomyocytes to hyperglycemia include metabolic abnormalities, altered gene expression, and increased apoptosis (13,14,15). It has been shown that the incidence of apoptosis increases in the hearts of patients with diabetes (16) and streptozotocin-induced diabetic animals (17).
The present study was undertaken to delineate the signaling pathway(s) by which β2AR stimulation evokes protective effects against Dox-induced apoptosis and to examine whether the protective effects of β2AR are impaired under high glucose environment. For this purpose, we studied Dox-induced apoptosis in a β2AR-dominant fetal rat cardiomyocyte cell line, H9c2 (18). The results of this study provide evidence that the antiapoptotic signaling via β2AR is mediated by transactivation of the platelet-derived growth factor receptor (PDGFR) and phosphoinositide 3-kinase (PI3K). Furthermore, the antiapoptotic effect is significantly suppressed under high ambient glucose conditions by inhibition of these signaling pathways.
Materials and Methods
Reagents
Unless otherwise specified, all materials were of reagent grade and were obtained from Sigma (St. Louis, MO).
Cell culture
H9c2 rat fetal cardiomyocytes were grown in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% (vol/vol) fetal bovine serum in a humidified atmosphere containing 5% CO2 at 37 C. Cells were grown to 70% confluence and synchronized overnight in serum-free medium before experiments.
Immunoblotting and antibodies
Protein expression was evaluated by Western blotting as previously described (19). Antibodies against β1AR, β2AR, p85α, PDGFR, and phosphotyrosine were purchased from Santa Cruz Biotechnology (sc-568, sc-570, sc-1637, sc-432 and sc-18182; Santa Cruz, CA). Antibody against cSrc was purchased from Biosource International (AHO1152; Camarillo, CA). Antibodies against IGF-I receptor (IGF-IR)-β, phosphorylated-Akt (Thr-308), total Akt, and caspase-3 were purchased from Cell Signaling Technology (3022, 9275, 9272 and 9662; Danvers, MA). Antibody against total actin was purchased from Millipore (MAB1501; Billerica, MA).
DNA fragmentation ELISA
After a 6- or 10-h cell culture in medium without serum, the amount of apoptosis that had occurred was examined by using a DNA fragmentation detection ELISA kit (Roche Diagnostics, Mannheim, Germany).
PI3K assay
H9c2 cell lysates were prepared as described (19), and protein concentrations were determined with the bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL). PI3K activity was determined by an in vitro immunoprecipitation lipid kinase assay as described previously (19,20).
Immunoprecipitation
Protein lysates (0.5 mg) from H9c2 cells were incubated with an antibody specific for PDGFR at 4 C for 4 h with continuous rotation. Protein G-Sepharose (GE Healthcare, Piscataway, NJ) was added, and the mixture was incubated overnight at 4 C. After washing, 40 μl of 2× Laemmli sample buffer were added. The sample was heated in boiling water for 5 min and quenched on ice for 2 min. After vortex and centrifuge, 20 μl of the supernatant were resolved on a 7.5% SDS-PAGE gel.
Stable transfection
To inhibit Gβγ, a βAR kinase-1 C terminus minigene (βARKct; kindly provided by Dr. R. J. Lefkowitz, Duke University, Durham, NC) was inserted into pUSEamp(+) (pUSE; Upstate Biotechnology, Lake Placid, NY) and stably transfected into H9c2 cells as described (19). Some cells were transfected with a vector expressing a dominant-negative Akt1, which was engineered by inserting a K179M mutant dominant-negative Akt1 cDNA (21) into the multiple cloning site of pUSE. Another set of cells were transfected with a vector expressing an activated mutant Src, which was engineered by inserting a Y529F mutant cDNA (22) into the multiple cloning site of pUSE. For the above experiments, control H9c2 cells were transfected with pUSE.
To induce expression of a constitutively active PI3Kα, a 4.4-kb iSH2-p110 fusion gene was excised from a pCMV6-iSH2-p110-MT plasmid (kindly provided by Dr. T. F. Franke, Columbia University, New York, NY) and inserted into the multiple cloning site of pEGFP-N1 vector (CLONTECH, Mountain View, CA). H9c2 cells were then stably transfected with pEGFP-N1 or the vector expressing iSH2-p110. All stable transfection was performed using Lipofectamine 2000 (Invitrogen). Individual single cells were selected for neomycin resistance. The stably transfected cells were used in biochemical assays as described above.
Intracellular cAMP measurement
Intracellular cAMP levels were measured by enzymatic immunoassay (EIA) using cAMP Biotrak EIA System (GE Healthcare). Briefly, cells were seeded in standard 96-well microplates (Corning 3595; Corning Inc., Corning, NY) with a density of 104/well (100 μl). After incubating the plate overnight, cells were deprived of serum and incubated overnight again to make cells quiescent. Cells were then incubated under normal (100 mg/dl) or high (450 mg/dl) glucose conditions and treated with isoproterenol (ISO) with the indicated duration (0–30 min). Cell lysates were collected for cAMP assay according to the manufacturer’s instruction.
Src activity assay
Kinase activities in H9c2 cell lysates (1 mg) were determined by measuring phosphorylation of the specific Src substrate (KVEKIGEGTYGVVYK) using a Src assay kit (Millipore) as described previously (20).
Transfection of small interfering RNA (siRNA) for cSrc
Two complementary hairpin siRNA oligonucleotides containing 21 nucleotides target sequences of the mouse cSrc tyrosine kinase, (5′-AAG TAC AAC TTC CAT GGC ACT-3′) were ligated into pSilencer 5.1-H1 Retro vector (Ambion, Austin, TX). The vector was then stably transfected into H9c2 cells as described previously (19).
Statistical analysis
Statistical significance of differences among groups was analyzed by the paired Student’s t test or one-way ANOVA followed by Newman-Keuls test. All data were represented as the mean ± se of three different experiments. A probability of P < 0.05 was considered to represent a significant difference.
Results
βAR subtypes in H9c2 cells
H9c2 cardiomyocytes are known to express both β1AR and β2AR with the β2AR being the predominant subtype (β1AR: β2AR = ∼3:7) (18). This estimate has been based solely on the experimental data generating from radioligand receptor binding studies (18). Because we aimed to delineate the anti-apoptotic signaling cascades mediated by βAR, we further compared the density of β1AR and β2AR in H9c2 cells at the protein level. Western blotting, applying an even amount (50 μg) of protein on each lane, showed that a very low amount of β1AR was expressed in H9c2 cells and that more β2AR was expressed in H9c2 cells than rat left ventricles (Fig. 1).
Figure 1.
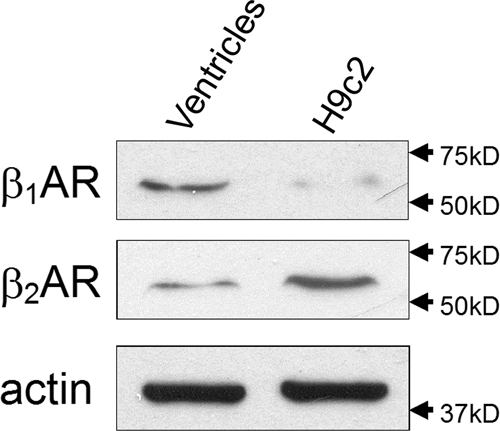
β2AR is the major subtype of βAR in H9c2 cells. Western blotting was performed on lysates (50 μg) prepared from quiescent H9c2 cells or adult (3 months old) rat ventricles, respectively, using anti-β1 and anti-β2AR antibodies. Total actin immunoblotting was used as a loading control.
β2AR stimulation attenuates Dox-induced apoptosis in H9c2 cardiomyocytes
H9c2 cells were treated with Dox (1 nm to 10 μm) for 10 h, and the cell lysates were subject to Western blotting using an anticaspase-3 antibody. Cleavage of caspase-3, a good indicator of cellular apoptosis, was seen in groups treated with 10 nm or higher doses of Dox (Fig. 2A, upper panel). We next used DNA fragmentation assay to assess cell apoptosis quantitatively. A dose-dependent effect on apoptosis was observed as the levels of DNA fragmentation increased significantly with 10 nm or higher doses of Dox, and the peak response was found in the 1 μm Dox group (Fig. 2A, lower panel). Based on these results, we chose a dose of 1 μm Dox for all the subsequent experiments.
Figure 2.

β2AR stimulation attenuates Dox-induced apoptosis in H9c2 cells. H9c2 cells were incubated with indicated concentrations of Dox for 10 h under serum-free condition. After incubation, cell lysates (50 μg) were resolved on 10% SDS-PAGE, transferred to polyvinyl difluoride membrane, and immunoblotted with anticaspase-3 antibody. Blotting with anti-total actin antibody served as loading control (A, upper panel). Some cell lysates were used for DNA fragmentation assay (A, lower panel). *, P < 0.05 vs. 1 nm Dox group. B, H9c2 cells were incubated for 10 h with vehicle, Dox (1 μm) alone, Dox in combination with ISO, or Dox/ISO with 1 h pretreatment of propranolol (10 μm), CGP20712 (1 μm), or ICI118,551 (1 μm). Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as above. *, P < 0.05 vs. Dox alone group.
To confirm that β2AR stimulation induces an antiapoptotic effect, H9c2 cells were pretreated with propranolol (10 μm), a nonselective βAR antagonist, or the highly selective antagonists against β1AR (CGP20712, 1 μm) or β2AR (ICI118,551, 1 μm) for 1 h before adding ISO (10 μm) and Dox. After a 10-h incubation, apoptosis was assessed by caspase-3 immunoblotting and DNA fragmentation ELISA. Results from both assays confirmed that βAR stimulation with ISO significantly reduced Dox-induced apoptosis and that the antiapoptotic effect was completely abrogated by pretreatment with propranolol or ICI118,551 but not CGP20712 (Fig. 2B). These data demonstrate that the antiapoptotic effect of ISO is mediated by the β2AR signaling pathway.
β2AR-mediated antiapoptotic effect is Gαi and Gβγ dependent but Gαs independent
β2AR stimulation activates both the Gαs-adenylyl cyclase-cAMP-protein kinase A and the Gαi-Gβγ signaling pathways in vivo (5). To differentiate the role of these two pathways on β2AR-mediated antiapoptosis, H9c2 cells were pretreated with forskolin (1 μm, 5 min) before Dox treatment. This pretreatment did not reverse the apoptosis induced by Dox (Fig. 3A). Inhibition of protein kinase A with H89 (10 μm, 1 h pretreatment) or inhibition of AC with 2′,5′-dideoxyadenosine (2′,5′-DOA, 100 μm, 1 h pretreatment) did not affect the βAR-mediated antiapoptotic effect (Fig. 3A). In contrast, pretreatment with pertussis toxin (PTX; 1 μg/ml, 3 h) significantly attenuated the βAR-mediated antiapoptotic effect (Fig. 3A). PTX alone did not effect on apoptosis. These data suggest that βAR-mediated antiapoptosis is Gαi dependent and Gαs independent.
Figure 3.

β2AR-mediated antiapoptotic effect in H9c2 cells is Gαi and Gβγ dependent but Gαs independent. A, H9c2 cells were treated with vehicle, Dox (1 μm) alone, or Dox in combination with ISO (10 μm) for 10 h under serum-free environments. Some sets of Dox/ISO-treated cells were pretreated with H89 (10 μm; 1 h), 2′,5′-DOA (100 μm; 1 h), or PTX (1 μg/ml; 3 h). Some cells were treated with H89, 2′,5′-DOA, forskolin, or PTX alone similarly but without Dox or ISO. Another set of the Dox-treated cells was pretreated with forskolin for 5 min. Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05 vs. Dox-alone group. B, H9c2 cells were stably transfected with an empty vector, pUSE, or the vector containing a minigene expressing a Gβγ sequestrant peptide derived from the C terminus of βAR kinase-1 (βARKct). Cells were treated with vehicle, Dox (1 μm) alone, or Dox in combination with ISO (10 μm) for 10 h. Caspase-3 immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05.
We next examined whether the Gβγ subunits of G protein are required for β2AR stimulation-dependent antiapoptosis. H9c2 cells were stably transfected with an empty vector (pUSE) or a vector expressing a βARKct minigene that inhibits Gβγ (23). As expected, in cells transfected with pUSE, βAR stimulation reversed Dox-induced apoptosis (Fig. 3B). In contrast, in H9c2 cells overexpressing βARKct, βAR stimulation failed to reduce Dox-induced apoptosis (Fig. 3B). These data suggest that the Gβγ subunits of G protein are required for the βAR-mediated antiapoptotic effect.
Involvement of PI3K signaling pathway in β2AR-mediated antiapoptosis
To confirm the role of PI3K signaling pathway in β2AR-mediated antiapoptosis in H9c2 cardiomyocytes, we conducted a series of experiments using different approaches. First, cells were cultured with Dox (1 μm) with or without ISO (10 μm) for 10 h. Some cells treated with both Dox/ISO were pretreated with wortmannin (20 nm, 1 h), a PI3K inhibitor. Treatment with wortmannin alone, at the dose chosen, did not induce apoptosis in H9c2 cells. Pretreatment with wortmannin, however, effectively abrogated the β2AR-mediated protection against Dox-induced apoptosis (Fig. 4A).
Figure 4.

β2AR-mediated antiapoptotic effect in H9c2 cell requires PI3K signaling pathway. A, H9c2 cells were treated for 10 h with vehicle, wortmannin (20 nm) alone, Dox (1 μm) alone, Dox in combination with ISO, or Dox/ISO plus pretreatment with wortmannin (1 h). B, H9c2 cells were stably transfected with an empty vector (pEGFP-N1) or the vector containing a constitutively active PI3Kα (iSH2–p110). Cell lysates (500 μg) from nontransfected wild-type cells, empty vector-transfected cells, and iSH2–p110 transfected cells were subjected to in vitro immunoprecipitation lipid kinase assay for PI3K activity as described. PIP, Phosphoinositide 3-phosphate, the phosphorylated end product. C, Empty vector (pEGFP-N1) or constitutively active PI3Kα (iSH2–p110)-transfected H9c2 cells were treated with vehicle, Dox (1 μm) alone, or Dox in combination with ISO (10 μm) with or without PTX (1 μg/ml) for 10 h. D, H9c2 cells transfected with the empty vector or iSH2–p110 were treated for 10 h with vehicle, Dox (1 μm) alone, Dox in combination with ISO, or Dox/ISO with pretreatment of Akt inhibitor (10 μm). E, H9c2 cells were transfected with an empty vector (pUSE) or a dominant-negative Akt1 (K179M; DN-Akt1) cDNA and treated with vehicle, Dox (1 μm) alone, or Dox in combination with ISO for 10 h. F, H9c2 cells were cotransfected with empty vectors (pUSE and pEGFP-N1) or iSH2-p110 and βARKct minigene. The cotransfected cells were treated with vehicle, Dox (1 μm) alone, or Dox in combination with ISO for 10 h. For all the above experiments (except for B), caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05.
Second, a constitutively active PI3K cDNA (iSH2-p110) was stably transfected into H9c2 cells. In vitro lipid kinase assay of these transfected cells showed significantly increased PI3K activity in the basal/nonstimulated state, compared with wild-type (nontransfected) and control (empty vector transfected) cells (Fig. 4B). As seen above in nontransfected cells, Dox-induced apoptosis was blocked by ISO treatment in the empty vector transfected cells and the effect of ISO was reversed by cotreatment with PTX (1 μg/ml, 3 h) (Fig. 4C). Overexpression of PI3K, however, completely abrogated Dox-induced apoptosis, and pretreatment of PTX showed no reversal effect on ISO stimulation (Fig. 4C). Because these PI3K-overexpressing cells were resistant to Dox-induced apoptosis, further βAR stimulation resulted in no further changes in caspase-3 cleavage or DNA fragmentation. Moreover, when these PI3K-overexpressing cells were cotreated with an Akt inhibitor [1L6-hydroxymethyl-chiro- inositol-2-(R)-2-O-methyl-3-O-octadecyl-sn-glycerocarbonate; Calbiochem, San Diego, CA], the βAR mediated protection against apoptosis disappeared (Fig. 4D). In cells transfected with the empty vector, Dox-induced apoptosis was effectively attenuated by ISO, and such attenuation was reversed by cotreatment with Akt inhibitor. These data confirm the important role of PI3K-Akt signaling pathway in cell protection exerted by βAR.
Third, a dominant-negative Akt1 cDNA was stably transfected into H9c2 cells. As was seen above, in cells transfected with pUSE, the empty vector, treatment with ISO blocked Dox-induced apoptosis (Fig. 4E). In contrast, in cells overexpressing the dominant-negative Akt1, βAR stimulation had no effect on Dox-induced apoptosis (Fig. 4E).
Fourth, H9c2 cells were cotransfected with iSH2-p110 and βARKct minigene. We have shown ovexpression of βARKct abrogates β2AR mediated antiapoptotic effect (Fig. 3B). In cells cotransfected with βARKct and iSH2-p110, however, the degree of apoptosis was equivalent to that of empty vectors (pUSE + pEGFP-N1)-transfected, ISO-treated cells (Fig. 4F). This was true with or without ISO treatment. In other words, the presence of the constitutively active PI3K negated the proapoptotic effect of βARKct. Taken together, these series of experiments demonstrate that β2AR-mediated resistance to Dox-induced apoptosis is dependent on PI3K/Akt signaling pathway, which is functioning downstream of Gαi and Gβγ.
β2AR-mediated antiapoptotic effect in H9c2 cells is dependent on Src family tyrosine kinase
In a previous study, we have shown that the Src-family tyrosine kinase is involved in βAR-mediated transactivation of PI3K in H9c2 cells (19). We do not know, however, whether Src is required for the antiapoptotic effect mediated by βAR stimulation. To explore the role of Src in β2AR-mediated antiapoptosis, a series of experiments were initiated. First, cells were cultured with vehicle, Dox alone, or Dox in combination with ISO for 10 h. Some cells were pretreated with 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d] pyrimidine (PP2; 10 μm; Tocris Bioscience, Ellisville, MO), a specific Src family kinase inhibitor, for 1 h before start of Dox/ISO treatment. The β2AR-induced protective effect against Dox-induced apoptosis was totally blocked by PP2 (Fig. 5A).
Figure 5.

β2AR-mediated antiapoptotic effect in H9c2 cells is dependent on Src family tyrosine kinase. A, H9c2 dells were treated for 10 h with vehicle, Dox (1 μm) alone, Dox in combination with ISO, or Dox/ISO with 1 h pretreatment of PP2 (10 μm). B, H9c2 cells were stably transfected with scrambled oligonucleotides or cSrc siRNA and treated with vehicle, Dox alone, or Dox in combination with ISO for 10 h. C, H9c2 cells were stably transfected with an empty vector (pUSE) or the vector containing a constitutively active Src cDNA (Y529F). The pUSE-transfected cells were treated with vehicle, Dox alone, or Dox and ISO with or without pretreatment of PTX (1 μg/ml, 3 h) for 10 h. The active Src-transfected cells were treated for 10 h with vehicle, Dox alone, or Dox with pretreatment of PTX (1 μg/ml, 3 h) or wortmannin (20 nm, 1 h). D, H9c2 cells were stably cotransfected with active Src cDNA (Y529F) and βARKct minigene. Cells were treated with vehicle, Dox alone, or Dox in combination with ISO for 10 h. For all the above experiments, caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05.
Second, cSrc was knocked down by siRNA transfection. We have shown effective knockdown of Src at gene and protein levels in H9c2 cardiomyocytes with this approach (19). In cells stably transfected with a scramble oligo, βAR stimulation effectively reversed Dox-induced apoptosis (Fig. 5B). After knockdown of Src, however, the protective effect of βAR stimulation on Dox-induced apoptosis was abrogated (Fig. 5B).
Third, a constitutively active Src cDNA (Y529F) was stably transfected into H9c2 cells. These mutant Src-overexpressing cells showed resistance to Dox-induced apoptosis (Fig. 5C). In empty vector (pUSE)-transfected cells, pretreatment with PTX abrogated the antiapoptosis of ISO (Fig. 5C). In contrast, active Src-overexpressing cells were resistant to Dox-induced apoptosis. Moreover, whereas pretreatment with PTX had no effects, pretreatment with wortmannin (20 nm, 1 h) completely reverse the βAR-mediated resistance to apoptosis in active Src-overexpressing cells (Fig. 5C).
Fourth, H9c2 cells were cotransfected with active Src cDNA (Y529F) and βARKct minigene. As was seen previously in which overexpression of constitutively active PI3K abrogated the proapoptotic effect of βARKct (Fig. 4F), overexpression of active Src was sufficient to abrogate the proapoptotic effect of βARKct (Fig. 5D). These observations established the fact that Src is a critical factor in the β2AR-mediated antiapoptotic effect by functioning downstream of Gαi and Gβγ and upstream of PI3K signaling pathway.
β2AR mediated antiapoptotic effect in H9c2 cells involves PDGFR transactivation
We have shown that PDGFR is required in βAR-mediated transactivation of PI3K (19). To more carefully observe activation of PDGFR after βAR stimulation, H9c2 cells were treated with ISO for 0, 0.5, 1, 2, 5, 10, 15, or 30 min, and cell lysates were subjected to immunoprecipitation with an antibody against PDGFR, followed by immunoblotting using anti-PDGFR and an antibody specific for phosphotyrosine (pY). We found that the peak PDGFR phosphorylation occurred at 2 min after βAR stimulation and then returned to baseline levels after 30 min (Fig. 6A). In contrast, similar experiment using anti-IGF-IRβ failed to induce tyrosine phosphorylation of IGF-IR after ISO stimulation (Fig. 6B).
Figure 6.

Transactivation of PDGFR is required for β2AR-mediated antiapoptotic effect. A, Time course of βAR stimulation-induced tyrosine phosphorylation of PDGFR. H9c2 cells were treated with ISO (10 μm) for the indicated durations. Cell lysates were immunoprecipitated (IP) with an anti-PDGFR and immunoblotted (IB) with anti-pY. The same membrane was stripped and reblotted with an anti-PDGFR as a loading control. The line graphs show the densitometric scanning result from three individual experiments. Data represent mean ± se of percent change in tyrosine phosphorylation relative to that of time zero control. *, P < 0.05 vs. time zero control. B, Time course of βAR stimulation-induced tyrosine phosphorylation of IGF-IR. H9c2 cells were treated with ISO (10 μm) for the indicated durations. Cell lysates were immunoprecipitated with an anti-IGF-IR and immunoblotted with anti-pY. The same membrane was stripped and reblotted with an anti-IGF-IR as a loading control. C, H9c2 cells were treated for 10 h with vehicle, Dox alone, Dox in combination with ISO, or Dox/ISO with 1 h pretreatment of AG1296 (10 μm). Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05. D, Scrambled oligonucleotides- and cSrc siRNA-transfected cells were treated with vehicle or ISO (10 μm) for 2 min. Cell lysates were immunoprecipitated with anti-PDGFR and immunoblotted with anti-pY. The same membrane was stripped and reblotted with anti-PDGFR antibody as a loading control. The bar graphs show the densitometric scanning results from three individual experiments. Data represent means ± se of percent change in tyrosine phosphorylation relative to that of similarly transfected vehicle control. *, P < 0.05. E, H9c2 cells were transfected with pEGFP-N1 empty vector or a constitutively active PI3K minigene (iSH2p110) as described and treated for 10 h with vehicle, Dox alone, Dox in combination with ISO, Dox with 1 h pretreatment of AG1296 (10 μm), or Dox/ISO with 1 h pretreatment of AG1296. Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05. F, H9c2 cells were stimulated with ISO (10 μm) for 0, 0.5, 1, 1.5, and 2 min, and then the cell lysates were immunoprecipitated with anti-cSrc antibody. The precipitated proteins were resolved by 10% SDS-PAGE, transferred to polyvinyl difluoride membrane, and immunoblotted with an anti-cSrc, anti-PDGFR, anti-p85α, or anti-Akt antibody.
To examine the role of PDGFR in βAR-mediated antiapoptosis, H9c2 cells were treated with vehicle, Dox alone, Dox in combination with ISO or Dox/ISO plus pretreatment with a selective PDGFR antagonist, AG1296 (10 μm, 1 h). Pretreatment with AG1296 completely abrogated the antiapoptotic effect induced by ISO (Fig. 6C). We have shown the importance of Src in βAR-mediated antiapoptosis using Src siRNA in H9c2 cells (Fig. 5B). As shown above, Src plays critical roles in βAR-mediated antiapoptosis by functioning upstream of PI3K (Fig. 5C). To further examine whether Src is required for PDGFR transactivation in βAR-mediated antiapoptosis, scrambled oligo- or Src siRNA-transfected cells were treated with vehicle or ISO, and cell lysates were subject to immunoprecipitation with an antibody against PDGFR, followed by immunoblotting using anti-PDGFR and anti-pY antibodies. In cells transfected with scrambled oligos, βAR stimulation significantly increased tyrosine phosphorylation of PDGFR. In contrast, in cells with knockdown of cSrc, βAR stimulation no longer induced PDGFR activation (Fig. 6D). These results suggest that Src is required for activation of PDGFR in H9c2 cells.
With transfection of a constitutively active PI3Kα, we have shown the critical roles of PI3K in β2AR-mediated antiapoptosis (Fig. 4, C and D). These transfected cells were further treated with Dox in combination with AG1296. As expected, in cells transfected with the empty vector, inhibition of PDGFR with AG1296 completely reversed the β2AR-mediated antiapoptotic effect (Fig. 6E). In contrast, overexpression of PI3K rendered cells resistant to Dox-induced apoptosis, and inhibition of PDGFR did not alter the effect of β2AR stimulation (Fig. 6E). Data shown in Fig. 6, D and E, firmly establish PDGFR as an important factor in β2AR-madiated antiapoptotic effect by functioning downstream of Src and upstream of PI3K.
As shown in Fig. 6A, transactivation of PDGFR after ISO stimulation in H9c2 cells occurred as early as at 0.5 min and peaked by 2 min. To explain the quickness of the cross talk, we tried to confirm coprecipitation of the signaling factors after ISO stimulation. H9c2 cells were stimulated with ISO (10 μm) for 0.5, 1, 1.5, and 2 min. The cell lysates were immunoprecipitated with anti-cSrc antibody and immunoblotted with anti-cSrc, anti-PDGFR, anti-p85α (a regulatory subunit of PI3K), and anti-Akt antibodies. All these signaling factors showed coprecipitation as early as 1–1.5 min after ISO stimulation (Fig. 6F). The quick formation of the cell signaling complex is consistent with the quick PDGFR transactivation shown in Fig. 6A and in our previous studies (19). Taken together, these data suggest that PDGFR transactivation plays a critical role in β2AR-induced antiapoptotic effect by functioning downstream of Src and upstream of PI3K signaling pathway.
β2AR stimulation attenuates hydrogen peroxide-induced apoptosis in H9c2 cardiomyocytes
To examine whether β2AR mediated antiapoptotic signaling pathway is generalized, we explored the preventive effects of ISO against hydrogen peroxide (H2O2)-induced apoptosis. We first established the time course of H2O2 treatment. H2O2 (1 mm) induced apoptosis can be observed as early as 1.5 h, and the peak response was seen at 6 h (Fig. 7A).
Figure 7.

β2AR stimulation attenuates hydrogen peroxide-induced apoptosis in H9c2 cardiomyocytes. A, Time course of apoptosis induction by hydrogen peroxide (H2O2) in H9c2 cells. Cells were incubated with H2O2 (1 mm) for 1.5, 3, 4, 6, and 10 h under serum-free condition. Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05. B, H9c2 cells were incubated for 6 h with vehicle, hydrogen peroxide (H2O2, 1 mm) alone; H2O2 in combination with ISO; or H2O2/ISO with pretreatment of PTX (1 μg/ml, 3 h), PP2 (10 μm, 1 h), AG1296 (10 μm, 1 h), wortmannin (20 nm, 1 h), or Akt inhibitor (10 μm, 1 h). Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05 vs. H2O2 alone group.
Second, H9c2 cells were incubated for 6 h with vehicle; H2O2 (1 mm) alone; H2O2 in combination with ISO; or H2O2/ISO with pretreatment of PTX (1 μg/ml), PP2 (10 μm), AG1296 (10 μm), wortmannin (20 nm), or Akt inhibitor (10 μm). All inhibitors or antagonists used for pretreatment reversed the effect of ISO against H2O2-induced apoptosis (Fig. 7B). These results suggest the existence of a common signaling pathway for β2AR mediated protection against Dox- and H2O2-induced apoptosis.
High ambient glucose attenuates the preventive effect of ISO against Dox-induced apoptosis
To determine whether high ambient glucose alters β2AR-mediated antiapoptotic effect, H9c2 cells were incubated with vehicle, Dox (1 μm) alone, or Dox in combination with ISO (10 μm) for 10 h under various concentrations of glucose. We demonstrated that the degree of ISO-induced antiapoptotic effect was proportionally decreased with increasing concentration of glucose (Fig. 8A). When glucose concentration reached 700 mg/dl, ISO-induced antiapoptotic effect was completely abolished. In a time-course study, we compared the change in ISO-induced antiapoptotic effect under normal (100 mg/dl) and high glucose (450 mg/dl) conditions. In ISO-treated cells, there was a significant increase in apoptosis in the high glucose concentration at the 10-h time point (Fig. 8B). After 16 h, both glucose concentration groups had maximal apoptosis, and there were no differences between the two groups. Based on these results, we chose 450 mg/dl as a concentration of high glucose and 10 h as the time of incubation for all subsequent normal/high glucose experiments.
Figure 8.

High ambient glucose attenuates ISO-mediated protection against Dox-induced apoptosis. A, H9c2 cells were treated with vehicle, Dox (1 μm) alone, or Dox in combination with ISO under various concentrations of ambient glucose (100, 200, 450, 700, and 1000 mg/dl). Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05. B, Time course of alteration in preventive effect of ISO against Dox-induced apoptosis under normal and high ambient glucose. Cells were incubated with Dox (1 μm) in combination with ISO (10 μm) under normal (100 mg/dl) or high (450 mg/dl) ambient glucose concentrations for 2, 6, 10, and 16 h. Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05.
High ambient glucose attenuates β2AR-mediated activation of Src, PDGFR, PI3K signaling pathway, and antiapoptosis
To explore the impact of high ambient glucose on β2AR-mediated anti-apoptosis, H9c2 cells were cultured in normal (100 mg/dl) or high (450 mg/dl) glucose conditions. H9c2 cells were incubated with vehicle, Dox alone, or Dox in combination with ISO for 10 h. Normal glucose medium (100 mg/dl = 5.6 mm) supplemented with glycerol (19.4 mm) was used for isoosmotic control for the high glucose medium (25 mm). The levels of apoptosis were assessed by immunoblotting for anticaspase-3 and DNA fragmentation ELISA as described above. Dox treatment induced apoptosis in H9c2 cells cultured under normal glucose as well as under high glucose conditions (Fig. 9A). High ambient glucose, however, significantly attenuated the β2AR-mediated resistance to Dox-induced apoptosis (Fig. 9A). In contrast, in both the normal glucose and isoosmotic control groups, Dox-induced apoptosis was effectively attenuated by βAR stimulation (Fig. 9A). This suggests that high ambient glucose inhibits the β2AR-induced antiapoptotic effect by osmotic pressure-independent mechanisms.
Figure 9.

High ambient glucose attenuates β2AR-mediated antiapoptotic effect via inhibition of Src, PDGFR, and PI3K signaling pathway. A, H9c2 cells were treated with vehicle, Dox (1 μm) alone, or Dox in combination with ISO under physiologically normal (N; 100 mg/dl) or high (H; 450 mg/dl) glucose conditions for 10 h. In a Dox/ISO treatment group, normal glucose medium (5.6 mm) was supplemented with glycerol (19.4 mm) and used as an isoosmotic control for high glucose medium (25 mm). Caspase-3 and total actin immunoblotting (upper panel) and DNA fragmentation assay (lower panel) were carried out as described. *, P < 0.05. B, Quiescent cells were incubated with vehicle (▴) or ISO (10 μm) under normal (⋄, 100 mg/dl) or high (▪, 450 mg/dl) ambient glucose for 0, 1, 2, 5, 10, and 30 min. Cell lysates were subjected to EIA for measurement of cAMP levels. *, P < 0.05 vs. vehicle-incubated control. C, Cells were treated with vehicle or ISO for indicated durations under normal (N) or high (H) glucose conditions. Glycerol was added to achieve isoosmosis. Cell lysates were subjected to in vitro lipid kinase assay for PI3K activity as described. PIP, Phosphoinositide 3-phosphate, the phosphorylated end product. Values are mean ± se of percent change from 0 min control. *, P < 0.05. D, Cells were treated with vehicle or ISO for the indicated durations under normal (N) or high (H) glucose conditions. Cell lysates were subjected to Western blotting using antibodies against phospho-Akt (Thr-308) and total Akt. The bar graphs show the densitometric scanning results from three individual experiments. Data represent mean ± se of percent change in protein phosphorylation relative to that of control. *, P < 0.05.
To explore whether high glucose alters Gs signaling, we measured the levels of intracellular cAMP at 0, 1, 2, 5, 10, and 30 min after ISO (10 μm) stimulation. Vehicle-treated cells had no changes in intracellular cAMP levels (Fig. 9B). Significant intracellular accumulation of cAMP emerged as early as 1 min and peaked at 2 min and then declined gradually to basal levels at 30 min after ISO stimulation (Fig. 9B). The experiments were performed under normal (100 mg/ml) and high (450 mg/dl) ambient glucose, and no significant difference was observed between the two groups (Fig. 9B). These results suggest that Gs signaling does not play a critical role in the different antiapoptotic response between the two glucose conditions.
Because we have shown a critical role for PI3K signaling in the β2AR-induced antiapoptotic effect, it is likely that the effect of high ambient on attenuation of antiapoptotic effect is via inhibition of PI3K signaling pathway. To investigate this possibility, H9c2 cells were treated with vehicle or ISO under normal and high glucose environment for 2–15 min, and cell lysates were subjected to immunoprecipitation in vitro lipid kinase assay for PI3K activity and Western blotting for Akt. βAR stimulation significantly increased PI3K activity, which peaked at 2 min after treatment, declined gradually, and returned to baseline levels 15 min later in both normal and high glucose conditions (Fig. 9C). The increase in PI3K activity, however, was significantly attenuated under high ambient glucose condition (Fig. 9C). Akt phosphorylation (Thr-308) also peaked at 2 min after βAR stimulation. In high ambient glucose cultured cells, however, the degree of Akt phosphorylation was consistently decreased compared with normal glucose cultured cells (Fig. 9D).
The effect of high ambient glucose on βAR-induced Src activity was also examined. H9c2 cells were treated with vehicle or ISO for 0.5–15 min under normal or high glucose conditions, and cell lysates were subjected to Src kinase activity assay. ISO treatment significantly increased Src activity. In both glucose conditions, peak Src activity was seen at 1.5 min after ISO treatment (Fig. 10A). The peak Src activity, however, was significantly lower in cells under high glucose condition than those under normal glucose condition (Fig. 10A). We next examined the influence of high ambient glucose on β2AR-induced tyrosine phosphorylation of PDGFR. Cells were treated with vehicle or ISO for 2 min under normal or high glucose environment, and the levels of PDGFR phosphorylation were assessed as described. β2AR stimulation induced a significant increase in PDGFR phosphorylation in both the normal and high glucose conditions. The level of phosphorylation, however, was significantly lower in cells under high glucose condition than those under normal glucose condition (Fig. 10B). Taken together, these results suggest that high ambient glucose inhibits β2AR-induced antiapoptotic effect via concomitant inhibition of Src, PDGFR and PI3K signaling (Fig. 11).
Figure 10.

High ambient glucose reduces Src activity and tyrosine phosphorylation of PDGFR. A, H9c2 cells were treated with vehicle or ISO (10 μm) for the indicated durations under normal (N) or high (H) glucose conditions. Cell lysates (1 mg) were subjected to Src activity assay as described. Data represent mean ± se of counts per minute from three different experiments. *, P < 0.05 vs. 0 min control. B, H9c2 cells were treated for 2 min with vehicle or ISO (10 μm) under normal (N) or high (H) glucose conditions. Cell lysates were immunoprecipitated (IP) with anti-PDGFR and immunoblotted (IB) with anti-pY and anti-PDGFR as a loading control. The bar graphs show the densitometric scanning results from three individual experiments. Data represent mean ± se of percent change in tyrosine phosphorylation relative to that of vehicle control of the same glucose condition. *, P < 0.05.
Figure 11.

A hypothetical pathway for β2AR-mediated antiapoptotic effect in H9c2 cardiomyocytes. Stimulation of β2AR by ISO leads to sequential activation of Gαi/Gβγ, Src, and PDGFR and results in transactivation of PI3K signaling pathway. Activation of PI3K and its downstream kinases, including Akt, leads to resistance to apoptosis. Ambient high glucose reverses the β2AR-mediated antiapoptotic effect by suppressing the activities of Src, PDGFR, PI3K, and Akt. P, Phosphorylated tyrosine.
Discussion
Doxorubicin is an anthracycline antibiotic and has been a mainstay of cancer chemotherapy for more than 30 yr. Unfortunately, the development of a cardiomyopathy often limits the cumulative dose that can be delivered (11). The chemotherapeutic benefits of Dox and other anthracyclines appear to be derived from induction of apoptosis in malignant tissues (24). Anthracyclines, however, also induce apoptosis in normal cells and often result in cardiomyocyte loss in the heart (11). Because cardiomyocytes do not proliferate in adult heart, the loss of cardiomyocytes eventually leads to compromised cardiac function. Myocyte death associated with a decrease in myocardial performance and ventricular dilation has been demonstrated (16,17). Hence, understanding the mechanisms underlying Dox-induced apoptosis in cardiomyocytes and developing preventive strategy for the side effect is of particular importance for more effective neoplastic treatments.
In a cardiac context, inhibition of apoptosis through β2AR and induction of apoptosis by β1AR in cardiomyocytes has been reported (6). There are multiple signaling factors contributing to apoptosis and antiapoptosis induced by GPCRs, including Ca2+-calmodulin-dependent protein kinase, matrix metalloproteinases, β1 integrin, c-Jun NH2-terminal kinase, arrestin, and PI3K (25,26,27,28). Although it is well known that mitochondrial apoptosis cascade leads to eventual proteolytic activation of caspase 3, the immediate downstream factors involved after β2AR stimulation are not well documented. In a recent study, we reported that acute β2AR stimulation activates PI3K signaling pathway through sequential activation of Gαi-Gβγ-Src-PDGFR in H9c2 cardiomyocytes (19). The goals of the present study were to delineate the signaling cascade responsible for β2AR-mediated resistance to Dox-induced apoptosis and determine the impact of high glucose on the cardioprotection effect mediated by β2AR stimulation. Our data provide convincing evidence that β2AR signaling is critical in mediating resistance against Dox-induced apoptosis and that this antiapoptotic effect is accomplished via transactivation of PDGFR and PI3K. We further demonstrate the critical roles of the Gαi-Gβγ subunits of G protein and the Src family tyrosine kinases in β2AR-mediated cardioprotection. Our observation is the first demonstration of a more detailed signaling cascade and the immediate downstream factors, Src and PDGFR in particular, that are involved in the β2AR-mediated antiapoptotic effect.
Src is important for GPCR-mediated signal transduction (29). We show that β2AR stimulation significantly induces Src activity in H9c2 cells. We also demonstrate the involvements of Gαi and Gβγ protein in β2AR-induced antiapoptotic effect in the cells. Both Gαs and Gαi, but not Gβγ, directly interact with and activate Src (29). Overexpression of Gβγ in vitro, however, results in an increase in cSrc autophosphorylation (30). This suggests that although there is no direct interaction, Gβγ is still able to regulate Src activity, a notion consistent with the findings in the present study.
Shutting down Src activity by either pharmacological inhibition with PP2 or siRNA is sufficient to abrogate β2AR-induced resistance to Dox-induced apoptosis in H9c2 cells. We have shown that PP2 alone blocks βAR-mediated activation of PDGFR in H9c2 cells (19). Overexpression of constitutively active Src renders H9c2 cells resistant to Dox-induced apoptosis even without β2AR stimulation. Such resistance against apoptosis, however, is reversed when PI3K signaling is inhibited. From these experiments we conclude that Src must function upstream of PI3K in the β2AR-mediated antiapoptosis. Activation of PDGFR involves receptor autophosphorylation, which results in activation of other signaling systems including PI3K (31). We demonstrated that tyrosine phosphorylation of PDGFR is increased shortly after β2AR stimulation. Because inhibition of Src, by either pharmacological inhibition with PP2 or siRNA, abolishes β2AR-mediated increase in tyrosine phosphorylation of PDGFR and β2AR-mediated resistance against Dox-induced apoptosis, it is likely Src acts upstream of PDGFR. Moreover, because Gβγ is required for the βAR-mediated increase in PI3K activity but does not interact with Src (30), it is likely there are intermediate signaling molecule(s) between Gβγ and Src.
Dox-induced apoptosis is believed to be mainly initiated by oxidative DNA damage (32,33). In the present study, we showed that ISO prevented H2O2-induced apoptosis by similar manners to those seen in Dox-induced apoptosis. The results suggest that in cardiomyocyte there exists common β2AR-mediated antiapoptotic signaling pathways disregard the types of apoptotic insults. This notion, however, needs to be confirmed in other system, such as hypoxic condition in neonatal cardiomyocyte.
Cardiomyopathy is a common cardiac disorder that has been widely observed in diabetic patients. Several studies have shown that enhanced apoptosis is associated with the development of cardiomyopathy. At the cellular level, much is known about the deleterious effects of high ambient glucose concentrations. Some studies have demonstrated oxidative and nitrosative stress in cells after exposure to high ambient glucose (34). High glucose causes disturbances in apoptosis-related mitochondrial proteins and the bcl-2 family of apoptosis regulators (35). In addition, high glucose-induced apoptosis can be prevented by activation of IGF-I (36). This finding is interesting because the majority of data to date have suggested that the antiapoptotic effect of IGF-I is mediated predominantly by the PI3K signaling pathway (36). In the present study, however, we failed to show evidence of IGF-IR transactivation by β2AR stimulation (Fig. 6B). Based on this result, we assumed that IGF-IR did not take part in the cross talk between β2AR and PI3K in this H9c2 cell. In our recent studies, we showed transactivation of PI3K signaling pathway by β2AR stimulation in H9c2 cardiomyocyte (19). The transactivation was mediated by cross talk with the receptor tyrosine kinase, PDGFR, but not IGF-IR. In this study, we demonstrate that high ambient glucose attenuates β2AR-mediated antiapoptotic effect by inhibiting Src activity, PDGFR activation, and PI3K signaling pathway. Based on these findings, we propose a hypothetical pathway for β2AR-mediated cardioprotection and the impact of high ambient glucose on such cardioprotection effect (Fig. 11).
In summary, we have demonstrated novel intracellular signaling mechanisms immediately downstream of β2AR stimulation that result in a protection effect against Dox-induced apoptosis in H9c2 cardiomyocytes. We have also provided a molecular insight into the influence of high ambient glucose. Although the Dox-induced apoptosis might not mimic the apoptosis that is naturally induced by hyperglycemia in diabetic patients, our data, at least, suggest that ambient high glucose could disrupt the regulation of apoptosis in cardiomyocytes. Further study of these molecular pathways should lead to a better understanding of the causes of the two common forms of cardiomyopathy and the identification of new targets for intervention.
Footnotes
This work was supported by a grant from the National Institutes of Health Grant 1-P20-RR-018728-01 (to Y.-T.T., T.C.Z., and J.F.P.) and a collaboration grant from Tokai University (to N.Y.).
Disclosure Statement: The authors have nothing to disclose.
First Published Online August 21, 2008
Abbreviations: β2AR, β2-Adrenergic receptor; βARKct, βAR kinase-1 C terminus minigene; 2′,5′-DOA, 2′,5′-dideoxyadenosine; Dox, doxorubicin; EIA, enzymatic immunoassay; GPCR, G protein-coupled receptor; IGF-IR, IGF-I receptor; ISO, isoproterenol; PDGFR, platelet-derived growth factor receptor; PI3K, phosphoinositide 3-kinase; PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d] pyrimidine; PTX, pertussis toxin; pY, phosphotyrosine; siRNA, small interfering RNA.
References
- Nagatomo T, Ohnuki T, Ishiguro M, Ahmed M, Nakamura T 2001 β-Adrenoreceptors: three-dimensional structures and binding sites for ligands. Jpn J Pharmacol 87:7–13 [DOI] [PubMed] [Google Scholar]
- Frielle T, Collins S, Daniel KW, Caron MG, Lefkowitz RJ, Kobilka BK 1987 Cloning of the cDNA for the human β1-adrenergic receptor. Proc Natl Acad Sci USA 84:7920–7924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, Frielle T, Dohlman HG, Bolanowski MA, Dixon RA, Keller P, Caron MG, Lefkowitz RJ 1987 Delineation of the intronless nature of the genes for the human and hamster β2-adrenergic receptor and their putative promoter regions. J Biol Chem 262:7321–7327 [PubMed] [Google Scholar]
- Emorine LJ, Marullo S, Briend-Sutren MM, Patey G, Tate K, Delavier-Klutchko C, Strosberg AD 1989 Molecular characterization of the human β3-adrenergic receptor. Science 245:1118–1121 [DOI] [PubMed] [Google Scholar]
- Xiao RP, Ji X, Lakatta EG 1995 Functional coupling of the β2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol 47:322–329 [PubMed] [Google Scholar]
- Communal C, Singh K, Sawyer DB, Colucci WS 1999 Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation 100:2210–2212 [DOI] [PubMed] [Google Scholar]
- Wang JJ, Cortes E, Sinks LF, Holland JF 1971 Therapeutic effect and toxicity of adriamycin in patients with neoplastic disease. Cancer 28:837–843 [DOI] [PubMed] [Google Scholar]
- Gottlieb JA, Gutterman JU, McCredie KB, Rodriguez V, Frei III E 1973 Chemotherapy of malignant lymphoma with adriamycin. Cancer Res 33:3024–3028 [PubMed] [Google Scholar]
- Nohl H, Gille L, Staniek K 1998 The exogenous NADH dehydrogenase of heart mitochondria is the key enzyme responsible for selective cardiotoxicity of anthracyclines. Z Naturforsch [C] 53:279–285 [DOI] [PubMed] [Google Scholar]
- Green PS, Leeuwenburgh C 2002 Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. Biochim Biophys Acta 1588:94–101 [DOI] [PubMed] [Google Scholar]
- Sawyer DB, Fukazawa R, Arstall MA, Kelly RA 1999 Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazoxane. Circ Res 84:257–265 [DOI] [PubMed] [Google Scholar]
- Sobel BE, Schneider DJ 2005 Cardiovascular complications in diabetes mellitus. Curr Opin Pharmacol 5:143–148 [DOI] [PubMed] [Google Scholar]
- Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH 2003 Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcN acylation. J Biol Chem 278:44230–44237 [DOI] [PubMed] [Google Scholar]
- Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, Davies PJ, Taegtmeyer H 2000 Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol 32:985–996 [DOI] [PubMed] [Google Scholar]
- Shizukuda Y, Reyland ME, Buttrick PM 2002 Protein kinase C-Δ modulates apoptosis induced by hyperglycemia in adult ventricular myocytes. Am J Physiol Heart Circ Physiol 282:H1625–H1634 [DOI] [PubMed] [Google Scholar]
- Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P 2000 Myocardial cell death in human diabetes. Circ Res 87:1123–1132 [DOI] [PubMed] [Google Scholar]
- Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, Kajstura J 2000 Myocyte death in streptozotocin-induced diabetes in rats in angiotensin II-dependent. Lab Invest 80:513–527 [DOI] [PubMed] [Google Scholar]
- Dangel V, Giray J, Ratge D, Wisser H 1996 Regulation of β-adrenoceptor density and mRNA levels in the rat heart cell-line H9c2. Biochem J 317:925–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano N, Ianus V, Zhao TC, Tseng A, Padbury JF, Tseng YT 2007 A novel signaling pathway for β-adrenergic receptor-mediated activation of phosphoinositide 3-kinase in H9c2 cardiomyocytes. Am J Physiol Heart Circ Physiol 293:H385–H393 [DOI] [PubMed] [Google Scholar]
- Yano N, Suzuki D, Endoh M, Zhao TC, Padbury JF, Tseng YT 2007 A novel phosphoinositide 3-kinase-dependent pathway for angiotensin II/AT-1 receptor-mediated induction of collagen synthesis in MES-13 mesangial cells. J Biol Chem 282:18819–18830 [DOI] [PubMed] [Google Scholar]
- Feng LX, Ravindranath N, Dym M 2000 Stem cell factor/c-kit up-regulates cyclin D3 and promotes cell cycle progression via the phosphoinositide 3-kinase/p70 S6 kinase pathway in spermatogonia. J Biol Chem 275:25572–25576 [DOI] [PubMed] [Google Scholar]
- Chan TO, Tanaka A, Bjorge JD, Fujita DJ 1990 Association of type I phosphatidylinositol kinase activity with mutationally activated forms of human pp60c-src. Mol Cell Biol 10:3280–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Inglese J, Stone WC, Lefkowitz RJ 1993 The binding site for the βγ subunits of heterotrimeric G proteins on the β-adrenergic receptor kinase. J Biol Chem 268:8256–8260 [PubMed] [Google Scholar]
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L 2004 Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharm Rev 56:185–229 [DOI] [PubMed] [Google Scholar]
- Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, Singh K, Colucci WS 2003 β-Adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ Res 92:136–138 [DOI] [PubMed] [Google Scholar]
- Revankar CM, Vines CM, Cimino DF, Prossnitz ER 2004 Arrestins block G protein-coupled receptor-mediated apoptosis. J Biol Chem 279:24578–24584 [DOI] [PubMed] [Google Scholar]
- Menon B, Singh M, Ross RS, Johnson JN, Singh K 2006 β-Adrenergic receptor-stimulated apoptosis in adult cardiac myocytes involves MMP-2-mediated disruption of β1 integrin signaling and mitochondrial pathway. Am J Physiol Cell Physiol 290:C254–C261 [DOI] [PubMed] [Google Scholar]
- Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP 2007 Activation of CaMKIIδC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem 282:10833–10839 [DOI] [PubMed] [Google Scholar]
- Ma YC, Huang J, Ali S, Lowry W, Huang XY 2000 Src tyrosine kinase is a novel direct effector of G proteins. Cell 102:635–646 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ 1996 Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gβγ subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem 271:19443–19450 [DOI] [PubMed] [Google Scholar]
- Tallquist M, Kazlauskas A 2004 PDGF signaling in cells and mice. Cytokine Growth Factor Rev 15:205–213 [DOI] [PubMed] [Google Scholar]
- Jung K, Reszka R 1999 Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev 49:87–105 [DOI] [PubMed] [Google Scholar]
- Mizutani H, Tada-Oikawa S, Hiraku Y, Kojima M, Kawanishi S 2005 Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci 76:1439–1453 [DOI] [PubMed] [Google Scholar]
- Cellek S, Qu W, Schmidt AM, Moncada S 2004 Synergistic action of advanced glycation end products and endogenous nitric oxide leads to neuronal apoptosis in vitro: a new insight into selective nitrergic neuropathy in diabetes. Diabetologia 47:331–339 [DOI] [PubMed] [Google Scholar]
- Pampfer S, Cordi S, Vanderheyden I, Van Der Smissen P, Courtoy PJ, Van Cauwenberge A, Alexandre H, Donnay I, De Hertogh R 2001 Expression and role of Bcl-2 in rat blastocysts exposed to high d-glucose. Diabetes 50:143–149 [DOI] [PubMed] [Google Scholar]
- Fernández M, Sánchez-Franco F, Palacios N, Sánchez I, Fernández C, Cacicedo L 2004 IGF-I inhibits apoptosis through the activation of the phosphatidylinositol 3-kinase/Akt pathway in pituitary cells. J Mol Endocrinol 33:155–163 [DOI] [PubMed] [Google Scholar]