

Abstract
Regulation of classic cadherins plays a critical role in tissue remodeling during development and cancer; however, less attention has been paid to the importance of desmosomal cadherins. We previously showed that EGFR inhibition results in accumulation of the desmosomal cadherin, desmoglein 2 (Dsg2), at cell–cell interfaces accompanied by inhibition of matrix metalloprotease (MMP)-dependent shedding of the Dsg2 ectodomain and tyrosine phosphorylation of its cytoplasmic domain. Here, we show that EGFR inhibition stabilizes Dsg2 at intercellular junctions by interfering with its accumulation in an internalized cytoplasmic pool. Furthermore, MMP inhibition and ADAM17 RNAi, blocked shedding and depleted internalized Dsg2, but less so E-cadherin, in highly invasive SCC68 cells. ADAM9 and 15 silencing also impaired Dsg2 processing, supporting the idea that this desmosomal cadherin can be regulated by multiple ADAM family members. In contrast, ADAM10 siRNA enhanced accumulation of a 100-kDa Dsg2 cleavage product and internalized pool of Dsg2. Although both MMP and EGFR inhibition increased intercellular adhesive strength in control cells, the response to MMP-inhibition was Dsg2-dependent. These data support a role for endocytic trafficking in regulating desmosomal cadherin turnover and function and raise the possibility that internalization and regulation of desmosomal and classic cadherin function can be uncoupled mechanistically.
INTRODUCTION
The ability of cells to modulate their contacts with each other and the underlying matrix is essential for epithelial remodeling that occurs in development and cancer progression (Behrens, 1999; Thiery, 2003; Kramer et al., 2005). In particular, members of cadherin family of calcium-dependent intercellular adhesion molecules have been demonstrated to both suppress (Frixen et al., 1991) and promote (Islam et al., 1996) cell migration and invasion. Although classic cadherins assemble into intercellular adhesive structures known as adherens junctions that associate with the cortical actin cytoskeleton, desmosomal cadherins, including desmogleins and desmocollins, make up the adhesive core of desmosomes, which anchor intermediate filaments (IF) to sites of strong intercellular adhesion (Green and Simpson, 2007). Anchorage to the IF cytoskeleton is established with the cooperation of the desmosomal cadherin–associated proteins plakoglobin and plakophilins, which in turn link the IF-associated protein desmoplakin (DP) to the membrane complex. Desmosomes provide mechanical integrity to epithelial and heart tissues by redistributing the forces of mechanical stress (Godsel et al., 2004). In addition, desmosomal cadherins have more recently emerged as playing a role in tissue morphogenesis (Runswick et al., 2001; Chidgey and Dawson, 2007; Dusek et al., 2007).
In spite of desmosomes' importance in tissue function, most studies have focused on the contribution of classic cadherins to epithelial remodeling. Classic cadherins engage in bidirectional signaling with receptor tyrosine kinases (RTKs), whereby they are both responsive to, and can trigger, signals that regulate cell behavior (Wheelock and Johnson, 2003; McLachlan and Yap, 2007). E-, N-, and VE-cadherin ectodomains are also subject to shedding via matrix metalloproteases (MMPs; Lochter et al., 1997; Llorens et al., 1998; Noe et al., 2001), including the ADAM (a disintegrin and metalloprotease) family of transmembrane sheddases (Maretzky et al., 2005; Reiss et al., 2005; Najy et al., 2008; Schulz et al., 2008). Both phosphorylation of the cadherin complex and MMP-dependent processing of cadherins can also regulate the availability of a pool of nonjunctional catenins, which propagate signals that can regulate cell growth, differentiation, and motility (Conacci-Sorrell et al., 2002; Nelson and Nusse, 2004).
Although our understanding of the role of desmosomal modulation in tissue homeostasis and remodeling is not well understood, changes in desmosomal cadherin expression, phosphorylation, and function are known to occur during tissue morphogenesis and tumor progression (Runswick et al., 2001; Chidgey and Dawson, 2007; Dusek et al., 2007; Green and Simpson, 2007; Muller et al., 2008). Our previous work suggested that epidermal growth factor receptor (EGFR), which is overexpressed in many tumor types (Rogers et al., 2005; Kalyankrishna and Grandis, 2006), regulates desmosome assembly and function in squamous cell carcinoma (SCC) cells, by decreasing the level and cell surface localization of desmosomal cadherins (Lorch et al., 2004). The resulting weakened intercellular adhesive strength was accompanied by phosphorylation of desmoglein 2 (Dsg2) and associated proteins (which occurred preferentially on desmosomal and not classic cadherins), and MMP-dependent shedding of the Dsg2 ectodomain. EGFR inhibition reversed these effects. However, the mechanism by which EGFR inhibition stabilizes intercellular desmosomal cadherins is not understood.
Endocytic trafficking of classic cadherins has emerged as an important mechanism to regulate adhesion during epithelial remodeling. E-cadherin trafficking can occur through clathrin-dependent, clathrin-independent, and caveolae-dependent pathways and can be regulated by RTKs (Lu et al., 2003; Bryant and Stow, 2004). In addition to regulating interactions with the cytoskeleton, RTK-dependent phosphorylation of cadherins and their associated proteins can contribute to the internalization and/or diversion of cadherins to a lysosomal degradation pathway (Fujita et al., 2002; d'Azzo et al., 2005; McLachlan and Yap, 2007). However, the contribution of desmosomal cadherin regulation to epithelial remodeling via endocytic trafficking has not been addressed, nor is the involvement of RTKs or MMPs understood. The importance of RTKs and MMPs in epithelial remodeling processes is highlighted by the well-established role of erbB family members in tumor progression and recent recognition that ADAM family members are aberrantly expressed in certain cancers (Borrell-Pages et al., 2003; Rocks et al., 2008).
Here, we ask whether EGFR inhibition stabilizes intercellular Dsg2 by regulating endocytic trafficking of this molecule. Our results indicate that EGFR regulates Dsg2 trafficking, and does so in part through an ADAM-dependent pathway. Although E-cadherin internalization is also regulated by EGFR in SCC68 cells, responsiveness to MMP inhibitors is muted compared with Dsg2. Importantly, this differential responsiveness is mirrored by functional data showing that intercellular adhesive strength in control cells increases in response to both EGFR and MMP inhibition, but Dsg2 is specifically required for the response to MMP inhibitors. Collectively, these data support the idea that different modes of regulation exist for these cadherin family members.
MATERIALS AND METHODS
Cell Culture and Inhibitors
SCC68 squamous cell carcinoma cells (a gift from J. Rheinwald, Harvard Medical School, Boston, MA) were cultured in keratinocyte serum-free media (0.09 mM Ca2+) supplemented with 1 ml of bovine pituitary extract and 0.3 ng/ml epidermal growth factor (EGF; Invitrogen, Carlsbad, CA). Cells were propagated or used in experiments after reaching 70–80% confluency. For most experiments, cells were incubated overnight in 0.25 mM CaCl2 with the following reagents: 1% dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO), 7 μm PKI166 (PKI; gift from Novartis AG, Basel, Switzerland), 50 μm TAPI-0 (TAPI; EMD Chemicals, Gibbstown, NJ), 25 μm GM6001 (Chemicon International, Temecula, CA) or 200 ng/ml PMA (Calbiochem, San Diego, CA). For experiments requiring EGFR stimulation, cells were incubated overnight in starvation media containing 0.25 or 1.0 mM CaCl2 and treated with 10 ng/ml EGF (Sigma-Aldrich) for 1 h before processing the cells.
siRNA Transfections
SCC68 cells grown to 30% confluency were transfected with small interfering RNA (siRNA) for human ADAM 9, -10, -15, and -17, Dsg2, E-cadherin, or GAPDH (Dharmacon, Lafayette, CO) at a final concentration of 100 nM using DharmaFECT-1 transfection reagent (Dharmacon) according to manufacturer's recommendations. For Western blot analysis and biotinylation experiments, cells were processed 96 h after transfection. Cells used in antibody internalization assays were processed 48 h after transfection.
Antibodies
The following primary antibodies were used: mouse-monoclonals 6D8 (gift from M. Wheelock, K. Johnson, and J. Wahl, University of Nebraska Medical Center, Omaha, NE), 4B2 (Getsios et al., 2004), G129 (Progen, Heidelberg, Germany) and Dg3.1 (Research Diagnostics, Concord, MA) against Dsg2, HECD-1 (gift from M. Takeichi) against E-cadherin. Ab12 and 13 against EGFR (Lab Vision, Fremont, CA), E7 and 12G10 against β- and α-tubulin, respectively (University of Iowa Developmental Studies Hybridoma Bank, Iowa City, IA). Clone H68.4 against transferrin (Zymed, Carlsbad, CA) and nonspecific IgG (Sigma-Aldrich); rabbit polyclonals ab9845 against GAPDH (Abcam, Cambridge, England), AB19026 against ADAM10 (Chemicon), AB19027 and sc-25782 against ADAM17 (Chemicon and Santa Cruz Biotechnology, Santa Cruz, CA, respectively), ab11531 against ADAM9 (Abcam), AB19036 against ADAM15 (Chemicon) and horseradish peroxidase (HRP)-conjugated PY99 against phosphotyrosine (Santa Cruz Biotechnology). The following secondary antibodies were used: HRP-conjugated goat anti-mouse and anti-rabbit (Kirkegaard & Perry Laboratories, Gaithersburg, MD), Alexa-Fluor 488 goat anti-mouse and Alexa Fluor 568 goat anti-rabbit (Molecular Probes, Eugene, OR) and goat anti-mouse (Thermo Fisher Scientific, Rockford, IL).
Immunoblotting
Whole cell lysates were generated in urea sample buffer, and total protein was quantified using the amido black assay (Sheffield et al., 1987). Equal amounts of protein were fractionated on a 7.5% SDS-polyacrylamide gel, and immunoblotting was performed as described (Kowalczyk et al., 1997). Antibodies were used at the following dilutions: 4B2 (1:1000), 6D8 (1:1000), Dg3.1 (1:1000), HECD-1 (1:1000), Ab12 (1:500), AB19026 (1:1000), AB19027 (1:1000), ab11531 (1:1000), AB19036 (1:1000), clone H68.4 (1:1000), E7 (1:500), 12G10 (1:1000), ab9845 (1:1000), PY99 (1:200), and HRP-conjugated goat anti-mouse and anti-rabbit (1:5000). Proteins were visualized using enhanced chemiluminescence.
Immunoprecipitation
Cells were lysed in RIPA buffer (10 mM Tris, pH 7.5, 140 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 5 mM EDTA, and 2 mM EGTA) containing EDTA-free protease inhibitor (Roche Diagnostics, Mannheim, Germany), and lysates were centrifuged at 14,000 rpm for 30 min at 4°C. Immunoprecipitations were performed by incubating cleared supernatant with 3–5 μl of the appropriate antibody followed by 40 μl gamma-bind Sepharose (Amersham Biosciences, Uppsala, Sweden) overnight at 4°C. Immune complexes were released using reducing Laemmli buffer at 95°C and analyzed by immunoblot.
Immunofluorescence
Cells grown on glass coverslips were rinsed in phosphate-buffered saline (PBS), fixed in anhydrous methanol for 2 min at −20°C, and incubated with 6D8 (1:100), HECD-1 (1:100), or sc-25782 (1:500). In some experiments, cells were fixed in 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 or methanol. Primary antibodies were visualized using Alexa Fluor 488 goat anti-mouse IgG or Alexa Fluor 568 goat anti-rabbit IgG (1:400). Coverslips were examined with a Leica upright microscope (model DMR; Deerfield, IL), and images were captured using Hamamatsu Orca digital camera (model C4742-95; Bridgewater, NJ) and MetaMorph 6.1 imaging software (Universal Imaging, West Chester, PA). To quantify border staining, 75 borders were examined, and the length of the border occupied by Dsg2 or E-cad staining was scored as follows: one-third (black), two-thirds (gray), or the entire (white) border.
Generation of Desmoglein 2-GFP Virus and Live Cell Imaging
A cDNA construct encoding human Dsg2 in pBluescript R3 BptII SR (p594; gift from Drs. W. Franke and S. Schafer, German Cancer Research Center, Heidelberg, Germany) was used to generate a Dsg2-FLAG-EGFP chimera. The stop codon was replaced with a C-terminal FLAG epitope followed by a BamHI restriction site. This FLAG-tagged human Dsg2 cDNA was subcloned in frame with the enhanced green fluorescent protein (EGFP) sequence of pEGFP-N2 (Clontech Laboratories, Mountain View, CA) using BamHI, and the resulting plasmid was termed p1105. The entire Dsg2-FLAG-EGFP chimeric cDNA was then subcloned into the LZRS-Linker retroviral vector (p989; Denning et al., 2002) in the forward orientation using XhoI and NotI and named p1106. Retroviral supernatants containing the full-length Dsg2-FLAG-EGFP were generated using Phoenix amphotropic cell lines provided by Gary Nolan (Stanford University Medical School, Palo Alto, CA) and SCC68 cell lines transduced as previously described (Getsios et al., 2004).
Cells expressing Dsg2-GFP were seeded on Lab-Tek chambered coverglass slides (Nunc, Naperville, IL) at 70–80% confluency. The next day, cells were cultured in 0.25 mM calcium-containing medium in the presence of DMSO, TAPI, or PKI at 37°C for 7–8 h before imaging. Time-lapse images were obtained from a single plane at 3-s intervals using 100× (HCX PLAN APO, NA 1.35) with an Application Solution Multidimensional Workstation (ASMDWQ, Leica), equipped with a DMIRE2 inverted microscope with a Coolsnap HQ camera (Roper Scientific, Tucson, AZ) and 37°C climate chamber. Images were processed using ASMDW and Metamorph 6.1 (Universal Imaging) imaging software.
Biotinylation Assay
Cells were rinsed in ice-cold PBS, at the appropriate calcium concentration, and incubated with 2 mg/ml EZ-Link Sulfo-NHS-SS-biotin (Pierce Chemical, Rockford, IL) for 30 min at 4°C. Excess biotin was removed with PBS (three times), and internalization of biotin-labeled surface proteins was allowed to proceed at 37°C for the indicated times in prewarmed culture media containing inhibitors, when indicated. Cells were returned to 4°C and washed with PBS (three times). Residual biotin was stripped from the cell surface by washing with 100 mM sodium 2-mercaptoethanesulfonic acid (Sigma-Aldrich) in 50 mM Tris-HCl, pH 8.6, 100 mM NaCl, 1 mM EDTA, 0.2% bovine serum albumin (BSA; Sigma-Aldrich) three times for 20 min. Cells were washed with 120 mM iodoacetamide (Sigma-Aldrich) one time for 10 min, and PBS (three times) to quench residual MESNA and lysed in RIPA buffer containing protease inhibitors. Lysates were vortexed for 1 min, centrifuged at 14,000 rpm for 30 min, and recovered supernatants were normalized for total protein using the amido black assay. Twenty microliters of lysate was removed to serve as an input. Biotinylated proteins were retrieved with 25–40 μl UltraLink immobilized streptavidin (Pierce) by end-over-end rotation at 4°C overnight and washed with RIPA buffer (four times). Complexes were released using reducing Laemmli buffer at 95°C and analyzed using immunoblot analysis as described above. Densitometric analysis of band intensity was performed using ImageJ (National Institutes of Health; http://rsb.info.nih.gov/ij/). To quantify internalization, the ratio of cytoplasmic/surface intensity was determined after normalizing to tubulin.
Antibody Internalization Assay
Cells grown on coverslips were incubated at 4°C for 1 h with 6D8 (1:00) or HECD-1 (1:100) in KSFM containing 3% BSA and 20 mM HEPES. Unbound antibody was removed by washing with PBS (three times), at the appropriate calcium concentration. Internalization of surface-bound antibody was stimulated at 37°C for the indicated times in prewarmed culture media containing inhibitors, when indicated. After returning to 4°C, coverslips were washed with PBS (three times), and residual antibody was stripped from the cell surface by acid washing (0.5 mM NaCl, 0.5 mM acetic acid) four times for 15 min. Cells were fixed in 4% paraformaldehyde and unwashed antibody was quenched with goat anti-mouse (1:10). Coverslips were washed with PBS (three times) and processed using indirect immunofluorescence as described above. To quantify internalization, the cytoplasmic fluorescence of 75–100 cells was measured for each time point using Metamorph Imaging. Data are expressed as average cytoplasmic intensity ± SEM.
Mechanical Strength (Dispase) Assay
Confluent cell cultures were washed with PBS and incubated with 2.4 U/ml dispase for 30 min at 37°C. Released monolayers were either analyzed directly without applying mechanical stress or transferred to a 15-ml conical tube and washed with PBS (two times). Tubes were inverted (0.09 mM = 0 inversions, 0.50 mM = 5–10 inversions, 1.0 mM = >10 inversions), and fragments were counted with a dissecting scope (Leica, MZ6) as described (Hudson et al., 2004) or imaged with a Hamamatsu Orca digital camera (model C4742-95) and analyzed using MetaVue imaging software (Universal Imaging). Data are expressed as average number of fragments ± SD. Statistical analysis was determined using a Student's t test.
RESULTS
EGFR and MMP Inhibition Increase Dsg2 at Cell–Cell Borders and Strengthen Intercellular Adhesion
Our previous results showed that EGFR inhibition interferes with MMP-dependent shedding of the desmosomal cadherin Dsg2, while also resulting in its redistribution from the cytoplasm to intercellular junctions, even in reduced calcium where desmosomes do not typically form. Classic cadherins have previously been shown to be depleted from the cell surface by endocytic internalization (Le et al., 1999). This suggested to us that EGFR inhibition may be able to override signals that promote internalization of desmosomal cadherins under reduced calcium conditions. Here we ask whether EGFR decreases cell surface Dsg2 by regulating endocytic trafficking and explore the role MMP-dependent shedding has in this process.
To compare the relative abilities of EGFR and MMP inhibitors to stabilize Dsg2 at cell–cell interfaces, SCC68 cells in 0.25 mM calcium were treated with vehicle only (DMSO), the broad spectrum MMP inhibitor TAPI, or the ErbB1/2 inhibitor PKI and analyzed by conventional immunofluorescence microscopy. Although similar results were seen at all calcium concentrations, an intermediate concentration was selected from a range between 0.09 and 1.0 mM calcium as optimal for observing Dsg2 cleavage and internalization. At lower concentrations Dsg2 levels were decreased because of its instability, and at higher concentrations cleavage/internalization was somewhat attenuated most likely because of junctional stabilization. Importantly, however, as discussed below, similar results were seen at physiological calcium concentrations (Supplemental Figure S1).
Like PKI and the mAb inhibitor C225 (Lorch et al., 2004), TAPI treatment led to a dramatic redistribution of Dsg2 to intercellular borders (Figure 1, A and B). In contrast, there was little impact on the extent to which cell–cell interfaces were occupied by E-cadherin, although both inhibitors resulted in a more organized appearance of this classic cadherin (Figure 1, A and B). To test whether Dsg2 redistribution correlated with an increase in intercellular adhesive strength, epithelial sheets treated with DMSO, PKI, or the broad spectrum MMP inhibitors GM6001 (Figure 1C) or TAPI (not shown) were released with dispase and were subjected to mechanical shear stress as described (Hudson et al., 2004). Like EGFR inhibition, MMP inhibition increased epithelial sheet integrity in a calcium-dependent manner, even at the highest concentration, supporting the physiological relevance of MMP-dependent effects on intercellular adhesion (Figure 1C).
Figure 1.

MMP inhibition promotes Dsg2 accumulation at cell–cell borders and strengthens intercellular adhesion. (A) SCC68 cells were treated overnight with DMSO, TAPI-0 (TAPI), or PKI166 (PKI) in medium containing 0.25 mM CaCl2. Indirect immunofluorescence was used to determine the cellular localization of desmoglein 2 (Dsg2) and E-cadherin (E-cad). Bar, 20 μm. (B) Staining along the border was quantified as follows: one-third border occupied (black), two-thirds (gray), or entire (white) border. TAPI- and PKI-treated cells showed increased intercellular Dsg2, but not E-cad, localization. (C) Confluent monolayers of SCC68 cells were cultured overnight in the presence of DMSO, GM6001 (GM), or PKI in 0.09, 0.50, or 1.0 mM CaCl2. Intercellular adhesion was measured by counting the number of fragments generated from dispase-released monolayers subjected to a defined amount of mechanical stress. At 0.50 mM and 1.0 mM calcium, significantly fewer fragments were produced in the TAPI- and PKI-treated monolayers (p < 0.05).
Dsg2 Is Constitutively Internalized in SCC68 Cells
To begin to address the mechanism of Dsg2 cell surface stabilization, we first asked whether Dsg2 and E-cadherin are internalized in SCC68 cells using two complementary approaches (Le et al., 1999; Figure 2). Cells were incubated at 4°C either with antibodies directed against the extracellular domain of Dsg2 or E-cadherin or with biotin and then switched to 37°C to allow internalization. At time points beginning at 5 min, internalization was analyzed either by stripping the surface label and subsequently analyzing the internal pool using labeled secondary antibody or by streptavidin pulldown followed by immunoblotting, respectively.
Figure 2.

Dsg2 is constitutively internalized. SCC68 cells were cultured in the presence of 0.25 mM CaCl2 overnight. (A) Dsg2 and E-cadherin (E-cad) were labeled with monoclonal antibodies directed against their extracellular domains at 4°C. Internalization of the surface labeled proteins was allowed for 5, 15, 30, or 60 min at 37°C. Residual antibody was stripped from the cell surface using an acid wash, and internalized Dsg2 and E-cad were visualized using indirect immunofluorescence. Bar, 20 μm. (B) Cells were incubated with biotin at 4°C and then switched to 37°C to allow internalization to proceed for 5, 15, 30, 60, 90, or 120 min or 3, 4, 6, 8, or 24 h. After stripping residual biotin from the cell surface, biotinylated protein samples were pulled down using immobilized streptavidin and analyzed by immunoblotting for Dsg2 (top panel) and E-cad (middle panel; note that this blot was reprobed without stripping, resulting in retention of bands representing full-length and processed Dsg2). Tubulin immunoblotting of RIPA lysates (RCL) was used to normalize samples. (C) Densitometric analysis was performed using ImageJ to determine the ratio of cytoplasmic/surface intensity at each time point. Dsg2 accumulated in the cytoplasm as early as 5 min.
As shown in Figure 2A, under baseline conditions where EGFR is active, Dsg2 and E-cadherin were rapidly internalized at 37°C. Both cadherins were detected as early as 5 min and accumulated over time, the levels decreasing beginning at 90 min presumably due to degradation of the internalized pool and/or recycling back to the surface (Figure 2, B and C). Both full-length and a 100-kDa cell-retained Dsg2 fragment, previously shown to result from MMP-dependent processing of Dsg2 (Lorch et al., 2004), appeared in the internalized pool over a similar time course. Importantly, internalization occurred at calcium concentrations ranging from 0.09 to 1 mM, supporting the physiological relevance of the process (Figures 2 and Supplemental Figure S1 and not shown).
Both Dsg2 and E-Cadherin Undergo EGFR-dependent Internalization, But Dsg2 Is More Sensitive to MMP Inhibition
To test whether the redistribution of Dsg2 in response to TAPI and PKI was due to interference with the accumulation of the internalized pool of cadherins, antibody internalization and biotinylation assays were performed in cells pretreated with PKI (or the mAb C225) or TAPI, to block EGFR and MMP activities, respectively. PKI and the mAb C225 interfered with the appearance of both Dsg2 and E-cadherin, but not transferrin, in an internalized pool (Figure 3, A and B, and not shown). However, Dsg2 appeared to be more responsive than E-cadherin to TAPI inhibition in both antibody internalization and biotinylation assays (Figure 3, A–D). The extent to which the responsiveness to TAPI differed depended somewhat on the conditions of the experiment, but differences were observed at all calcium concentrations tested (Figure 3 and Supplemental Figure S1). GM6001 also interfered with internalization triggered by acute treatment with EGF following cell starvation at a higher calcium concentration (Supplemental Figure S1A).
Figure 3.

MMP and EGFR inhibition interfere with the accumulation of an internalized pool of Dsg2. (A) SCC68 cells were cultured overnight with DMSO, TAPI, or PKI in 0.25 mM CaCl2. Dsg2 and E-cadherin (E-cad) monoclonal antibodies were used to measure internalization after 60 min. Bar, 20 μm. (B) Internalization was quantified by measuring the cytoplasmic fluorescence intensity using Metamorph Imaging. (C) SCC68 cells were incubated with DMSO, TAPI, or PKI overnight in the presence of 0.09 mM calcium, and cell surface biotinylation was performed as described to measure internalization after 60 min. (D) The ratio of cytoplasmic/surface intensity was used to measure internalization. PKI effectively blocked the accumulation of Dsg2 and E-cad, whereas TAPI was more effective inhibiting the appearance of internalized Dsg2.
To corroborate these findings, we examined the behavior of a C-terminally green fluorescent protein (GFP)-tagged Dsg2 in control, PKI-, or TAPI-treated living cells using time-lapse live cell imaging (Figure 4 and Supplemental Videos). GFP-labeled Dsg2 particles exhibited rapid movements in the cortical region of the cells treated with vehicle, DMSO (Figure 4A and Supplemental Video 1). Although some particles appeared loosely tethered to the plasma membrane, they were also observed to uncouple from the membrane and move in a retrograde direction into the cytoplasm (Figure 4, arrows, and Supplemental Video 1). An increase in Dsg2 was observed at cell–cell interfaces in the TAPI- (Figure 4 and Supplemental Video 2) and PKI-treated (Figure 4 and Supplemental Video 3) cells, and fluorescent puncta appeared to be more stably associated with cell–cell interfaces, as one would expect for more mature junctions. Collectively, these experiments strongly support the idea that EGFR and MMPs destabilize Dsg2 at cell–cell interfaces by elevating the level of an internalized pool of cadherin at the expense of the adhesion-competent junctional pool.
Figure 4.

EGFR and MMP inhibition alter the dynamic behavior of Dsg2. SCC68 cells expressing Dsg2-GFP were cultured for 8 h with DMSO, TAPI, or PKI in 0.25 mM calcium-containing medium. Cells were imaged at 3-s intervals for 20 min. Bar, 10 μm. Arrows indicate one of several Dsg2 vesicles that appear to move away from cell–cell interfaces in control cells. Dsg2 was more stably associated with cell–cell interfaces in TAPI and PKI-treated cells. (see also Supplemental Videos S1, S2, and S3).
Dsg2 Is Required for Responsiveness to MMP Inhibition
As shown in Figure 1C, alterations in cadherin trafficking resulting from EGFR or MMP-inhibition are accompanied by increased intercellular adhesive strength. To assess the specific contribution of Dsg2, we examined cellular responses to PKI or TAPI in Dsg2-deficient dispase-released epithelial sheets without further mechanical stress applied to the cultures. Under these conditions control cultures held together, whereas Dsg2 deficient cultures dissociated (Figure 5, A and B), even though E-cadherin was still present (Figure 5C). Importantly, although PKI treatment of the Dsg2-deficient cultures protected them from dissociation, loss of Dsg2 resulted in failure of the cultures to respond to TAPI treatment. These data support the idea that the sensitivity of Dsg2 trafficking to TAPI-treatment translates into a physiologically relevant increase in adhesive strength, specifically because of its action on this desmosomal cadherin. These data do not rule out a contribution for EGFR-dependent regulation of Dsg2 trafficking in adhesion, but suggest that the presence of classic cadherins is sufficient for a cellular response to EGFR inhibition.
Figure 5.

Dsg2 is required to mediate increased intercellular adhesion in MMP-inhibited cells. SCC68 cells were transfected with GAPDH or Dsg2 siRNA. Ninety-six hours after transfection, cells were cultured overnight in DMSO, TAPI, or PKI in 0.25 mM calcium-containing medium. (A) Confluent monolayers of SCC68 cells were released using dispase. (B) Intercellular adhesion was measured by counting the number of fragments generated without applying mechanical stress. (C) Western blot analysis was performed on whole cell lysates using antibodies against Dsg2, E-cadherin, GAPDH, and tubulin. Fragmentation was observed in the TAPI-treated monolayers when Dsg2 was knocked down.
Dsg2 Processing and Trafficking Is ADAM-dependent
Based on the observed role for MMPs in destabilizing intercellular Dsg2, we set out to further characterize Dsg2 processing and identify the responsible MMP. We previously showed that MMP-dependent Dsg2 processing generates a cell-associated 100-kDa band that is reduced by EGFR inhibition and an ectodomain fragment shed into the medium (Lorch et al., 2004). Immunoprecipitation of endogenous Dsg2 with antibodies recognizing specific extra- and intracellular domains of Dsg2, and subsequent immunoblotting with a mAb directed against the terminal portion of the cytoplasmic domain indicated that the 100-kDa cleavage product comprises the entire Dsg2 cytoplasmic tail, transmembrane domain, and an extracellular juxtamembrane domain (Supplemental Figure S2). Dsg2 shedding occurred in a panel of SCC lines (not shown), including SCC13 cells constitutively overexpressing ectopic EGFR but not in their control counterparts (McCawley et al., 1997). Desmosomal cadherin shedding was greatest in the SCC68 line, which was thus chosen for use throughout this study.
In addition to being blocked by TAPI, Dsg2 proteolysis was enhanced by PMA treatment, which is known to stimulate the activity of ADAM family member proteases (Figures 6 and 7). ADAM family members have been shown to cleave classic cadherins, and Dsg2 was also recently identified as a substrate of ADAM10 and 17 (Bech-Serra et al., 2006). To test whether either of these ADAM family members is responsible for Dsg2 cleavage in SCC68 cells, we introduced ADAM10 or 17 small interfering RNA (siRNA) into SCC68 cells. As shown in Figure 6, A and B, ADAM17 and 10 knockdown reduced the active forms of these proteases, but only ADAM17 silencing blocked production of the 100-kDa Dsg2 cleavage product. Furthermore, TAPI and ADAM17 silencing blocked cleavage and accumulation of the 100-kDa fragment even in PMA- and EGF-stimulated cells under which conditions EGFR activity remained high (Figures 6B and 7 and not shown). Unexpectedly, ADAM10 knockdown resulted in increased levels of the 100-kDa fragment, raising the possibility that ADAM10 may be involved in further processing of this fragment. To determine the specificity of the ADAM17 siRNA and to test for the possible involvement of ADAM9 or ADAM15, both of which have recently been linked to classic cadherin regulation (Hirao et al., 2006; Najy et al., 2008), we compared cells treated with ADAM9, 10, 15, and 17 RNAis for Dsg2 processing (Supplemental Figure S3). Interestingly, ADAM15, and to a lesser extent ADAM9, also impaired cleavage of Dsg2, without having any off target effects on the mature forms of the other ADAM family members, indicating that Dsg2 shedding may be regulated by multiple ADAMs.
Figure 6.

ADAM17 knockdown blocks the proteolysis of Dsg2. (A) SCC68 cells were transfected with reagent alone, GAPDH, ADAM10, or ADAM17 siRNA in 0.25 mM calcium-containing medium. Western blot analysis using antibodies against Dsg2, ADAM10, ADAM17, GAPDH, and tubulin was performed on whole cell lysates prepared 96 h after transfection. ADAM17, but not ADAM10, knockdown inhibited the cleavage of Dsg2. (B) Before harvesting, SCC68 cells were treated with DMSO, PMA, or TAPI overnight. The PMA-induced increase in Dsg2 cleavage was prevented in response to ADAM17 knockdown.
Figure 7.

Relationship between ADAM17-mediated Dsg2 cleavage and EGFR activity. (A) SCC68 cells were treated overnight with DMSO, PMA, GM, TAPI, PKI, or PMA, and PKI in starvation media containing 0.25 mM calcium. When indicated, cells were also stimulated with EGF for 1 h before lysis. Whole cell lysates were analyzed by immunoblotting for Dsg2, phosphotyrosine, EGFR, and tubulin. (B) SCC68 cells were transfected with GAPDH, ADAM10, or ADAM17 siRNA and analyzed by Western blotting 96 h after transfection for Dsg2, phosphotyrosine, EGFR, ADAM10, ADAM17, and tubulin. Dsg2 cleavage was blocked in MMP inhibited and ADAM17-deficient cells despite robust EGFR phosphorylation.
Recently it was shown that EGFR regulates ADAM17 by increasing the half-life of ADAM17 protein and consequently the level of mature ADAM17 in A431 cells (Santiago-Josefat et al., 2007). Therefore, we were surprised when we observed that EGFR inhibition by PKI increased ADAM17 (both pro- and mature forms) in SCC68 cells despite decreased cleavage of Dsg2 under these conditions (Supplemental Figure S4A). To reconcile these findings we investigated ADAM17 distribution in PKI-treated cultures. Although Dsg2 accumulated at cell–cell interfaces in PKI-treated cells, ADAM17 staining remained predominantly in the cell center, presumably at the dorsal cell surface and/or in intracellular vesicles (Supplemental Figure S4B). Thus, EGFR inhibition shifts the pool of Dsg2 spatially away from the ADAM17-rich compartment, which likely explains at least in part why Dsg2 cleavage is diminished in spite of the increased levels of protease, but not completely abrogated, in PKI-treated cells. Differences in our results from those reported by Santiago-Josefat may be due to cell type specific differences in the mechanism responsible for regulating ADAM17.
To test whether inhibition of ADAM17 interferes with accumulation of an internalized pool of Dsg2, we carried out antibody and biotinylation assays after transfection with ADAM17 siRNA. Although there was some variability in the level to which the pro- and mature forms of ADAM17 were reduced by knockdown, we consistently observed the loss of a dispersed cytoplasmic pool of ADAM17 that partially overlapped with Dsg2 staining, with more variable retention of a resistant particulate pool (Figure 8C, bottom panel). This particulate pool remained cytoplasmic, largely distinct from Dsg2, raising the possibility that the desmosomal cadherin is thus inaccessible and protected from further degradation. Consistently, ADAM17 knockdown increased the general cell surface staining for Dsg2 (Figure 8A, bottom left) and effectively blocked the appearance of an internalized pool of Dsg2, but not transferrin (Figure 8, A–E, and not shown). In contrast, ADAM10 knock down not only resulted in an accumulation of the 100-kDa Dsg2 cleavage product in SCC68 cells, it also led to an increase in the internalized pool of Dsg2 (Figure 8, D and E). Collectively, our data suggest that the balance of different MMP activities is likely to play an important role in controlling the extent to which specific cadherins are stabilized at the cell surface and in intercellular junctions during epithelial remodeling.
Figure 8.

ADAM17 knockdown blocks the accumulation of an internalized pool of Dsg2. (A) SCC68 cells were transfected with GAPDH or ADAM17 siRNA in media containing 0.25 mM CaCl2 and analyzed 48 h after transfection. Cell surface Dsg2 was labeled with antibody, and internalization was assessed after 60 min at 37°C. ADAM17 localization was assessed using indirect immunofluorescence. Bar, 20 μm. (B) The average cytoplasmic intensity was measured using Metamorph Imaging. (C) Western blot analysis of Dsg2, ADAM17, and tubulin. ADAM17 was reduced by 65% percent under these conditions (C, top panel), and the residual ADAM17 exhibits a particulate pattern that appears distinct from Dsg2 staining (C, bottom right panel). (D and E) Cell surface biotinylation was performed to measure internalization after 60 min in GAPDH, ADAM10, or ADAM17 knockdown cells 96 h after transfection. Tubulin blot from RIPA lysates is shown to demonstrate equal starting material for all the samples. Cytoplasmic Dsg2 staining was absent in ADAM17 knockdown cells.
DISCUSSION
Although recent studies have identified classic cadherin trafficking as a potential mechanism for regulating tumor cell behavior (Bryant and Stow, 2004; D'Souza-Schorey, 2005; Yap et al., 2007), the possibility that altered trafficking of desmosomal cadherins contribute to loss of tumor cell adhesion had not been addressed previously. Here we show that the desmosomal cadherin, Dsg2, is constitutively internalized in SCC tumor cells. Our results suggest furthermore that ErbB1/2 or MMP inhibition results in an accumulation of junctional Dsg2 and depletion of the internalized pool, and this redistribution is accompanied by increased intercellular adhesion. The importance of such a posttranslational mechanism in regulating Dsg2 expression is supported by 35S-pulse labeling experiments showing that neither TAPI nor PKI alter Dsg2 protein synthesis (J. Chen, unpublished results). Our data indicate furthermore that Dsg2 trafficking is more sensitive to MMP inhibitors than E-cadherin trafficking and that Dsg2 is required for increased adhesive strength observed in MMP-inhibited epithelial sheets in SCC68 cells.
We have previously demonstrated that Dsg2 shedding occurs in an MMP-dependent manner (Lorch et al., 2004). Subsequently, ADAM10 was shown to shed N- and E-cadherin from the surface and reduce adhesion of neuronal and epithelial cells (Maretzky et al., 2005; Reiss et al., 2005), and Dsg2 was recently identified by a proteomics approach as a substrate for both ADAM10 and 17 (Bech-Serra et al., 2006). However, a possible role for ADAM10 or 17 in endocytic turnover of cadherins had not been previously examined. Our current data support the idea that stabilization of Dsg2 at cell–cell junctions in TAPI-treated and ADAM17-deficient cells occurs through regulation of Dsg2 endocytic trafficking. The inhibition of Dsg2 cleavage observed in ADAM9- and 15-deficient cells raises the possibility that a spectrum of sheddases may regulate Dsg2 trafficking and accumulation in intercellular junctions. ADAM10 knockdown does not prevent Dsg2 cleavage, but results in an increase in the accumulation of a Dsg2 100-kDa fragment, suggesting it may be involved in further processing of this fragment. The increase in this cell bound fragment correlates with an increase in internalized Dsg2, supporting the idea that its presence may contribute to regulation of Dsg2 trafficking (Figure 9). Although it is clear that ADAM17 silencing effectively blocks both Dsg2 processing and accumulation of internalized Dsg2, it is not known whether Dsg2 cleavage is solely responsible for ADAM17's role in Dsg2 trafficking. Although the complexity of previously characterized ADAM cleavage sites has made developing cleavage resistant mutant substrates challenging, in the future, such an approach might be possible once the precise site of cleavage is mapped (Hattori et al., 2000).
Figure 9.
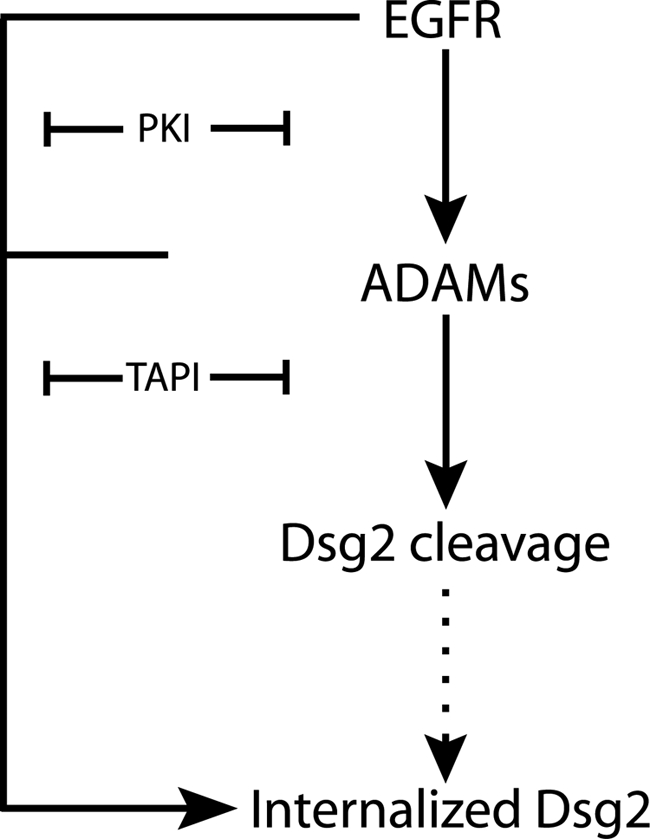
EGFR and MMPs cooperate to regulate the cleavage and endocytic trafficking of Dsg2. EGFR acts upstream of MMPs to promote cleavage and internalization of Dsg2. EGFR-mediated tyrosine phosphorylation of the Dsg2 cytoplasmic domain and adaptor proteins may also facilitate internalization.
Our data are consistent with a model in which EGFR and ADAMs cooperate to promote ectodomain shedding, internalization and, ultimately, degradation of Dsg2 (Figure 9). Supporting the existence of an interdependent pathway is the observed decrease in ADAM-dependent Dsg2 fragment production in PKI-treated cells. Our data suggest that this effect is due to recruitment of Dsg2 to intercellular borders where it is protected from further degradation, despite a concomitant increase in ADAM17 protease under these conditions. Because EGFR is still active in TAPI- and ADAM17 siRNA-treated cultures, it seems unlikely that the reverse is true, i.e., ADAM17 regulates Dsg2 internalization by generating erbB family ligands.
Importantly, PKI interferes with Dsg2 internalization under conditions where ADAM17 is still active, supporting the idea that ADAM-dependent proteolysis is not solely responsible for regulating accumulation of the internalized pool and suggesting that other EGFR-dependent events are involved. It has been shown that HGF-dependent tyrosine phosphorylation of E-cadherin can lead to recruitment of enzymes that ubiquitinate the cadherin complex, which in turn regulates its internalization or degradation (Fujita et al., 2002; Palacios et al., 2005). Likewise, the previously observed tyrosine phosphorylation of Dsg2 and its associated proteins (Lorch et al., 2004) may be required for efficient internalization. One model that is consistent with our data is that an EGFR-dependent event, such as phosphorylation of the desmosomal cadherin complex, regulates internalization, whereas the presence of processed Dsg2 in the desmosomal cadherin complex regulates the commitment to a degradative versus recycling pathway. In this scenario, either PKI or MMP inhibition would be predicted to interfere with accumulation of the internal pool of Dsg2, as shown here. Although posttranslational modification of the Dsg2 tail could theoretically also regulate ectodomain cleavage, removal of most of the ∼500-amino acid Dsg2 tail, including the EGFR-dependent phosphorylation sites, does not prevent ectodomain cleavage, making this possibility seem less likely (Chen and Green, unpublished data).
Internalization of classic cadherins occurs via both clathrin-dependent and -independent pathways (Lu et al., 2003; Paterson et al., 2003; Bryant and Stow, 2004; D'Souza-Schorey, 2005; Bryant et al., 2007). However, little is known about trafficking pathways in which desmosomal cadherins are engaged. Pathogenic antibodies from patients with the cutaneous blistering disease, pemphigus vulgaris, drive the internalization of desmoglein 3 in epidermal keratinocytes (Calkins et al., 2006) accompanied by its depletion from desmosomes (Yamamoto et al., 2007), but the mechanism of internalization and turnover has not been elucidated. Our preliminary data are consistent with the internalization of Dsg2 into a nonconventional pool that codistributes poorly if at all with common early endosomal or lysosomal markers (not shown). The possibility that desmosomes are turned over from the plasma membrane through a nonconventional route has been suggested by early studies in which acute, EGTA-mediated, calcium depletion led to clathrin-independent half desmosome internalization into a compartment that appeared distinct from typical endosomes and lysosomes (Burdett, 1993; Holm et al., 1993). However, mechanisms of internalization stimulated by these acute treatments may differ considerably from those responsible for the EGFR- and MMP-dependent internalization shown here.
In conclusion, our data establish novel pathways for regulating desmosomal cadherin trafficking and desmoglein-dependent adhesion. The possibility that these pathways affect E-cadherin and Dsg2 differently underscores the importance of understanding the specific roles of desmosomal cadherins in epithelial remodeling and cancer, and suggests new strategies for therapeutic intervention based on tissue-specific variation in cadherin, RTK, and MMP status.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. A. Kowalczyk and members of the Kowalczyk and Green labs for helpful discussion and critical reading of the manuscript. Thanks also go to Novartis AG for PKI166 and the University of Iowa Developmental Hybridoma Bank for tubulin antibodies, and W.W. Franke for a Dsg2 cDNA. This work was supported in part by the J. L. Mayberry Endowment and NIH grants R01 CA122151 and R01AR043380 to K.G.; J.K. was supported in part by T32 CA80621, and B.D. was supported in part by the Tiecke-Sowers Fellowship in Pathology.
Abbreviations used:
- ADAM
a disintegrin and metalloprotease
- Dsg2
desmoglein 2
- EGFR
epidermal growth factor receptor
- MMP
matrix metalloprotease
- TAPI
TNF-α protease inhibitor
- SCC
squamous cell carcinoma.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-04-0356) on November 5, 2008.
REFERENCES
- Bech-Serra J. J., Santiago-Josefat B., Esselens C., Saftig P., Baselga J., Arribas J., Canals F. Proteomic identification of desmoglein 2 and activated leukocyte cell adhesion molecule as substrates of ADAM17 and ADAM10 by difference gel electrophoresis. Mol. Cell. Biol. 2006;26:5086–5095. doi: 10.1128/MCB.02380-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J. Cadherins and catenins: role in signal transduction and tumor progression. Cancer Metastasis Rev. 1999;18:15–30. doi: 10.1023/a:1006200102166. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages M., Rojo F., Albanell J., Baselga J., Arribas J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 2003;22:1114–1124. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D. M., Kerr M. C., Hammond L. A., Joseph S. R., Mostov K. E., Teasdale R. D., Stow J. L. EGF induces macropinocytosis and SNX1-modulated recycling of E-cadherin. J. Cell Sci. 2007;120:1818–1828. doi: 10.1242/jcs.000653. [DOI] [PubMed] [Google Scholar]
- Bryant D. M., Stow J. L. The ins and outs of E-cadherin trafficking. Trends Cell Biol. 2004;14:427–434. doi: 10.1016/j.tcb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Burdett I.D.J. Internalisation of desmosomes and their entry into the endocytic pathway via late endosomes in MDCK cells. J. Cell Sci. 1993;106:1115–1130. doi: 10.1242/jcs.106.4.1115. [DOI] [PubMed] [Google Scholar]
- Calkins C. C., Setzer S. V., Jennings J. M., Summers S., Tsunoda K., Amagai M., Kowalczyk A. P. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J. Biol. Chem. 2006;281:7623–7634. doi: 10.1074/jbc.M512447200. [DOI] [PubMed] [Google Scholar]
- Chidgey M., Dawson C. Desmosomes: a role in cancer? Br. J. Cancer. 2007;96:1783–1787. doi: 10.1038/sj.bjc.6603808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M., Zhurinsky J., Ben-Ze'ev A. The cadherin-catenin adhesion system in signaling and cancer. J. Clin. Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Azzo A., Bongiovanni A., Nastasi T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic. 2005;6:429–441. doi: 10.1111/j.1600-0854.2005.00294.x. [DOI] [PubMed] [Google Scholar]
- D'Souza-Schorey C. Disassembling adherens junctions: breaking up is hard to do. Trends Cell Biol. 2005;15:19–26. doi: 10.1016/j.tcb.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Denning M. F., Wang Y., Tibudan S., Alkan S., Nickoloff B. J., Qin J. Z. Caspase activation and disruption of mitochondrial membrane potential during UV radiation-induced apoptosis of human keratinocytes requires activation of protein kinase C. Cell Death Differ. 2002;9:40–52. doi: 10.1038/sj.cdd.4400929. [DOI] [PubMed] [Google Scholar]
- Dusek R. L., Godsel L. M., Green K. J. Discriminating roles of desmosomal cadherins: beyond desmosomal adhesion. J. Dermatol. Sci. 2007;45:7–21. doi: 10.1016/j.jdermsci.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Frixen U. H., Behrens J., Sachs M., Eberle G., Voss B., Warda A., Lochner D., Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 1991;113:173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y., Krause G., Scheffner M., Zechner D., Leddy H. E., Behrens J., Sommer T., Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- Getsios S., Amargo E. V., Dusek R. L., Ishii K., Sheu L., Godsel L. M., Green K. J. Coordinated expression of desmoglein 1 and desmocollin 1 regulates intercellular adhesion. Differentiation. 2004;72:419–433. doi: 10.1111/j.1432-0436.2004.07208008.x. [DOI] [PubMed] [Google Scholar]
- Godsel L. M., Getsios S., Huen A. C., Green K. J. The molecular composition and function of desmosomes. In: Starke K., editor. Handbook of Experimental Pharmacology. Vol. 165. Berlin: Springer-Verlag; 2004. pp. 137–193. [DOI] [PubMed] [Google Scholar]
- Green K. J., Simpson C. L. Desmosomes: new perspectives on a classic. J. Invest. Dermatol. 2007;127:2499–2515. doi: 10.1038/sj.jid.5701015. [DOI] [PubMed] [Google Scholar]
- Hattori M., Osterfield M., Flanagan J. G. Regulated cleavage of a contact-mediated axon repellent. Science. 2000;289:1360–1365. doi: 10.1126/science.289.5483.1360. [DOI] [PubMed] [Google Scholar]
- Hirao T., Nanba D., Tanaka M., Ishiguro H., Kinugasa Y., Doki Y., Yano M., Matsuura N., Monden M., Higashiyama S. Overexpression of ADAM9 enhances growth factor-mediated recycling of E-cadherin in human colon cancer cell line HT29 cells. Exp. Cell Res. 2006;312:331–339. doi: 10.1016/j.yexcr.2005.10.032. [DOI] [PubMed] [Google Scholar]
- Holm P. K., Hansen S. H., Sandvig K., van Deurs B. Endocytosis of desmosomal plaques depends on intact actin filaments and leads to a nondegradative compartment. Eur. J. Cell Biol. 1993;62:362–371. [PubMed] [Google Scholar]
- Hudson T., Fontao L., Godsel L., H.-Choi J., Huen A., Borradori L., Weis W., Green K. In vitro methods for investigating desmoplakin-intermediate filament interactions and their role in adhesive strength. In: Omary B., Coulombe P., editors. Methods in Cell Biology. Vol. 78. New York: Elsevier; 2004. pp. 757–786. [DOI] [PubMed] [Google Scholar]
- Islam S., Carey T. E., Wolf G. T., Wheelock M. J., Johnson K. R. Expression of N-cadherin by human squamous carcinoma cells induces a scattered fibroblastic phenotype with disrupted cell-cell adhesion. J. Cell Biol. 1996;135:1643–1654. doi: 10.1083/jcb.135.6.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyankrishna S., Grandis J. R. Epidermal growth factor receptor biology in head and neck cancer. J. Clin. Oncol. 2006;24:2666–2672. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- Kowalczyk A. P., Bornslaeger E. A., Borgwardt J. E., Palka H. L., Dhaliwal A. S., Corcoran C. M., Denning M. F., Green K. J. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J. Cell Biol. 1997;139:773–784. doi: 10.1083/jcb.139.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer R. H., Shen X., Zhou H. Tumor cell invasion and survival in head and neck cancer. Cancer Metastasis Rev. 2005;24:35–45. doi: 10.1007/s10555-005-5046-2. [DOI] [PubMed] [Google Scholar]
- Le T. L., Yap A. S., Stow J. L. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J. Cell Biol. 1999;146:219–232. [PMC free article] [PubMed] [Google Scholar]
- Llorens A., Rodrigo I., Lopez-Barcons L., Gonzalez-Garrigues M., Lozano E., Vinyals A., Quintanilla M., Cano A., Fabra A. Down-regulation of E-cadherin in mouse skin carcinoma cells enhances a migratory and invasive phenotype linked to matrix metalloproteinase-9 gelatinase expression. Lab. Invest. 1998;78:1131–1142. [PubMed] [Google Scholar]
- Lochter A., Galosy S., Muschler J., Freedman N., Werb Z., Bissell M. J. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular interactions that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 1997;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorch J. H., Klessner J., Park J. K., Getsios S., Wu Y. L., Stack M. S., Green K. J. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J. Biol. Chem. 2004;279:37191–37200. doi: 10.1074/jbc.M405123200. [DOI] [PubMed] [Google Scholar]
- Lu Z., Ghosh S., Wang Z., Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4:499–515. doi: 10.1016/s1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
- Maretzky T., Reiss K., Ludwig A., Buchholz J., Scholz F., Proksch E., de Strooper B., Hartmann D., Saftig P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. USA. 2005;102:9182–9187. doi: 10.1073/pnas.0500918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCawley L. J., O'Brien P., Hudson L. G. Overexpression of the epidermal growth factor receptor contributes to enhanced ligand-mediated motility in keratinocyte cell lines. Endocrinology. 1997;138:121–127. doi: 10.1210/endo.138.1.4844. [DOI] [PubMed] [Google Scholar]
- McLachlan R. W., Yap A. S. Not so simple: the complexity of phosphotyrosine signaling at cadherin adhesive contacts. J. Mol. Med. 2007;85:545–554. doi: 10.1007/s00109-007-0198-x. [DOI] [PubMed] [Google Scholar]
- Muller E. J., Williamson L., Kolly C., Suter M. M. Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J. Invest. Dermatol. 2008;128:501–516. doi: 10.1038/sj.jid.5701248. [DOI] [PubMed] [Google Scholar]
- Najy A. J., Day K. C., Day M. L. The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J. Biol. Chem. 2008;283:18393–18401. doi: 10.1074/jbc.M801329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson W. J., Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe V., Fingleton B., Jacobs K., Crawford H. C., Vermeulen S., Steelant W., Bruyneel E., Matrisian L. M., Mareel M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 2001;114:111–118. doi: 10.1242/jcs.114.1.111. [DOI] [PubMed] [Google Scholar]
- Palacios F., Tushir J. S., Fujita Y., D'Souza-Schorey C. Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol. Cell Biol. 2005;25:389–402. doi: 10.1128/MCB.25.1.389-402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson A. D., Parton R. G., Ferguson C., Stow J. L., Yap A. S. Characterization of E-cadherin endocytosis in isolated MCF-7 and chinese hamster ovary cells: the initial fate of unbound E-cadherin. J. Biol. Chem. 2003;278:21050–21057. doi: 10.1074/jbc.M300082200. [DOI] [PubMed] [Google Scholar]
- Reiss K., Maretzky T., Ludwig A., Tousseyn T., de Strooper B., Hartmann D., Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005;24:742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks N., Paulissen G., El Hour M., Quesada F., Crahay C., Gueders M., Foidart J. M., Noel A., Cataldo D. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie. 2008;90:369–379. doi: 10.1016/j.biochi.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Rogers S. J., Harrington K. J., Rhys-Evans P., O-Charoenrat P., Eccles S. A. Biological significance of c-erbB family oncogenes in head and neck cancer. Cancer Metastasis Rev. 2005;24:47–69. doi: 10.1007/s10555-005-5047-1. [DOI] [PubMed] [Google Scholar]
- Runswick S. K., O'Hare M. J., Jones L., Streuli C. H., Garrod D. R. Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat. Cell Biol. 2001;3:823–830. doi: 10.1038/ncb0901-823. [DOI] [PubMed] [Google Scholar]
- Santiago-Josefat B., Esselens C., Bech-Serra J. J., Arribas J. Post-transcriptional up-regulation of ADAM17 upon epidermal growth factor receptor activation and in breast tumors. J. Biol. Chem. 2007;282:8325–8331. doi: 10.1074/jbc.M608826200. [DOI] [PubMed] [Google Scholar]
- Schulz B., Pruessmeyer J., Maretzky T., Ludwig A., Blobel C. P., Saftig P., Reiss K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ. Res. 2008;102:1192–1201. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield J. B., Graff D., Li H. P. A solid-phase method for the quantitation of protein in the presence of sodium dodecyl sulfate and other interfering substances. Anal. Biochem. 1987;166:49–54. doi: 10.1016/0003-2697(87)90544-6. [DOI] [PubMed] [Google Scholar]
- Thiery J. P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Wheelock M. J., Johnson K. R. Cadherins as modulators of cellular phenotype. Annu. Rev. Cell Dev. Biol. 2003;19:207–235. doi: 10.1146/annurev.cellbio.19.011102.111135. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Aoyama Y., Shu E., Tsunoda K., Amagai M., Kitajima Y. Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J. Biol. Chem. 2007;282:17866–17876. doi: 10.1074/jbc.M607963200. [DOI] [PubMed] [Google Scholar]
- Yap A. S., Crampton M. S., Hardin J. Making and breaking contacts: the cellular biology of cadherin regulation. Curr. Opin. Cell Biol. 2007;19:508–514. doi: 10.1016/j.ceb.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.