

Abstract
Timely degradation of regulatory proteins by the ubiquitin proteolytic pathway (UPP) is an established paradigm of cell cycle regulation during the G2/M and G1/S transitions. Less is known about roles for the UPP during S phase. Here we present evidence that dynamic cell cycle–dependent changes in levels of UbcH7 regulate entrance into and progression through S phase. In diverse cell lines, UbcH7 protein levels are dramatically reduced in S phase but are fully restored by G2. Knockdown of UbcH7 increases the proportion of cells in S phase and doubles the time to traverse S phase, whereas UbcH7 overexpression reduces the proportion of cells in S phase. These data suggest a role for UbcH7 targets in the completion of S phase and entry into G2. Notably, UbcH7 knockdown was coincident with elevated levels of the checkpoint kinase Chk1 but not Chk2. These results argue that UbcH7 promotes S phase progression to G2 by modulating the intra-S phase checkpoint mediated by Chk1. Furthermore, UbcH7 levels appear to be regulated by a UPP. Together the data identify novel roles for the UPP, specifically UbcH7 in the regulation of S phase transit time as well as in cell proliferation.
INTRODUCTION
The eukaryotic cell cycle is typically divided into four major phases: G1, S, G2, and M. Acquiring an understanding of how transitions between phases of the cell cycle are regulated is crucial for understanding cell proliferation and organogenesis and is essential to enable discovery and design of drugs to control these processes. Transitions between the phases of the cell cycle have often been associated with the timed destruction of cell cycle regulatory proteins. Many of these are degraded via the ubiquitin proteolytic pathway (UPP; Yew, 2001; Peters, 2002; Pines and Lindon, 2005). Protein ubiquitination is the posttranslational covalent attachment of one or more molecules of ubiquitin, a highly conserved 8.5-kDa polypeptide, to substrate proteins. This is accomplished through the sequential activities of three groups of enzymes. First ubiquitin is activated via formation of a thiol ester with the ubiquitin-activating enzyme or E1. Ubiquitin is then transferred to an E2/Ubc (ubiquitin-conjugating enzyme), also through a thiol ester linkage. Transfer of ubiquitin to the target protein is accomplished in conjunction with an E3 enzyme (ubiquitin ligase). The variety and combinations of E2 and E3 enzymes allow for substrate specificity. A major role of ubiquitination is to target proteins for proteasomal degradation (reviewed in Hershko and Ciechanover, 1998; Pickart and Fushman, 2004).
Most research regarding cell cycle regulation by the UPP has focused on the G1/S and G2/M transitions and on the specific roles in these transitions of two types of multisubunit E3 ligases: 1) Skp1, Cullin, F-box complexes (SCF) that ubiquitinate the G1 phase CDK inhibitors, p21 (Yu et al., 1998; Bornstein et al., 2003) and p27 (Carrano et al., 1999; Tsvetkov et al., 1999), thereby regulating the G1-to-S transition, and 2) the anaphase promoting complex/cyclosome (APC/C), which mediates degradation of cyclin A, cyclin B, and securin, thereby allowing progression through M phase (reviewed in Peters, 2002). Far less is known concerning UPP components and protein substrates that regulate the progression through S phase. Additionally, relatively few studies have addressed functions and regulation of E2/Ubcs during the cell cycle, other than describing roles for Cdc34/Ubc3, together with the SCF, in regulating the G1-to-S transition, and UbcH10, in conjunction with the APC, regulating the G2/M transition and progress through G1 (Bastians et al., 1999; Rape and Kirschner, 2004; Rape et al., 2006).
A single report indicates that the E2, UbcH7, is essential for embryonic development (Harbers et al., 1996), but essential postnatal functions have not been elucidated. Here, using HeLa cells and human lens epithelial cells, we demonstrate that levels of UbcH7 protein decrease dramatically in S phase but are fully restored by G2. Moreover, in multiple cell types, the duration of S phase is inversely related to the level of UbcH7. Importantly, levels of the intra-S phase checkpoint kinase Chk1 are increased in UbcH7-depleted cells, whereas the level of phosphorylated phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is decreased. Additionally, we provide evidence that UbcH7 gene expression is not altered during cell cycle progression, but UbcH7 is itself a substrate for UPP-dependent degradation. Considered together, these data support a novel role for UbcH7 in the regulation of S phase and progression to G2, potentially through associated degradation of Chk1.
MATERIALS AND METHODS
Cells
Human lens epithelial (HLE) cells were obtained and grown as described (Liu et al., 2004). HeLa cells were obtained from American Type Culture Collection (Manassas, VA) and maintained in αMEM or DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 50 U/ml penicillin, and 50 μg/ml streptomycin. COS and HEK-293 cells were obtained from ATCC and maintained in DMEM supplemented as above. For synchronization with hydroxyurea (HU; Sigma, St. Louis, MO) HeLa and HLE cells were grown in medium containing 2 mM HU for 18 h. After treatment, cells were washed twice with PBS, and medium without drugs was added. At time points indicated, cells were washed twice with PBS, treated with 0.5% trypsin-EDTA to disrupt the monolayer, and then collected by centrifugation. For Western blot analysis, cells were lysed in 10 mM Tris-HCL (pH 7.6), 50 mM EDTA, 1% NP-40, 0.1% SDS, 20 mM N-ethylmaleimide, and 2 mM 4-(2-aminoethyl)-benzene-sulfonylfluoride. For extract preparation, cells were lysed in 10 mM HEPES, 10 mM KCl, 1 mM 4-(2-aminoethyl)-benzene-sulfonylfluoride, and 1 mM DTT. After lysis for >30 min on ice, samples were centrifuged, and soluble supernatants were saved. For experiments analyzing phosphoproteins, a phosphatase inhibitor cocktail (Sigma) was added to the lysis buffer and to the PBS used for washing the cells before lysis. For fluorescence-activated cell sorting (FACS) analysis of DNA content, trypsinized cells were resuspended in 70% ethanol and stored at −20°C. Cells were rehydrated in PBS containing 100 ng/ml RNAse A for 15 min, centrifuged, and resuspended in PBS containing 50 μg/ml propidium iodide. DNA content was analyzed with a FACScalibur Flow cytometry system (Becton-Dickinson Immunocytometer Systems, San Jose, CA) and ModFit LT 3.0 software (Verity Software house, Topsham, ME).
Antibodies
Anti-E1 was produced in this laboratory (Shang et al., 2001); anti-UbcH7 polyclonal, anti-UbcH10, and anti-Ubc2 were obtained from Boston Biochem (Boston, MA). Monoclonal anti-UbcH7 and a nonspecific isotype matched control IgG were obtained from BD Biosciences (San Jose, CA). Anti-Ubc3 and anti-Chk1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-PTEN, anti-Akt, anti-Phospho Ser 380 PTEN, anti-Phospho Ser 380/Thr382/383 PTEN, and anti-Phospho Ser 280 Chk1 were obtained from Cell Signaling Technologies (Danvers MA). Anti-E6-AP was obtained from Affinity Bioreagents (Golden, CO). Anti-tubulin was obtained from Sigma. Secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA).
Quantitative RT-PCR
HLE cells were synchronized by contact inhibition as described (Liu et al., 2004). Cells were replated at lower density and allowed to reenter the cell cycle. At time points indicated, cells were harvested and parallel samples were processed for FACS analysis and mRNA isolation. Quantitative RT-PCR was performed and UbcH7 mRNA levels were normalized to GAPDH levels.
Plasmid Construction
A UbcH7 expression plasmid was constructed from a UbcH7-pET3b vector kindly provided by Martin Scheffner (University of Cologne, Germany). The coding region was amplified by PCR using the primers forward: 5′ GAATTCTACATATGGCGGCCAGCAGG 3′ and reverse: 5′ CCTTTCGGGCTTTGTTAGCAG 3′. The forward primer was engineered to contain an EcoRI restriction site to facilitate cloning. The resulting product was digested with EcoRI and BamHI and subcloned into pADTrack for expression in mammalian cells.
RNA Interference
Double-stranded small interfering RNAs (siRNAs) specific for UbcH7 were obtained from Qiagen (Valencia, CA). The following sequences were used: oligo “3,” AAGGGCTTATTGTTCCTGACA; oligo “4,” AATCCGCAAATGTCCCATGAA; oligo “5,” AATTCAGAGCCAGCAATGCCT (Verma et al., 2004), and nonspecific (NS) siRNA AATTCTCCGACGTGTCACGT. For most experiments, cells were seeded at 1 × 105 per well in six-well culture plates on day −1. On day 0, RNA interference (RNAi) and RNAifect were added. For immunofluorescence experiments, cells were seeded at 1 × 104 per chamber in a four-well chamber slide.
MTS Assay
HeLa cells were plated into 96 well plates at a density of 2 × 103 cells per well at day –1. On day 0, siRNA specific for UbcH7 or NS siRNA were added. After 72 or 96 h of knockdown, an MTS assay was performed according to manufacturer's directions (Promega, Madison, WI).
Immunofluorescence
Cells were grown on four-well chamber slides (BD Biosciences). Before staining, monolayers were washed with PBS and treated for 10 min with PBS containing 4% paraformaldehyde. Subsequently, the monolayers were washed and either stored at 4°C in PBS or immediately processed for immunofluorescence. For staining, slides were treated with PBS containing 0.1% Triton X-100 (TX-100) for 3 min and then blocked with PBS containing TX-100 and 0.3% bovine serum albumin (BSA) for 30 min. Primary and secondary antibodies were diluted in PBS containing TX-100 and BSA. Staining with primary monoclonal antibodies was for >2h, and the slides were then washed with PBS and incubated with the secondary anti-mouse antibody for 30 min−1h. Slides were again washed with PBS and treated with Hoescht dye for 5–10 min, rinsed, and briefly dried before mounting with Anti-fade (Molecular Probes, Eugene, OR).
In Vivo Degradation
HeLa cells were treated with 50 μg/ml cycloheximide in the presence or absence of 40 μM MG132 for the times indicated. An untreated sample was used to compare the amount of UbcH7 remaining after treatment. Samples were harvested by scraping and lysed as described above.
In Vitro Degradation
Degradation of E2s was determined using rabbit reticulocyte lysate (Green Hectares, Oregon, WI) or lysates prepared from HeLa cell cultures isolated at various stages of the cell cycle using drug synchronization. Recombinant proteins were radiolabeled with 125I-Na using the chloramine T method (Tashtoush et al., 2001) and purified by G25 Sephadex column chromatography. For ATP-dependent degradation, purified 125I-E2s were incubated at 37°C for 2 h in a final reaction volume of 25 μl containing 50 mM Tris-HCl, pH 7.6, 6 mM MgCl2, 1.2 mM DTT, 2.7 mM ATP, 17 mM creatine phosphate, and 4.7 U creatine phosphokinase in the presence or absence of cell lysate, Ubc4, or MG132. ATP-independent degradation of UbcH7 was determined as above but in buffer not containing ATP, creatine phosphate, or creatine phosphokinase. Degradation was terminated and TCA-soluble cpm were quantified using a Cobra II gamma counter. Percent degradation was calculated as [(experimental soluble cpm) − (buffer control soluble cpm)/(total cpm)] × 100.
In Vitro Ubiquitination
Ubiquitin conjugation assays of UbcH7 were performed using rabbit reticulocyte lysate. Purified 125I-UbcH7 was incubated at 37°C for 1 h in a final reaction volume of 30 μl containing 40 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 40 μM MG132, 2 μM ubiquitin aldehyde, 5 mM ATP, 75 μM ubiquitin, and 2 mM DTT in the presence or absence of cell lysate, ubiquitin, and Ubc4. After incubation, Laemmli buffer was added to the assay sample and boiled for 5 min, and the proteins were resolved by 12% SDS-PAGE. Ubiquitin-UbcH7 conjugates were visualized by autoradiography.
RESULTS
UbcH7 Is Regulated in a Cell Cycle–dependent Manner
To further investigate prior observations that UbcH7 levels increased as HLE cells progressed from S phase into G2/M (Liu et al., 2004), HeLa and HLE cells were synchronized by treatment with HU. After 18 h treatment with HU, the DNA profile suggests that the cells are at the G1/S boundary (Figure 1A, top). Within 4 h after release from HU, 100% of the cells are in S phase (middle), and by 8 h the vast majority are in G2/M (bottom). Notably, when the cells are 100% in S phase both HeLa cells (Figure 1B, left) and HLE cells (right) show a striking decrease (19-fold decrease in this experiment for HeLa cells and 2.4-fold decrease for HLE cells) in the amount of UbcH7. When the cells progress to G2/M, the level of UbcH7 recovers. UbcH7 levels remain high as the cells enter the next G1 phase (data not shown). It is highly unlikely that the change in UbcH7 levels is due to drug treatment, because cells synchronized by contact inhibition also show a decrease in UbcH7 as they enter S phase and a recovery of UbcH7 as they complete S phase and enter G2 (Liu et al., 2004). We also visualized the elevation of UbcH7 in mitotic cells by immunofluorescence. It is clear that the rounded up mitotic cells are far more fluorescent than the cells in interphase (Figure 1C). Similar changes in fluorescence were not observed with tubulin.
Figure 1.

UbcH7 is regulated in a cell cycle–dependent manner. (A) Cell cycle profile of cells synchronized by treatment with 2 mM HU. HeLa cells were treated for 18 h, HU-containing medium was washed out, and cells were allowed to resume cycling in medium without drugs. Mean fluorescence intensity relating to DNA content is plotted on the X axis, and cell number is plotted on the Y axis. (B) UbcH7 levels are low in S phase and rise in G2/M. HeLa (left panel) or HLE (right panel) cells were treated with HU for 18 h and then released from drug treatment to resume cycle. Lysates, prepared from samples at times after drug release, were blotted using α-UbcH7, α-UbcH10, α-Ubc3, α-Ubc2, and α-E1, the latter being used as a loading control. We and others have demonstrated no alterations in E1 levels during different phases of the cell cycle (Stephen et al., 1996; Liu et al., 2004). Because E2 levels from the same samples were assessed on different immunoblots, E1 controls are shown for each blot. The majority of cells (≥75%) were in the cell cycle phase indicated. Bottom, quantitation of gels shown above. (C) UbcH7 is high in mitotic cells. Immunofluorescent staining with α-UbcH7 (left), α-tubulin (middle), or isotype matched control antibody (right) on cells treated for 18 h with nocodazole. Mitotic cells are noted with a white arrow in all panels.
That this cell cycle related variation is specific to UbcH7 is indicated by examination of other mammalian E2 enzymes throughout the cell cycle. Levels of Ubc2 are invariant throughout these transitions, consistent with prior experiments, which showed that expression of a dominant negative mutant version of Ubc2 did not alter the cell cycle in the lens (Liu et al., 2006). Similarly, Ubc3 and UbcH10, the only E2s that are known to be involved in cell cycle regulation, do not show the cell cycle–dependent variation that is observed with UbcH7 (Figure 1B). Thus the decrease in S phase appears to be selective for UbcH7. The consistent levels of these E2s provides additional evidence that the drugs used for synchronization did not influence these other E2 levels as the cells progressed through the cell cycle.
Knockdown of UbcH7 Extends S phase
Given the relationship between UbcH7 levels and the cell cycle, it might be anticipated that there are relationships between levels of UbcH7, cell proliferation, the duration of S phase, and the cell cycle. To examine this possibility, UbcH7 was depleted in HeLa cells using siRNA. Decreasing UbcH7 (Figure 2A, top) in asynchronously growing cells resulted in an increase in the percentage of cells in S phase (Figure 2A, bottom). Furthermore, increasing the length of time during which cells were depleted of UbcH7 significantly increased the relative proportion of S phase cells when compared with UbcH7-replete cells (Figure 2B and Supplemental Figure S1, A—C). The increase in the S phase population upon UbcH7 knockdown was observed with three different siRNA sequences specific for UbcH7 (Supplemental Figure S1D), indicating that this result is not due to nonspecific or off-target effects of the siRNA. UbcH7 depletion in other cell types also increases the percentage of cells in S phase. In both HEK-293 cells and HLE cells, UbcH7 depletion resulted in a increase in S phase cells and a decrease in G1 cells (Figure 2C and Supplemental Figure S1E), similar to our observations in HeLa cells. A 30% increase in the percentage of cells in S phase was observed for HEK-293 cells and a 25% increase for HLE cells after 72 h of knockdown. These data indicate that UbcH7 is likely modifying a similar process in diverse cell types to control the duration of S phase of the cell cycle.
Figure 2.

Levels of UbcH7 levels determine the duration of S phase of the cell cycle. (A–C) Asynchronous HeLa cells were treated with siRNA specific for UbcH7 or a nonspecific siRNA for 48, 72, or 96 h as indicated. (A) Cell lysates were blotted for α-UbcH7 or α-E1 as a loading control (top panels). Cells were analyzed for DNA content (bottom panels). (B) Average ratio of S phase during increased time of knockdown. The percentage of cells in S phase in UbcH7-depleted samples was compared with the percentage in NS siRNA-treated cells. Average of three to seven experiments; *p < 0.01. (C) Depletion of UbcH7 results in an increase in S phase in HEK-293 and HLE cells. Cells were treated with siRNA for 72 h. (D) COS cells were transfected with UbcH7 to increase levels of the enzyme or empty plasmid for 48 h. The cell cycle profile was determined by FACS analysis, and the ratio of G1 or S phase cells from cells expressing UbcH7 compared with the empty vector was averaged from three independent experiments. Right panel, Western blot of lysates from cells expressing UbcH7 or empty plasmid.
Because depleting UbcH7 levels in cells results in an increased percentage of cells in S phase, we anticipated that increasing levels of UbcH7 might have the opposite effect. Cells were transiently transfected with a plasmid containing UbcH7 or an empty vector and the cell cycle profile was analyzed 48 h later. As shown in Figure 2D, COS cells expressing increased UbcH7 (see Western blot analysis, right) showed a decrease in the percentage of cells in S phase, with an increase in G1 phase. Consistent alterations in the percent of cells in G2/M upon UbcH7 overexpression were not observed. These data complement the UbcH7 depletion assays and indicate that UbcH7 is involved in the regulation of cell cycle progress through S phase.
To get a clearer idea if the proportionate increase of cells in S phase associated with limiting UbcH7 is due to an arrest or a delay in progression through S phase, we used synchronized siRNA-treated cells. Effective knockdown of UbcH7 was achieved in these HU-synchronized HeLa cells (Figure 3A). FACS analysis of the DNA content of cells treated with either the UbcH7 (Figure 3B, top) or NS (bottom) siRNA indicates that the cells are at the G1/S boundary immediately after release from HU, and that they progress synchronously to S phase 4 h after release from drug treatment. Eight hours after release from HU, NS siRNA-treated cells progressed to G2/M (Figure 3B, bottom, hatched bar). In contrast, the UbcH7-depleted cells were still in S phase (Figure 3B, top, black bar). Only 12 h after drug release did the UbcH7-depleted cells progress to G2/M. The cells in which UbcH7 was depleted remained delayed compared with NS siRNA-treated cells (see 18- and 24-h time points). UbcH7-depleted cells did eventually complete mitosis and enter the next cell cycle. Thus UbcH7 knockdown did not result in arrest of the cell cycle. Similar delays in progression from S-to-G2 phase were observed when other siRNA specific for UbcH7 were used (data not shown), corroborating that the delay in cell cycle progression is due to the knockdown of UbcH7.
Figure 3.
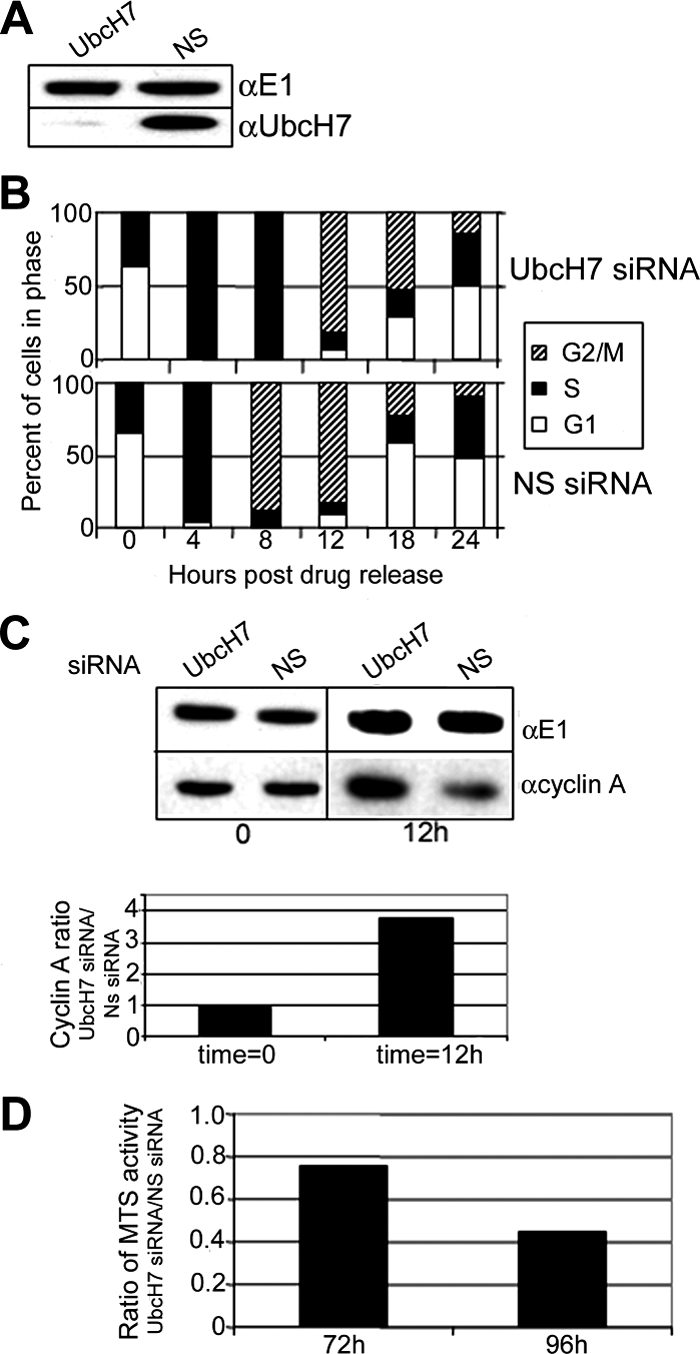
UbcH7 knockdown delays progression from S to G2 phase. HeLa cells were treated for 48 h with siRNA as indicated. Cells were then treated overnight with 2 mM HU for synchronization. (A) Immunoblot showing knockdown of UbcH7. (B) Cell cycle profile of cells at times after drug release. Cell cycle numbers are from duplicate samples. Similar delays in S phase progression to G2 were also observed with another UbcH7 siRNA sequence. (C) Top, cyclin A and E1 levels from samples treated with NS siRNA or UbcH7-specific siRNA immediately after drug release or 12 h after release. Bottom panel, quantitation of cyclin A levels normalized to E1 levels. Normalized cyclin A levels were compared between UbcH7 siRNA-treated cells and NS siRNA-treated cells at each time point. (D) Knockdown of UbcH7 decreases cell proliferation. Comparison of MTS OD between NS siRNA and UbcH7 siRNA at 72 and 96 h of knockdown.
Although the cell cycle profiles of the UbcH7 siRNA and the NS siRNA-treated samples at the 12-h time point are not differentiated using FACS analysis of DNA content, it appeared likely that the UbcH7 siRNA-treated cells have a higher proportion of cells in G2 rather than M phase compared with the UbcH7-replete cells. This idea is supported by observations that although at t = 0 when the cells appear to be at the G1/S boundary, the UbcH7-depleted and -replete cells have similar amounts of cyclin A (time 0; Figure 3C), 12 h later, UbcH7-depleted cells have nearly fourfold higher levels of cyclin A than do the cells replete with UbcH7. If cells are mostly in G2, there would be a higher level of cyclin A than if the cells were in M phase because cyclin A is degraded in early M phase. Thus, the increased cyclin A at the 12-h time point confirms that the UbcH7-depleted cells have a higher fraction of cells in G2 than do the UbcH7-replete cells.
A ramification of slower transit through S phase is decreased cell proliferation within a specific time frame. Consistent with an inverse relationship between UbcH7 levels and the length of S phase, MTS activity, which reflects cell number and viability (Cory et al., 1991), was decreased after 72 and 96 h of UbcH7 knockdown (Figure 3D). The decrease in cell proliferation was more profound after 96 h of knockdown than at 72 h. A decrease in absolute cell number was also observed with no differences in the amount of cell death (data not shown), indicating that the diminished proliferation reflects an increased transit time through cycle.
It is probable that these effects of limiting UbcH7 are minimal estimates because complete knockdown was not achieved. Attempts to completely deplete UbcH7 protein using two different siRNAs together or by adding siRNA on 2 subsequent days did not appreciably decrease the level of UbcH7 (data not shown).
UbcH7 Is Degraded by the Ubiquitin Proteasome Pathway
Because the protein levels of UbcH7 vary significantly and rapidly in a cell cycle–dependent manner, we asked whether the mRNA levels of UbcH7 change as cells progress through cycle. Quantitative RT-PCR analysis indicated no significant differences in mRNA levels as cells progress in cycle from G0 through S and G2/M phases (Supplemental Figure S2A). Therefore, we hypothesized that UbcH7 protein levels might be controlled by a proteolytic process. To determine whether UbcH7 protein levels were controlled by a proteasome-dependent proteolytic process, asynchronously growing HeLa cells were treated with cycloheximide to inhibit de novo protein synthesis in the presence or absence of MG132, a proteasome inhibitor. After 4 h nearly 60% of the UbcH7 protein disappeared in the presence of cycloheximide, whereas MG132 was able to stabilize the level of UbcH7 (Figure 4A). The half-life of UbcH7 was increased from 1 to more than 2 h by the addition of MG132 (Supplemental Figure S2B). These experiments demonstrate that UbcH7 protein levels are regulated by a proteasome-dependent proteolytic pathway.
Figure 4.

UbcH7 is a substrate of the ubiquitin proteasome pathway: (A) HeLa cells were treated for 4 h with cycloheximide in the presence or absence of MG132 as indicated (top). Levels of UbcH7 were normalized to E1 (bottom). (B) Ubiquitination reactions were performed with 125I-UbcH7, reticulocyte lysate, Ubc4, and ubiquitin as indicated. Bands for monoubiquitinated (bottom panel, short exposure) and polyubiquitinated (top panel, long exposure) UbcH7 are noted. A nonspecific band is indicated by an asterisk. As noted at the top of the gel in lane 2, some low background level of UbcH7 oligomerization or ubiquitination is occurring in the presence of lysate and Ubc4. (C) 125I-UbcH7 was added to reticulocyte lysates containing ATP, Ubc4, and MG132 as indicated. Degradation was calculated by comparing the TCA-soluble counts to total radioactivity after 2 h. (D) HeLa cells synchronized using HU as in Figure 1A were lysed, and the resulting extracts were used for degradation of 125I-UbcH7 in the presence of Ubc4, ATP, and ubiquitin. The data shown are the average of five experiments with independent extracts. *p < 0.05 S versus G2/M.
We further hypothesized that UbcH7 protein levels might be regulated in a ubiquitin-dependent manner. Conjugation to ubiquitin, particularly high-mass conjugates, provides strong evidence that a substrate is degraded in a UPP-dependent manner (Hough and Rechsteiner, 1986). To assess whether UbcH7 can be conjugated with ubiquitin, 125I-UbcH7 was incubated in the presence or absence of additional ubiquitin and/or Ubc4. High-mass forms of 125I-UbcH7 were readily observed when ubiquitin was added to the assay mixture (Figure 4B, lane 4) and were increased even further in the presence of added Ubc4, an E2 with broad specificity (Figure 4B, lane 3 vs. lane 4). Lower molecular mass conjugates of UbcH7 were also observed in the presence of Ubc4 and/or ubiquitin. In the absence of additional ATP, Ubc4, or ubiquitin, neither high-mass nor mono-Ub-UbcH7 species were observed (Figure 4B, lane 1). These data clearly indicate that UbcH7 is a substrate for ubiquitination.
Next we asked if UbcH7 was degraded in a ubiquitin- and proteasome-dependent manner in vitro. As shown in Figure 4C, in the absence of ATP, a hallmark of ubiquitin-dependent proteolysis, there is little if any degradation of UbcH7. However, degradation is doubled when ATP is added to the reaction, consistent with a role for ubiquitination in the degradation process (Hough and Rechsteiner, 1986). Degradation of UbcH7 was further increased fivefold when exogenous Ubc4 was added to assays containing ATP. In the absence of ATP, additional Ubc4 was without an effect. Proteasomal involvement in the degradation of UbcH7 was confirmed by the complete absence of degradation in the presence of MG132, an inhibitor of the proteasome. Taken together, these results confirm a role for the UPP in the turnover of UbcH7. To determine if the active site cysteine on UbcH7 is important for degradation, we assessed the level of degradation of a mutant UbcH7, which has a serine replaced for the cysteine. In contrast to the wild-type UbcH7 protein, there was no ATP-dependent degradation of the mutant UbcH7, suggesting that the active site cysteine may be important for the conjugation of ubiquitin to this substrate (Supplemental Figure S2C). Additionally, we determined the level of degradation of two other E2s. Similar to the mutant UbcH7, neither Ubc2 nor Ubc4 were degraded in an ATP-dependent manner (Supplemental Figure S2C). These data indicate that the ATP- and ubiquitin-dependent degradation that we observed is specific for UbcH7 and is not a property of all E2s.
Because UbcH7 levels are low in S phase (Figure 1B), we asked whether cell lysates prepared from cells in S phase showed differential ability to support degradation of UbcH7 relative to G1 or G2/M extracts. Indeed, as shown in Figure 4D, S phase extracts catalyze ATP-dependent UbcH7 degradation more efficiently than G1 or G2/M extracts. The level of ATP-dependent degradation of a control substrate mutated βB-crystallin (Q70E, Q162E, Supplemental Figure S2D) was not differentially regulated in the synchronized extracts. Additionally, another substrate, mutant αA crystallin also was not degraded differentially in the synchronized extracts (data not shown). Taken together, these data provide strong evidence that the decline of UbcH7 during S phase is due to a UPP-dependent process.
UbcH7 Knockdown Increases Chk1
Given that UbcH7 knockdown delays progression from S phase to G2, we asked if depletion of UbcH7 changed levels of specific proteins that participate in regulating the transition into G2/M. A screen for potential UbcH7 substrates indicated that Chk1 was up-regulated when UbcH7 is depleted (Figure 5A). This was of interest because Chk1 is involved in cell cycle checkpoints in both S and G2 phases after DNA damage (Liu et al., 2000; Sorensen et al., 2003). In contrast, Chk2, which is also activated after DNA damage, was not affected by UbcH7 knockdown (Figure 5A). Furthermore, putative UbcH7 partners, E6-AP (Nuber et al., 1998; Huang et al., 1999) and total PTEN (Waite and Eng, 2003) were not altered during UbcH7 knockdown (Figure 5B). Additionally, UbcH10, an E2 with a known role in cell cycle control in M and G1 phases, was not affected by UbcH7 knockdown (Figure 5B).
Figure 5.

UbcH7 knockdown increases Chk1 levels and decreases phosphorylated PTEN specifically. Asynchronously growing cells were treated with UbcH7 siRNA or NS siRNA for 72 h and were blotted for proteins indicated. All results were observed in at least two experiments each run in duplicate. (A) Samples from cells depleted of UbcH7 were blotted with α-Chk1, α-Chk2, α-E1, or α-UbcH7 as indicated. The E1 loading control is shown for each immunoblot. (B) Samples were blotted with α-E1, α-E6-AP, α-PTEN, α-UbcH10, or α-UbcH7 as indicated. (C) Left, HeLa cells were treated with siRNA specific for UbcH7 for 72 h and stained for α-Chk1 (top) or α-UbcH7 (bottom). Right, HeLa cells were treated with UbcH7 siRNA or NS siRNA for 72 or 96 h. Percentage of cells containing Chk1 foci were quantified from three separate experiments. *p < 0.05. (D) Samples were blotted with α-P-Ser 280 Chk1, α-P-Ser 380 PTEN (αP-PTEN), α-Akt, α-P-Ser 380/Thr 382/383 PTEN (αP3-PTEN), or α-E1 as indicated. (E) HeLa cells were synchronized at the G1/S boundary by treatment with 2 mM HU for 18 h. Cells were released from drug synchronization, and samples were obtained immediately after treatment (G1), 4 h after release into drug-free media (S phase), or 8 h after release (G2/M) and blotted with α-E1, α-P-Thr 382/383 PTEN (αP3-PTEN) or α-P-Ser 280 Chk1as indicated.
We also examined the subcellular localization of Chk1 after UbcH7 knockdown using immunofluorescence. Comparison of the images in Figure 5C (bottom left panels) confirms that UbcH7 levels were markedly reduced in the cells treated with UbcH7 siRNA. There was a marked increase in cells containing perinuclear cytoplasmic foci staining brightly for Chk1 (Figure 5C top left panel, arrows). In cells treated with the NS siRNA similar structures are observed much less frequently (Figure 5C, top right panel). Increased numbers of Chk1 foci were also observed after 96 h of knockdown (Figure 5C right panel). The appearance of these foci is consistent with the Akt- and PTEN-mediated cytoplasmic relocalization of Chk1 observed by Puc et al. (2005).
How might Chk1 levels be related to the UbcH7-directed regulation of the cell cycle? Chk1 is involved in the intra-S phase checkpoint. Thus, a ramification of Chk1 stabilization might be the retention of cells in S phase (Sorensen et al., 2003). Chk1 can be phosphorylated by both the ataxia telangiectasia mutated and rad3 related (ATR; Liu et al., 2000) and Akt kinases (Shtivelman et al., 2002). Akt phosphorylates Chk1 at Ser 280, resulting in its relocalization from the nucleus to the cytoplasm (Puc et al., 2005). UbcH7 knockdown increased the level of phospho Ser-280–modified Chk1, suggesting a role for the Akt pathway (α P-Ser280-Chk1, Figure 5D). Further evidence that UbcH7 may regulate Chk1 via the Akt pathway (see Figure 6) is gleaned by observing that in UbcH7-depleted cells, both mono- and triphosphorylated PTEN levels were decreased (P-PTEN and P3-PTEN; Figure 5D), although the level of total PTEN was not (Figure 5B). Phosphorylation of PTEN increases its stability while decreasing its phosphatase activity (Torres and Pulido, 2001). These data are consistent with a UbcH7-dependent regulation of the S-to-G2 transition via Chk1 and PTEN. There were no alterations in the level of Akt (Figure 5D) in UbcH7-depleted cells, and attempts to determine the level of phosphorylated Akt were unsuccessful.
Figure 6.

Model for UbcH7 regulation of Chk1 and PTEN. Dashed lines indicate proposed roles for UbcH7.
To ask if the increase in P-280 Chk1 and decrease in P3-PTEN were specifically due to alterations in UbcH7 rather than resulting from the alteration in cell cycle profile after UbcH7 depletion, HeLa cells were synchronized by treatment with 2 mM HU for 18 h. FACS analysis of DNA content indicated that the majority of cells were at the G1/S boundary immediately after HU treatment and that they progress to S and G2/M phase after 4 and 8 h in drug-free medium, respectively (data not shown). Both P-280 Chk1 and P3-PTEN protein concentrations are high immediately after drug release (Figure 5E, lane 1) but decrease when the cells are 100% in S phase (lane 2). Thus, the increase in P-280 Chk1 when UbcH7 is depleted (Figure 5D) cannot be accounted for by an increase in the fraction of S phase cells observed when UbcH7 is diminished because P-280 Chk1 is decreased in S phase. Rather, the increase in P-280 Chk1 appears to be the result of the decrease in UbcH7 levels.
Similarly, we asked if the decrease in P3-PTEN that was noted upon UbcH7 depletion (Figure 5D) was due to the decrease in UbcH7 rather than the extended S phase. Semiquantitative analysis suggests that UbcH7 depletion per se elicits an additional decrease in P3-PTEN beyond that which is due to the extension of S phase. Thus, there is a fivefold decrease in P3-PTEN when the cells go from the G1/S boundary (75% G1 and 25% S) to 100% in S phase (Figure 5E), but there is an eightfold decrease in P3-PTEN in UbcH7-depleted cells (Figure 5D). In the latter there is only a twofold increase in the percentage of S phase cells (Figure 2A and data not shown). Taken together these data indicate that decreases in UbcH7 per se are associated with increased levels of P-280 Chk1 and decreased levels of P3-PTEN that are at least partially independent of the cell cycle.
DISCUSSION
The cell cycle is tightly controlled by the timed destruction of cell cycle regulatory proteins via the UPP. Here we present novel evidence that UbcH7 controls the entry into and duration of S phase. The level of UbcH7 protein naturally varies in a cell cycle–dependent manner. When the level of UbcH7 is decreased, an increase in the S phase population is observed, due to a delay in the completion of S phase and progression to G2/M. Conversely, when UbcH7 is over expressed, there is a decrease in the proportion of cells in S phase with an apparent block in S phase entry. The regulation of UbcH7 levels appears to be through UPP-dependent proteolysis in S phase. Inverse relationships between levels of Chk1 and UbcH7 and direct relationships between levels of UbcH7 and P3-PTEN are consistent with UbcH7 mediating its control via the Akt pathway.
Cell cycle differences in UbcH7 protein levels were first noted using a lens epithelial cell line, where an increase in UbcH7 was observed as cells progress from S to G2/M (Liu et al., 2004). The present study clarifies and generalizes the relationship between UbcH7 levels and cell cycle progression. We observed that alteration of UbcH7 levels affects cell cycle progression in four different cell types, indicating that the role of UbcH7 in regulating the duration of S phase of the cell cycle is generalizable. Specifically, in HU-synchronized cells we noted a striking decrease in UbcH7 levels when all the cells were in S phase when compared with UbcH7 levels observed when the cells were at the G1/S boundary (Figure 1B). The UbcH7 levels recovered as the cells progressed into G2. The absence of a concerted change between UbcH7 protein levels and UbcH7 mRNA levels coupled with in vitro observations that UbcH7 is ubiquitinated and that in vivo degradation of UbcH7 in cells is stabilized by the proteasome inhibitor MG132 (Figure 4) suggest that the cell cycle–related modulation in UbcH7 protein levels are regulated via a UPP-dependent pathway. This is supported by the observation that degradation of UbcH7 in vitro is markedly increased in the presence of ubiquitin and Ubc4 and requires ATP. Additionally, these data suggest that Ubc4 is an E2 that can participate in the degradation of UbcH7. Furthermore, the inability of inactive UbcH7 to be degraded is consistent with a catalytic role for UbcH7 in its own degradation. Taken together, these data suggest that UbcH7 is unique with respect to other E2s (Rape and Kirschner, 2004) in that its activity is required for its own degradation and that degradation is enhanced by additional E2 activity.
We also sought mechanistic information about how UbcH7 depletion results in a delay in progression from S to G2 phase. Our discoveries that knockdown of UbcH7 results in an increase in Chk1 but not Chk2 (Figure 5A) and that this increase is coupled to an increase in P-280 Chk1 suggests involvement of the Akt pathway (Figure 6). A role for the Akt pathway in regulating P-280 Chk1 is corroborated by the marked decrease in P3-PTEN when UbcH7 is decreased (see Figures 5D and 6). In addition, our observation of increased P-280 Chk1 appears more consistent with modulation of Chk1 by the Akt pathway (Shtivelman et al., 2002; Puc et al., 2005) because phosphorylation via ATR would result in decreased Chk1 levels (Zhang et al., 2005). This is further supported by our observation of increased perinuclear cytoplasmic staining of Chk1 in UbcH7-depleted cells (Figure 5C). Chk1 relocalizes to the cytoplasm in PTEN-depleted cells (Puc et al., 2005), and PTEN modulates Chk1 via the Akt pathway. Furthermore, phosphorylation by Akt precludes phosphorylation by ATR, and therefore Chk1 is stabilized rather than targeted for degradation. Taken together the data indicate that the increase in Chk1 is due, at least in part, to an increase in signaling through Akt. Additionally, UbcH7 can catalyze the ubiquitination of Chk1 in vitro (Y.-W. Zhang, personal communication; Zhang et al., 2005) suggesting that Chk1 may be a direct UbcH7 target (see Figure 6).
It was recently demonstrated that UbcH7 can participate in the ubiquitination of PTEN in conjunction with NEDD 4.1 in cell free assays (Wang et al., 2007). However, it has also been shown that NEDD 4.1 is dispensable for the ubiquitination of PTEN (Fouladkou et al., 2008). Thus, the involvement of UbcH7 in controlling PTEN levels requires further investigation.
UbcH7 interacts with several E3 ligases, including Parkin (Shimura et al., 2000), Cbl (Yokouchi et al., 1999; Zheng et al., 2000), E6-AP (Nuber et al., 1998; Huang et al., 1999), NK-lytic–associated molecule (Fortier and Kornbluth, 2006), NEDD4 (Anan et al., 1998; Wang et al., 2007), TRIAD-1 (Marteijn et al., 2005), Smurf2 (Ogunjimi et al., 2005), TRAF6 (Geetha et al., 2005), and components of the SCF complex (Staropoli et al., 2003; Oh et al., 2004). Additionally UbcH7 can bind to the E3 ligase BRCA-1 but does not catalyze its self-ubiquitinating activity (Brzovic et al., 2003). Although UbcH7 working with one or more of these E3s may account for the S phase phenotype, at this point we cannot ascertain which of these associations explain our observations.
Only one other report related UbcH7 to cell proliferation. Our results differ from those of Pringa et al. (2000). They used a chronic knockdown model and found no relationship between cell cycle or proliferation and levels of UbcH7. However, it is difficult to determine whether their chronic knockdown could have allowed the outgrowth of cells with compensatory changes in gene expression that mask the function of UbcH7 as had been observed for acute versus chronic knockdown of the retinoblastoma (Rb) gene product (Sage et al., 2003). We have consistently seen that after 4 d (96 h) of UbcH7 depletion, there is an increase in the percentage of cells in S phase (Figure 2B and Supplementary Figure S3). After depletion for 8 d, an increase in S phase is still observed (Supplementary Figure S3). However, after 12 d of siRNA treatment, the level of UbcH7 recovers and the cell cycle profile normalizes. Thus, we conclude that the decrease in UbcH7 levels controls the increase in S phase percentage during this interval. We would predict that a longer knockdown of UbcH7 may result in the normalization of the cell cycle profile with compensatory changes in expression of other E2s or UbcH7 substrates.
In this report we provide a new paradigm for cell cycle control via the ubiquitin system describing a new role for UbcH7. We hypothesize that levels of UbcH7 must decrease in order for S phase to commence and rise for S phase to be completed. UbcH7 may be involved in controlling the levels of both phosphorylated and nonphosphorylated forms of Chk1 and phosphorylated PTEN. Additional understanding of how UbcH7 is activated and degraded will provide further insight into how this E2 is exerting control over cell cycle progression and may allow for more intelligent design of drugs to modulate proliferation for control of cancer, secondary cataract, and other proliferative diseases.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Drs. Martin Obin, Joan Ruderman, and Fu Shang for critical review of the manuscript and Wei Ren for excellent technical assistance. Financial support was received from National Institutes of Health Grant EY13250, USDA Grant 581950-5100-060-01A, and the Johnson and Johnson Focused Giving Award, American Health Assistance Award.
Abbreviations used:
- APC/C
anaphase-promoting complex/cyclosome
- ATR
ataxia telangiectasia and Rad3 related
- CDK
cyclin-dependent kinase
- HLE
human lens epithelial
- HU
hydroxyurea
- NS
nonspecific
- PTEN
phosphate and tensin homolog deleted on chromosome 10
- SCF
Skp1, Cul, F box complex
- Ubc
ubiquitin-conjugating enzyme
- UPP
ubiquitin proteasome pathway.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-01-0036) on October 22, 2008.
REFERENCES
- Anan T., et al. Human ubiquitin-protein ligase Nedd4, expression, subcellular localization and selective interaction with ubiquitin-conjugating enzymes. Genes Cells. 1998;3:751–763. doi: 10.1046/j.1365-2443.1998.00227.x. [DOI] [PubMed] [Google Scholar]
- Bastians H., Topper L. M., Gorbsky G. L., Ruderman J. V. Cell cycle-regulated proteolysis of mitotic target proteins. Mol. Biol. Cell. 1999;10:3927–3941. doi: 10.1091/mbc.10.11.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein G., Bloom J., Sitry-Shevah D., Nakayama K., Pagano M., Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem. 2003;278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- Brzovic P. S., Keeffe J. R., Nishikawa H., Miyamoto K., Fox D., 3rd, Fukuda M., Ohta T., Klevit R. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. USA. 2003;100:5646–5651. doi: 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A. C., Eytan E., Hershko A., Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- Cory A. H., Owen T. C., Barltrop J. A., Cory J. G. Use of an aqueous soluble tretrazolium/formazan assay for cell growth assays in culture. Cancer Comm. 1991;7:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- Fortier J. M., Kornbluth J. NK lytic-associated molecule, involved in NK cytotoxic function, is an E3 ligase. J. Immunol. 2006;176:6454–6463. doi: 10.4049/jimmunol.176.11.6454. [DOI] [PubMed] [Google Scholar]
- Fouladkou F., Landry T., Kawabe H., Neeb A., Lu C., Brose N., Stambolic V., Rotin D. The ubiquitin ligase Nedd4–1 is dispensable for the regulation of PTEN stability and localization. Proc. Natl. Acad. Sci. USA. 2008;105:8585–8590. doi: 10.1073/pnas.0803233105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geetha T., Kenchappa R. S., Wooten M. W., Carter B. D. TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J. 2005;24:3859–3868. doi: 10.1038/sj.emboj.7600845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbers K., Muller U., Grams A., Li E., Jaenisch R., Franz T. Provirus integration into a gene encoding a ubiquitin-conjugating enzyme results in a placental defect and embryonic lethality. Proc. Natl. Acad. Sci. USA. 1996;93:12412–12417. doi: 10.1073/pnas.93.22.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hough R., Rechsteiner M. Ubiquitin-lysozyme conjugates. Purification and susceptibility to proteolysis. The J. Biol. Chem. 1986;261:2391–2399. [PubMed] [Google Scholar]
- Huang L., Kinnucan E., Wang G., Beaudenon S., Howley P. M., Huibregtse J. M., Pavletich N. P. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2–E3 enzyme cascade. Science. 1999;286:1321–1326. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- Liu Q., et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Shang F., Guo W., Hobbs M., Valverde P., Reddy V., Taylor A. Regulation of the ubiquitin proteasome pathway in human lens epithelial cells during the cell cycle. Exp. Eye Res. 2004;78:197–205. doi: 10.1016/j.exer.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Liu Q., Shang F., Whitcomb E., Guo W., Li W., Taylor A. Ubiquitin-conjugating enzyme 3 delays human lens epithelial cells in metaphase. Invest. Ophthalmol. Vis. Sci. 2006;47:1302–1309. doi: 10.1167/iovs.05-0935. [DOI] [PubMed] [Google Scholar]
- Marteijn J. A., van Emst L., Erpelinck-Verschueren C. A., Nikoloski G., Menke A., de Witte T., Lowenberg B., Jansen J. H., van der Reijden B. A. The E3 ubiquitin-protein ligase Triad1 inhibits clonogenic growth of primary myeloid progenitor cells. Blood. 2005;106:4114–4123. doi: 10.1182/blood-2005-04-1450. [DOI] [PubMed] [Google Scholar]
- Nuber U., Schwarz S. E., Scheffner M. The ubiquitin-protein ligase E6-associated protein (E6-AP) serves as its own substrate. Eur. J. Biochem. 1998;254:643–649. doi: 10.1046/j.1432-1327.1998.2540643.x. [DOI] [PubMed] [Google Scholar]
- Ogunjimi A. A., Briant D. J., Pece-Barbara N., Le Roy C., Di Guglielmo G. M., Kavsak P., Rasmussen R. K., Seet B. T., Sicheri F., Wrana J. L. Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol. Cell. 2005;19:297–308. doi: 10.1016/j.molcel.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Oh K. J., Kalinina A., Wang J., Nakayama K., Nakayama K. I., Bagchi S. The papillomavirus E7 oncoprotein is ubiquitinated by UbcH7 and Cullin 1- and Skp2-containing E3 ligase. J. Virol. 2004;78:5338–5346. doi: 10.1128/JVI.78.10.5338-5346.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. M. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell. 2002;9:931–943. doi: 10.1016/s1097-2765(02)00540-3. [DOI] [PubMed] [Google Scholar]
- Pickart C. M., Fushman D. Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Pines J., Lindon C. Proteolysis: anytime, any place, anywhere? Nat. Cell Biol. 2005;7:731–735. doi: 10.1038/ncb0805-731. [DOI] [PubMed] [Google Scholar]
- Pringa E., Meier I., Muller U., Martinez-Noel G., Harbers K. Disruption of the gene encoding the ubiquitin-conjugating enzyme UbcM4 has no effect on proliferation and in vitro differentiation of mouse embryonic stem cells. Biochim. Biophys. Acta. 2000;1494:75–82. doi: 10.1016/s0167-4781(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Puc J., et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Rape M., Kirschner M. W. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature. 2004;432:588–595. doi: 10.1038/nature03023. [DOI] [PubMed] [Google Scholar]
- Rape M., Reddy S. K., Kirschner M. W. The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell. 2006;124:89–103. doi: 10.1016/j.cell.2005.10.032. [DOI] [PubMed] [Google Scholar]
- Sage J., Miller A. L., Perez-Mancera P. A., Wysocki J. M., Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- Shang F., Deng G., Obin M., Wu C. C., Gong X., Smith D., Laursen R. A., Andley U. P., Reddan J. R., Taylor A. Ubiquitin-activating enzyme (E1) isoforms in lens epithelial cells: origin of translation, E2 specificity and cellular localization determined with novel site-specific antibodies. Exp. Eye Res. 2001;73:827–836. doi: 10.1006/exer.2001.1091. [DOI] [PubMed] [Google Scholar]
- Shimura H., et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shtivelman E., Sussman J., Stokoe D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr. Biol. 2002;12:919–924. doi: 10.1016/s0960-9822(02)00843-6. [DOI] [PubMed] [Google Scholar]
- Sorensen C. S., Syljuasen R. G., Falck J., Schroeder T., Ronnstrand L., Khanna K. K., Zhou B. B., Bartek J., Lukas J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–258. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- Staropoli J. F., McDermott C., Martinat C., Schulman B., Demireva E., Abeliovich A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron. 2003;37:735–749. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- Tashtoush B. M., Traboulsi A. A., Dittert L., Hussain A. A. Chloramine-T in radiolabeling techniques. IV. Penta-O-acetyl-N-chloro-N-methylglucamine as an oxidizing agent in radiolabelling techniques. Anal. Biochem. 2001;288:16–21. doi: 10.1006/abio.2000.4832. [DOI] [PubMed] [Google Scholar]
- Torres J., Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem. 2001;276:993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- Tsvetkov L. M., Yeh K. H., Lee S. J., Sun H., Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999;9:661–664. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- Verma S., Ismail A., Gao X., Fu G., Li X., O'Malley B. W., Nawaz Z. The ubiquitin-conjugating enzyme UBCH7 acts as a coactivator for steroid hormone receptors. Mol. Cell. Biol. 2004;24:8716–8726. doi: 10.1128/MCB.24.19.8716-8726.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite K. A., Eng C. BMP2 exposure results in decreased PTEN protein degradation and increased PTEN levels. Hum. Mol. Genet. 2003;12:679–684. [PubMed] [Google Scholar]
- Wang X., et al. NEDD4–1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew P. R. Ubiquitin-mediated proteolysis of vertebrate G1- and S-phase regulators. J. Cell. Physiol. 2001;187:1–10. doi: 10.1002/1097-4652(2001)9999:9999<1::AID-JCP1049>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Yokouchi M., Kondo T., Houghton A., Bartkiewicz M., Horne W. C., Zhan H., Yoshimura A., Baron R. Ligand-induced ubiquitination of the epidermal growth factor receptor involves the interaction of the c-Cbl RING finger and ubcH7. J. Biol. Chem. 1999;274:31707–31712. doi: 10.1074/jbc.274.44.31707. [DOI] [PubMed] [Google Scholar]
- Yu Z. K., Gervais J. L., Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc. Natl. Acad. Sci. USA. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Otterness D., Chiang G., Xie W., Liu Y., Mercurio F., Abraham R. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Zheng N., Wang P., Jeffrey P. D., Pavletich N. P. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102:533–539. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.