

Abstract
Disruption of replication can lead to loss of genome integrity and increase of cancer susceptibility in mammals. Thus, a replication impediment constitutes a formidable challenge to these organisms. Recent studies indicate that homologous recombination (HR) plays an important role in suppressing genome instability and promoting cell survival after exposure to various replication inhibitors, including a topoisomerase I inhibitor, camptothecin (CPT). Here, we report that the deletion of RecQ helicase Recql5 in mouse ES cells and embryonic fibroblast (MEF) cells resulted in a significant increase in CPT sensitivity and a profound reduction in DNA replication after the treatment with CPT, but not other DNA-damaging agents. This CPT-induced cell death is replication dependent and occurs primarily after the cells had exited the first cell cycle after CPT treatment. Furthermore, we show that Recql5 functions nonredundantly with Rad51, a key factor for HR to protect mouse ES cells from CPT-induced cytotoxicity. These new findings strongly suggest that Recql5 plays an important role in maintaining active DNA replication to prevent the collapse of replication forks and the accumulation of DSBs in order to preserve genome integrity and to prevent cell death after replication stress as a result of topoisomerase I poisoning.
INTRODUCTION
Mammalian RecQ helicases share a high degree of homology with the RecQ helicase of Escherichia coli (Nakayama, 2002). These helicases have important roles in maintaining the integrity of the genomes and promoting cell survival in response to various types of genotoxic stress (Hickson, 2003). In addition, some of them have also been implicated in DNA replication (Oakley et al., 2002; Cheok et al., 2005). The only RecQ helicase in E. coli, RecQ, plays an important role in the recovery of genomic replication after DNA damage (Courcelle and Hanawalt, 1999, 2001). In both budding yeast and fission yeast, there exist also just a single RecQ helicase. In both species, the RecQ helicase is involved in a number of DNA transaction processes, including repair, recombination and chromosome segregation (Hickson, 2003). Interestingly, these enzymes do not appear to be essential for processive DNA synthesis. Rather, they are required to prevent genomic instability by coordinating replication and recombination events at stalled replication forks (Lambert et al., 2007). Unlike unicellular organisms which have a single RecQ helicase, mammals possess a family of helicases encoded by five different genes, i.e., human RECQL, BLM, WRN, RECQL4, and RECQL5 (Hickson, 2003). Each of these five genes encodes one or multiple polypeptides with distinctive C- and/or N-terminal domains, suggesting that they have related but nonoverlapping functions. In particular, mutations in three of the five RecQ homologues, RECQL4, WRN, and BLM give rise to three distinct genomic instability and cancer-prone genetic disorders: Rothmund-Thomson, Werner, and Bloom syndromes, respectively (Ellis et al., 1995; Yu et al., 1996; Kitao et al., 1999). However, their exact roles during DNA replication remain unclear (Hanawalt, 1966; Lambert et al., 2007).
Camptothecin (CPT) and its derivatives are potent inhibitors of DNA replications and are very useful for studying DNA replication in mammalian cells. They are a unique group of anticancer agents that target topoisomerase I (Topo I; Pizzolato and Saltz, 2003; Pommier et al., 2006). Recent studies have shown that camptothecins kill mammalian cells by interfering with the function of Topo I during DNA replication (Hsiang et al., 1985). Mammalian Topo I facilitates a highly reversible process of incision-religation in front of a replication fork to allow the rapid release of topological constrain. CPT and its derivatives can impede the function of Topo I by blocking the religation step of this process. As a consequence, CPT stabilizes the Topo I–DNA complex, which results in a persistent nick in front of a replication fork. Such a nick, when present on the leading strand of replication, can be converted into a double-strand break (DSB) by run-off synthesis, collapsing the fork (Strumberg et al., 2000). It is currently believed that the DSBs associated with such collapsed replication forks represent the main cytotoxic lesion induced by CPT (Ryan et al., 1991; Shao et al., 1999; Furuta et al., 2003). Consistent with this current hypothesis, it has been shown that homologous recombination repair (HRR) and the signal transduction cascade that is involved in this repair process both play critical roles in suppressing genome instability and promoting cell survival after CPT exposure (Arnaudeau et al., 2001a; Malik and Nitiss, 2004; Sorensen et al., 2005). Therefore, CPT has been widely used to interrogate the mechanisms involved in the stabilization, repair, or restoration of impeded replication forks in mammalian cells.
We recently reported that Recql5-deficient mouse cells were prone to gamma H2AX focus formation and gross chromosomal rearrangements (GCRs) after CPT treatment (Hu et al., 2007). These observations strongly suggest that Recql5 plays an important role in preventing the collapse of replication forks after CPT treatment. Here we show that Recql5 functions to ensure that replication forks remain active after CPT treatment. A significantly higher proportion of Recql5-deficient cells, compared with their wild-type counterpart, fail to incorporate bromodeoxyuridine (BrdU) when treated with CPT. Nonetheless, these CPT-treated mutant cells were able to resume replication and exit S phase after the removal of CPT. Moreover, the loss of replication capability upon CPT treatment in the absence of Recql5 is followed by a rapid increase in DSB accumulation, GCRs, and replication-dependent cell death. In addition, Recql5 and Rad51 have nonredundant roles in promoting cell survival after CPT treatment. Collectively, these data demonstrate that Recql5 plays an important role in maintaining the integrity of replication forks when replication is impeded as the result of Topo I poisoning, to preserve genome stability and promote cell survival.
MATERIALS AND METHODS
Establishment and Culture of Mouse ES Cells and MEFs
Culture and genetic manipulation of ES cells were performed as described (Hu et al., 2005). To generate ES cell clones expressing human RECQL5β, full-length RECQL5β cDNA was amplified by RT-PCR using total RNA prepared from HeLa cells (Sequences of primers used are available upon request). The PCR product was cloned into the pTRE2 vector (Clontech, Palo Alto, CA) to obtain pTRE-hQ5β, and the identities of the clones were confirmed by sequencing. PGK-RtTA was obtained by replacing the CMV promoter with a PGK promoter on the pTet-On vector (Clontech) for optimal expression in mouse cells. pTRE-hQ5β, PGK-RtTA, and PGKNeo were then coelectroporated into ES cells, and G418-resistant clones were obtained. Clones that contain both pTRE-hQ5β, PGK-RtTA were identified by PCR. They were then further screened by quantitative RT-PCR to identify those in which RECQL5β mRNA expression is regulated by doxycycline (Dox) treatment. The following primers were used in quantitative RT-PCR: hQ5e16f (5′-CAGACTGAGGAGTGCCTCAGG-3′) and hQ5e17r (5′-CTGCTGGGATCGAGGCCGCTT-3′). Gapdh was amplified as internal control by the following primers: GAPDH1 (5′-GTGCTGAGTATGTCGTGGAGT-3′) and GAPDH2 (5′-CACACACCCATCACAAACATG-3′). Rad51 knockdown mouse ES cell lines were generated using a construct expressing short hairpin RNA (shRNA) against mouse Rad51 obtained from Open Biosystems (Huntsville, AL; accession ID NM_011234, clone ID V2MM_77978). The construct was coelectroporated with PGKNeo into ES cells, and stable cell lines containing this vector were selected in G418-containing medium. G418-resistant clones were further screened by Western blot to identify clones with substantially reduced levels of Rad51. Primary MEFs cultures were derived from 13.5 days-post-coitus embryos of various genotypes as described (Hu et al., 2005; Mann et al., 2005). All MEF cells used in experiments were under passage 4. MEFs were cultured in a low-oxygen incubator (3% O2, 5% CO2, and 92% N2; Parrinello et al., 2003).
Clonogenic Survival Assay Experiments
ES cells of various genotypes were seeded onto six-well feeder plates at 1 × 104 cells/well density. Cells were treated with an individual drug or drug combinations for 24 h. CPT, methyl-methanesulfonate (MMS), and aphidicolin (APH) were obtained from Sigma-Aldrich (St. Louis, MO). For APH rescue experiments, 1 μM APH was added 1 h before the CPT treatment. After the treatments, drugs were removed, and treated cells were allowed to grow in drug-free medium for 8 d. Then number of colonies in each well was determined as described (Hu et al., 2005). For each treatment, the experiment was repeated three times, and the mean values derived from these three experiments were then used to construct the survival curves.
Cell Cycle Analysis and Flow Cytometry
A standard propidium iodide (PI) staining approach was used to analyze the cell cycle profile. Briefly, ES cells were seeded and then treated either with or without 100 nM CPT for 16 h. At different time points during the treatment, 1 × 106 ES cells were harvested and fixed with 70% ethanol at −20°C, washed with PBS, and resuspended in staining solution (50 μg/ml PI, Sigma; 200 μg/ml RNase A, Roche, Indianapolis, IN) for flow analysis. For S phase analysis, cells were initially treated as described above. After the treatment, cells were returned to fresh medium with 1 μg/ml nocodazole and harvested at various time points for analysis. Before each time point, cells were pulse-treated with 10 μM BrdU (Sigma) for 30 min and then processed for flow cytometry. BrdU-incorporated cells were detected using an FITC-conjugated anti-BrdU antibody (Becton Dickinson, San Diego, CA). TUNEL (terminal deoxynucleotidyl transferase [Tdt]-mediated nick end labeling) assay and annexin V staining experiments were performed with the APO-DIRECT and annexin V-FITC apoptosis detection kits (BD PharMingen, San Diego, CA), respectively. The experiments were carried out according to the manufacturer's instructions. All flow cytometry data were collected using a Coulter EPICS XL-MCL Cytometer (Beckman Coulter, Fullerton, CA) or a BD LSR I Cytometer (Becton Dickinson). Data were analyzed using FACScan (Becton Dickinson) and the WinMDI (J. Trotter, Scripps Institute) software packages.
Western Blot
Western blot experiments were performed with standard procedure. Specifically, the dilutions for each antibody were as follows: rabbit anti-RAD51 (Santa Cruz Biotechnology, Santa Cruz, CA), 1:1000; mouse anti-CHK1 (Santa Cruz), 1:500; rabbit anti-phospho-CHK1 (Ser 345, Cell Signaling Technology, Beverly, MA), 1:1000; and mouse anti-U1–70K (provided by Dr. Hua Lou, Case Western Reserve University, Cleveland, OH), 1:2000. HRP-conjugated goat anti-rabbit IgG and horse anti-mouse IgG (Cell Signaling Technology) were used as secondary antibodies.
Pulse Field Gel Electrophoresis
Pulse field gel electrophoresis (PFGE) experiments were carried out according to the protocol described in the user's manual of the PFGE instrument, with some modifications. Briefly, after the designed treatments, ES cells were embedded in 0.1 ml 1.0% low-melting-point agarose at a concentration of 5 × 106 cells/ml and incubated in 5 ml lysis buffer (100 mM EDTA, pH 8.0, 0.2% sodium deoxycholate, 1% sodium lauryl sarcosine, and 1 mg/ml proteinase K) at 50°C for 48 h. Plugs were inserted into 1% megabase agarose gel, and electrophoresis was performed in 0.5× TBE buffer using a CHEF-DR II system (Bio-Rad, Hercules, CA) with the following parameters: 4 V/cm, 120° angle and 60–240 switching time at 10°C for 28 h. Gels were stained in 0.5 μg/ml ethidium bromide (EtBr) for 30 min, and images were captured on an UV transilluminator equipped with a digital camera (Eastman Kodak, Rochester, NY). For quantification, DNA was transferred onto membranes following a standard Southern hybridization protocol. Membranes were probed with 32P-labeled whole mouse genomic DNA (DpnI digested) and then exposed in a phosphorimager screen cassette (Amersham Pharmacia, Piscataway, NJ). Images were captured using a Storm 820 scanner (Amersham) and analyzed using ImageJ software (http://rsb.info.nih.gov/ij/). Background in each lane was removed by subtracting the intensity of an empty lane, and then the signal intensity of each lane was normalized to total DNA in both well and lane. The relative amounts of damaged DNA in the nontreated lanes of both wild-type and mutant cells were set to one and the relative intensities of other lanes were expressed as folds increased.
RESULTS
Recql5-deficient Cells are Hypersensitive to CPT
We have reported recently that Recql5-deficient mouse cells exhibited a hyper-recombination phenotype that is accompanied by an increased accumulations of both gamma H2AX foci and GCRs in response to CPT-induced replication stress (Hu et al., 2007). In mammalian cells, HR plays a critical role in suppressing GCRs and promoting cell survival (Arnaudeau et al., 2001a; Saleh-Gohari et al., 2005). Thus, the observation that Recql5 deficiency resulted in an increase in the accumulation of gamma H2AX foci despite having an elevated rate of HR-mediated repair suggests that, in response to CPT treatment, Recql5 may act either upstream of or in parallel to HR in response to replication stress to prevent the collapse of replication forks and the accumulation of DSBs. To address this question, we examined the potential effects of Recql5 deletion on the cell survival response toward a number of replication inhibiting agents, including MMS, mitomycin C (MMC), etoposide, and CPT. We found that Recql5 deletion had no major impact on cell survival after treatment with any of these agents (Figure 1, A–C, and data not shown) except CPT (Figure 2). In particular, the difference in the clonogenic survival capacity between the Recql5-deficient mouse ES cells and their wild-type counterpart was more than 100-fold when cells were treated with 300 nM CPT (Figure 2A). Expression of human RECQL5β in Recql5-deficient cells restored the CPT resistance to a level similar to that of the wild-type cells (Figure 2B), demonstrating that this enhanced CPT sensitivity is due to the deletion of Recql5. Moreover, primary MEFs derived from Recql5-deficient mice were also significantly more sensitive to CPT than their wild-type counterpart (Figure 2C), indicating that Recql5 plays an important role in CPT resistance not only in ES cells, but also in primary MEFs. Together, these data provide definitive genetic evidence demonstrating that Recql5 has a specific role in promoting cell survival after CPT treatment.
Figure 1.
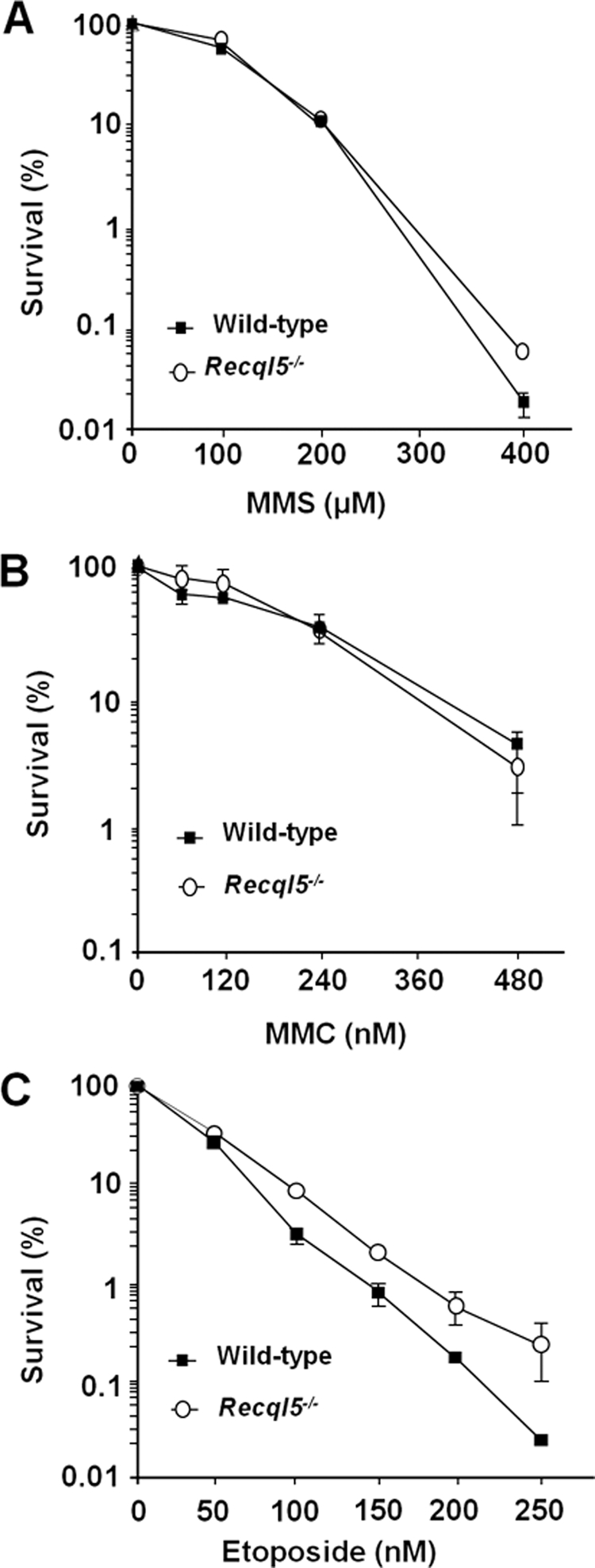
Clonogenic survival of mouse ES cells in response to DNA damage. Results of clonogenic survival assay of ES cells after exposure to a variety of DNA-damaging agents, including methyl-methanesulfonate (MMS; A), mitomycin C (MMC; B), etoposide (C), are shown. In each plot of clonogenic survival assay, the Y- and X-axis indicate percentages of cell survival and drug concentrations, respectively. The genotypes of cells used in each experiment are indicated. For all treatments, results from three sets of experiments were used to calculate the mean and SEM for each data point.
Figure 2.
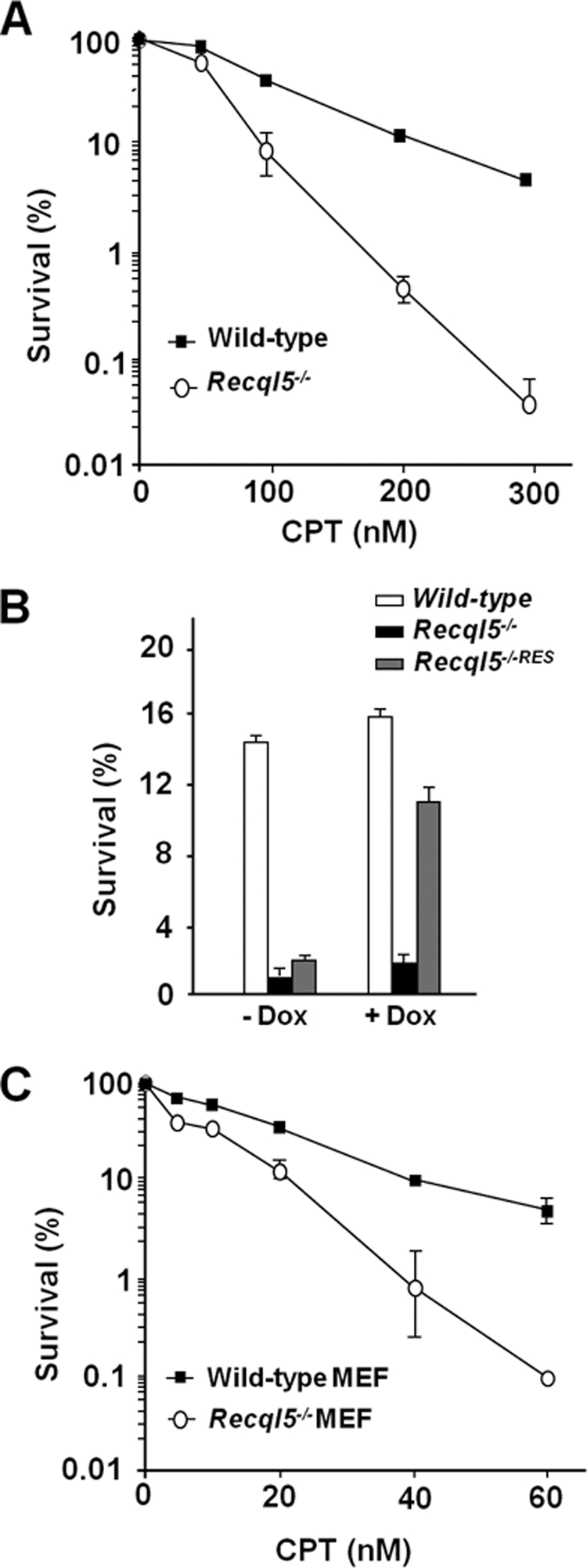
Recql5-deficient mouse ES and MEF cells are hypersensitive to CPT. Results of clonogenic survival assay of ES cells (A and B) or MEFs (C) after exposure to camptothecin (CPT) are shown. In each plot of clonogenic survival assay, the Y- and X-axis indicate percentages of cell survival and drug concentrations, respectively. The genotypes of cells used in each experiment are indicated. The Recql5−/−RES in B indicates a cell line derived from the Recql5−/− line by introducing a vector expressing human RECQL5β under the control of a doxycycline (Dox)-regulated promoter. The expression of human RECQL5β in this cell line was confirmed by Western blot (data not shown). For all treatments, results from three sets of experiments were used to calculate the mean and SEM for each data point.
A Significant Fraction of Recql5-deficient Cells Lost the Ability to Incorporate BrdU after CPT Treatment
It has been reported previously that the cytotoxic effect of CPT is dependent on the collapse of replication forks (Hsiang et al., 1985; Holm et al., 1989; Ryan et al., 1991) and that HR plays a critical role in promoting cell survival after CPT exposure (Arnaudeau et al., 2001a; Malik and Nitiss, 2004; Sorensen et al., 2005). Therefore, a defect in either preventing forks from collapsing or HR-mediated repair could account for the CPT hypersensitivity observed in Recql5-deficient cells. We showed previously that Recql5 deficiency led to an elevated frequency of HR (Hu et al., 2007), suggesting that a defect in replication, rather than HR-related repair is likely the culprit for the CPT hypersensitivity phenotype. This hypothesis is also consistent with a recent demonstration that homologues of Recql5 bind to replication fork-like structures with high affinities in vitro (Kanagaraj et al., 2006). To address this possibility, we examined the effect of Recql5 deletion on the capacity of DNA synthesis after CPT treatment using BrdU incorporation experiments. Specifically, ES cells were treated with 100 nM CPT, which is approximately equivalent to the IC50 for wild-type and IC95 for the mutant cells (Figure 2A), and then released into fresh medium containing nocodazole. CPT causes a cell cycle arrest in late S-phase in both wild-type and mutant cells (data not shown), which is consistent with previous observations (Zhou et al., 2002). Nocodazole-containing medium prevented cells from entering the next cell cycle to produce a new generation of S phase cells, which therefore allows for monitoring of the S phase cells that had been subjected to CPT treatment by standard BrdU pulse-labeling experiments. After pulse labeling with BrdU, the percentages of S phase cells (i.e., DNA content between 2n and 4n) as well as the percentages of BrdU-positive S phase cells (i.e., replicating) were determined by flow cytometry analyses. As expected, in untreated wild-type control, most (94.6%) of S phase cells were able to incorporate BrdU (Figure 3A). Interestingly, the percentage of such cells in Recql5-deficient cells was only 88.6%, slight lower than that of the wild-type control (Figure 3A). After the CPT treatment, the fraction of BrdU-positive S phase cells from the wild-type population was reduced by only 7%. In contrast, the same treatment to the mutant cells resulted in a 58% reduction in BrdU-positive S phase cells (Figure 3B). By 4 h after the removal of CPT, however, the percentages of BrdU incorporating S phase cells in both the wild-type and the mutant cells had returned to levels similar to those in respective untreated controls (Figure 3C). Meanwhile, the majority of both wild-type and mutant cells had a 4n DNA content in the absence of a substantial increase of the sub-G1 population (i.e., apoptotic cells; Figure 3C). These data clearly demonstrate that CPT treatment resulted in a substantial inhibition or loss of the ability to incorporate BrdU in Recql5-deficient cells, but not in wild-type control. However, once CPT was removed, both could resume DNA synthesis and traversed through S phase of the first cell cycle after the treatment without experiencing a substantial amount of apoptosis.
Figure 3.

A substantial reduction of BrdU-incorporating S phase population in Recql5-deficient ES cells after CPT treatment. (A) Untreated cells. (B) CPT treated cells at 0 h after released from the CPT treatment. (C) CPT treated cells at 4 h after release. For each data point, both a DNA content histogram indicating the profile of cell cycle distribution (on the left) and a BrdU incorporation plot showing the data from the BrdU incorporation experiment (on the right) are shown. For the DNA content plots, the X- and Y-axis indicate the DNA content measured by the intensity of PI staining and the number of cell count, respectively. In each of BrdU incorporation plots, the X- and Y-axis indicate the DNA content (measured by the intensity of PI staining) and the content of BrdU (the intensity of BrdU signal) in individual cell, respectively. The subpopulation of cells with a BrdU content above the background level is marked within a trapezoidal area and the percentage of this subpopulation among the total S phase population is also shown above the framed area.
CPT-treated Recql5-deficient Cells Underwent Replication-dependent Apoptosis
We showed above that although Recql5-deficient ES cells were hypersensitive to CPT and were unable to retain their ability to synthesize DNA after CPT treatment, they did not experience an extensive cell death within the first S phase after the treatment (Figure 3C). Previously, we have reported that a substantial increase of gamma H2AX focus formation occurred after a similar treatment with CPT, which peaked at ∼8 h after the treatment, when the cells had exited S phase (Hu et al., 2007). Together, these observations suggest that Recql5-deficient cells are hypersensitive to CPT because they are more susceptible to fork collapse than wild-type cells after CPT treatment. To formally test this idea, we first monitored the cell cycle progression and the accumulation of apoptotic cells after CPT treatment. DNA content analysis showed that consistent with the observations from BrdU incorporation experiments (Figure 3C), neither the wild-type nor the mutant ES cells exhibited a substantial increase in the sub-G1 population both during the 16-h incubation and within the first 8 h after CPT was removed and that the majority of the cells have exited S phase of the cell cycle (Figure 4A). However, by 16 h after the treatment an apparent increase in the sub-G1 population was observed in the mutant cells but not in the wild-type control (Figure 4A). TUNEL staining also confirmed that there was no significant increase in apoptosis in both wild-type and mutant cells within 8 h after the CPT treatment (Figure 4B). However, by 16 h after the treatment, ∼10% of mutant cells are TUNEL positive compared with only ∼2% in the wild-type control. At later time points, the levels of TUNEL-positive mutant cells continued to increase, whereas those in the wild-type controls remained relatively constant. For example, by 32 h after the treatment, the percentage of apoptotic mutant cells had reached ∼22%, whereas levels in the wild-type remained similar to the background level in the untreated control (Figure 4B). These data clearly show that Recql5 mutant cells suffered a delayed-onset type of apoptosis after CPT treatment. We then asked whether this delayed cell death was indeed due to demise in DNA replication. We reasoned that if the enhanced CPT cytotoxicity of Recql5 mutant cells is indeed due to a defect in DNA replication, the observed CPT cytotoxicity would be replication-dependent (Holm et al., 1989; Ryan et al., 1991). We found that cotreatment of CPT together with APH, an inhibitor of DNA replication (Ishii and Bender, 1980), alleviated the cytotoxic effect of CPT for both wild-type and Recql5-deficient ES cells (Figure 4C). Therefore, together, our data strongly suggest that the enhanced CPT cytotoxicity of Recql5-deficient cells is indeed caused by a defect in DNA replication.
Figure 4.

Delayed apoptotic response and replication-dependent cytotoxicity in CPT-treated Recql5-deficient ES cells. (A) Cell cycle profiles of mouse ES cells before and after CPT treatment. The time points (hours) after releasing from CPT treatment are shown to on the left of each plot. The relative positions of the 2n (G1) and 4n (G2/M) peaks of DNA content are shown and the sub-G1 peak in the mutant cells at 16 h after CPT treatment is indicated. (B) A histogram presentation showing the percentage of TUNEL-positive cells of both wild-type and mutant cells at various time points after CPT treatment. (C) A plot summarizing the results of clonogenic survival assay experiment showing the effect of cotreatment of aphidicolin (APH) on the cytotoxicity of CPT.
It is current believed that DSBs associated with collapsed replication forks is the main cytotoxic lesion generated by CPT. Thus, our finding that Recql5-deficient cells exhibited a replication-dependent hypersensitivity to CPT suggests that Recql5-deficient cells may experience a greater increase in replication fork collapse and DSB accumulation than their wild-type counterpart after the CPT treatment. To formally test this hypothesis, we performed PFGE experiments to measure the DNA fragmentation in both untreated and CPT-treated mouse ES cells. In the experiment, the ES cells were treated with the same low dose (100 nM) of CPT, which according to the data from the apoptosis analysis caused a substantial increase in apoptosis in the mutant but not the wild-type ES cells (Figure 4B). We found that this treatment caused a transient and small increase in the amount of fragmented DNA in the wild-type cells immediately after the CPT release. In contrast, the same experiment resulted in a more pronounced effect on the amount of fragmented DNA in the Recql5-deficient cells (Figure 5A). In particular, the amount of fragmented DNA was higher than the untreated control and was approximately twice that of the wild-type cells (Figure 5B, NT and 0 h). Moreover, the level of fragmented DNA continued to increase until it reached a peak level at approximate 12 h after the treatment (Figure 5B, 12 h). These data, therefore, support the idea that the enhanced CPT cytotoxicity in Recql5-deficient cells is due to, at least partly, the increase of replication fork collapse.
Figure 5.

DNA DSBs and Chk1 phosphorylation in CPT-treated ES cells. (A) An image of EtBr-stained gel from a representative pulse-field gel electrophoresis (PFGE) experiment. The size range of which fragmented DNA was quantified for individual samples was shown on the right (marked as Fragmented DNA). The sizes of mega-base (Mb) markers were indicated on the left. (B) A plot showing the relative amounts of fragmented DNA in control and CPT-treated ES cells. The vertical and the horizontal axes indicate the relative amount of fragmented DNA and the time after the release from the CPT treatment, respectively. The values on the vertical axis were derived by normalizing to the value of the untreated wild-type which was set as 1. (C) Western blot analysis of the levels of both phosphorylated (top panels) and total (middle panels) Chk1. The identities of the samples are shown on top of the Western blot. NT: untreated; R0, R2, R4, R8, and R16: 0, 2, 4, 8, and 16 h after releasing from the CPT treatment, respectively. Note that in the top panel, in addition to the specific Pser345-Chk1 band, a nonspecific band (indicated by a star sign) was also detected. Levels of U1–70K are shown as control in the bottom panels.
In human cells, cytotoxic doses of CPT activated CHK1-dependent intra-S checkpoint and inhibited both fork elongation and the initiation of new origins of replication (Seiler et al., 2007). Thus, we also analyzed the status of Chk1 in both wild-type and mutant cells before and after treating with 100 nM CPT. We found that 100 nM CPT caused only a modest increase in Chk1 phosphorylation in wild-type cells (Figure 5C, left panel). In contrast, the same treatment resulted in a much more pronounced increase in the levels of phosphorylated Chk1 in mutant cells. These relatively high levels of phosphorylated Chk1 persisted for up to 8 h after the release of the CPT treatment before subsiding at 16 h after the release (Figure 5C, right panel). A significant up-regulation of Chk1 phosphorylation in mutant cells but not in wild-type cells immediately after the CPT treatment is consistent with the significant reduction in the number of BrdU-incorporating S phase cells in the mutant but not wild-type control after the same treatment (Figure 3). In addition, the prolonged presence of high levels of phosphorylated Chk1 in the CPT-treated mutant cells is also consistent with the sustained high levels of fragmented DNA revealed by PFGE analysis (Figure 5A). Thus, in aggregate, the data presented thus far strongly suggest that Recql5 is required to prevent the collapse of CPT-induced stalled DNA replication fork to promote cell survival.
Recql5 Functions Nonredundantly with Rad51 to Promote Cell Survival after CPT Treatment
HR plays critical roles in suppressing genome instability and promoting cell survival after CPT exposure (Arnaudeau et al., 2001a; Malik and Nitiss, 2004; Sorensen et al., 2005). Thus, the conclusion reached above also suggests that Recql5 functions in a pathway that has a nonoverlapping role with HR with respect to cell survival after CPT treatment. We reasoned that if this is indeed the case, inhibition of HR should have similar effects on CPT sensitivity in both wild-type and mutant ES cells. To test this hypothesis, we inhibited HR by suppressing the expression of Rad51 in both wild-type and Recql5-deficient ES cells using shRNA-mediated knockdown (Lambert and Lopez, 2000; Stark et al., 2004) and then examined their effects on CPT sensitivity. Suppression of Rad51 expression resulted in an enhanced CPT sensitivity in wild-type cells (Figure 6, A and B), consistent with previous reports (Thacker and Ganesh, 1990; Arnaudeau et al., 2001b; Saintigny et al., 2001; Lundin et al., 2002). Meanwhile, a similar reduction in Rad51 expression in Recql5-deficient cells resulted in a further enhancement in CPT sensitivity (Figure 6, A and B). These data support our hypothesis that Recql5 contributes to cell survival after CPT treatment through a mechanism that is nonredundant to the Rad51-dependent mechanism (presumably HR; Figure 6C).
Figure 6.

Suppression of Rad51 expression resulted in similar enhancements in CPT cytotoxicity between Recql5-deficient and wild-type cells. The percentages of survival after CPT treatment were measured using clonogenic survival assay experiments. (A) The effects on suppressing Rad51 expression by shRNA on CPT sensitivity in wild-type and Recql5-deficient ES cells. (B) Western blot results showing the relative levels of Rad51 in different cell lines. The top and the bottom panels are results of Western blots detecting Rad51 and U1–70K (as a loading control), respectively. The relative levels of Rad51 expression after being normalized to that of the wild-type using U1–70K as a control are shown under the top panel. (C) A schematic illustration of a model by which Recql5 and HRR function to promote cell survival in response to topoisomerase I (Topo I) poisoning. First, a pair of arrows in opposite orientations is used to indicate the highly reversible nature of a Topo I–DNA complex associated with an on-going DNA replication fork (1F and 1R). However, camptothecin (CPT, shown as a C inside a black wedged-shape structure) can bind to both the DNA-bound Topo I and the DNA, creating a long-lived CPT-Topo I–DNA complex. This in turn hinders the release of tension in front of the fork (depicted by a thick twisted line), stalling the fork (2). This stalled fork can be stabilized by Recql5 and reactivated once CPT dissociates from the fork (Route 3–4-1R). Alternative, the stalled fork can collapse, creating a single-ended double-strand break (DSB; 3a). This DSB must be repaired by HR repair in order to promote cell survival. Otherwise, it could lead to genome instability or even cell death (4a).
DISCUSSION
We report here that Recql5-deficient mouse ES cells were hypersensitive to CPT in a replication-dependent manner. Yet, they were not more sensitive than their wild-type counterpart toward a number of other DNA-damaging agents that can also impede DNA replication. Moreover, this phenotype is also observed in Recql5-deficient MEF cells. Together, these data demonstrate the unique requirement of Recql5 for the optimal cell survival in both mouse ES and MEF cells after CPT treatment. In addition, we showed that after CPT treatment, a significant fraction of Recql5-deficient cells, but not their wild-type counterpart, lost their ability to incorporate BrdU in S phase. We noticed that this loss of replication competence during the S phase is followed by a rapid accumulation of DSBs and GCRs and then cell death. It has been demonstrated that CPT can cause the collapse of replication forks (Kohn and Pommier, 2000; Strumberg et al., 2000). DSB generated from such fork collapse is believed to represent the main cytotoxic lesion induced by CPT and its derivatives (Pommier et al., 2006). Therefore, our data strongly suggest that Recql5 is required for maintaining the replication competency when forks are impeded after CPT treatment.
We proposed that in CPT-treated wild-type cells, a Recql5-dependent mechanism functions to maintain the integrity and the activity of DNA replication to prevent the collapse of the impeded replication forks. The dilemma caused by the replication impediment can then be resolved either by HR or by other mechanism(s), preventing the accumulation of DSBs. In contrast, in Recql5-deficient cells, the impeded replication forks are more susceptible to collapse, resulted in a significant accumulation of DSBs. This extension is consistent with our previous observation that Recql5-deficient cells exhibited both an elevated HR-mediated repair and an increased in DSB accumulation after CPT treatment (Hu et al., 2005, 2007). Therefore, Recql5 has a nonredundant role with HR with respect to the optimal cell survival after CPT treatment. Thus, defects in either HR or the Recql5-related pathway can significantly increase the cells' sensitivity toward CPT and its derivatives. Importantly, the combined functions of these two mechanisms as well as other (Pommier et al., 2006) are required to provide an optimal protection against CPT cytotoxicity (Figure 6C).
It has been reported recently that under reconstituted conditions, camptothecin-induced Topo I–DNA complex formation resulted in the retardation of DNA uncoiling (Koster et al., 2007). Furthermore, in yeast, this retardation of DNA uncoiling is associated with the impediment of both transcription and replication in vivo (Koster et al., 2007). The implication that Recql5 can prevent impeded replication forks from collapsing after CPT treatment is in agreement with this recent finding and suggests that a similar scenario also exist in mammalian cells. A role of Recql5 in maintaining the integrity of stalled replication forks to prevent fork collapse is in agreement with previous reports that link this helicase (Garcia et al., 2004; Kanagaraj et al., 2006) and its homologues (Ozsoy et al., 2003) to DNA replication. Interestingly, we found that Recql5-deficient cells that were initially incapable of incorporating BrdU could eventually resume DNA synthesis and reenter the next phase of the cell cycle after CPT is removed. This temporally cessation of DNA synthesis could reflect either a delay in the reactivation of the impeded forks or the inability of impeded forks to resume DNA synthesis, and therefore cells must rely on the firing of new origins of replication in order to complete the duplication of the genome. Future experiments will be needed to distinguish these possibilities. Intriguingly, this resumption of DNA replication in CPT-treated Recql5-deficient cells occurred within 4 h after CPT removal, when the proper repair or restoration of the collapse forks had not yet been completed, which would be consistent with a current model that the stalled replication fork, but not the specific cues that cause the stalling, provides the necessary signal for intra-S cell cycle arrest in mammalian cells (Zachos et al., 2005). In any cases, this temporally cessation of DNA synthesis is associated with an increase in genome instability and a dramatic decrease in cell survival.
Human cells that are deficient in BLM or WRN and mouse cells deficient in Wrn are more sensitive to CPT and other DNA-damaging agents than their wild-type counterparts (Hook et al., 1984; Ogburn et al., 1997; Lebel and Leder, 1998; Poot et al., 1999; Rao et al., 2005). Intriguingly, to our knowledge, only Recql5-deficient cells are uniquely hypersensitive to CPT. The molecular basis underlying this unique role of Recql5 remains unclear. It should be noted that that the structures of the stalled forks elicited by CPT and other Topo I inhibitors are likely to be distinct from those induced by other DNA-damaging agents or by nucleotide deprivation, the two other major causes of replication stalling. For instance, when a replication fork is stalled by a DNA damage lesion or due to nucleotide deprivation, the progression of the DNA polymerase is impeded, but the MCM replication helicase complex associated with the fork can continue to advance, leading to the creation of an extended stretch of single-strand DNA (ssDNA) region and the activation of intra-S checkpoint (Liu et al., 2000; Byun et al., 2005). In contrast, CPT causes the stalling of replication fork by stabilizing DNA-Topo I covalent complexes in front of advancing replication forks. Thus, it impedes the advancement of both the DNA polymerase and MCM helicase complex and hence does not directly create excessive amount of ssDNA. Moreover, DNA-Topo I covalent complexes similar to those induced by CPT also exist under normal physiological conditions. Thus, the formation of a DNA-Topo I covalent complex per se may not be consequential. Indeed, CPT causes a peculiar late S–early G2 cell cycle arrest rather than the typical intra-S arrest triggered by many other types of replication-inhibiting agents or treatments (Zhou et al., 2002). Therefore, alternative molecular mechanism(s) must exist in order to maintain the integrity of the stalled forks induced by CPT. We hypothesize that Recql5 plays a critical role in such a mechanism. More importantly, when a replication fork is stalled by a DNA damage lesion, both the stabilization of the stalled fork and the subsequent successful repair of the damage lesion are likely to be necessary in order to preserve the integrity of the genome and to promote cell survival (Hook et al., 2007). In contrast, when a replication fork is stalled as the result of Topo I poisoning, the removal of the inhibition may be the only key event for reactivating the stalled fork. Thus, we propose that in the event of Topo I poisoning by CPT, stabilization of the stalled replication forks by Recql5 alone provides the critical time that is required for the dissociation of CPT from the stalled fork and the restoration of the fork before it collapses (Figure 6C, route 3-4-1R). Therefore, Recql5 is both necessary and sufficient for promoting cell survival after the exposure to CPT, but no other types of replication fork–stalling agents or treatments. However, CPT-induced stalled fork could also collapse, for instance, when it is not protected by Recql5 in Recql5 knockout cells or when the number of the stalled forks outpaced the capacity of Recql5. In this case, faithful HR repair is necessary for preserving the integrity of the genome and promoting survival. Failure to execute such a faithful repair can lead to genome instability or even cell death (Figure 6C, route 3a-4a).
CPT and its derivatives represent a group of very promising anticancer agents, two of which, Topotecan and Irinotecan, have recently been approved by the FDA for treating patients with several types of malignancies (Pizzolato and Saltz, 2003; Pommier et al., 2006). However, despite a broad range of indication in treating many types of cancers, these drugs, when used in single-agent therapy, cannot provide a satisfactory curative effect. Therefore, it has become increasingly evident that combinatorial therapies involving multiple agents and/or treatment modalities is necessary in order to fully realize the potential of these drugs in anticancer treatments (Pommier et al., 2006). A thorough understanding of the molecular determinants of drug sensitivity as well as the molecular pathways that are specific to camptothecins and other Topo I poisoning drugs will be critical in formulating such combinatorial treatment regimens. The mouse Recql5 and human RECQL5 are highly conserved (Ohhata et al., 2001). Thus, it is expected that mutations in RECQL5 in human cells should also resulted in an hypersensitivity to CPT. In this context, the finding that Recql5 is an important determinant of CPT resistance can have important implication in improving the use of CPT derivatives and other Topo I inhibitors in anticancer treatment. In particular, we have shown that deletion of Recql5 in mice resulted in a significant increase in cancer susceptibility (Hu et al., 2007). Thus, it is possible that RECQL5 mutations may also be associated with some human cancers. In that case, RECQL5 may be a valuable biomarker for determining whether Topo I inhibitors should be used in a patients' treatment regimen. Furthermore, we have noticed that the combination of Recql5 inactivation and CPT exposure is similar to the effect of PARP inhibitors, that is, both render mammalian cells more dependent on HR for survival in response to agents that can cause replication fork collapse. HR deficient cancer cells, for example, those with BRCA1 or BRCA2 deficiency, are hypersensitive to PARP inhibitors, and PARP inhibitors have been proven very effective in treating tumors that are deficient in either BRCA1 or BRCA2 (Bryant and Helleday, 2004; Bryant et al., 2005; Farmer et al., 2005). Thus, RECQL5 may be a potential target for developing new anticancer drugs that can enhance the efficacy of Topo I inhibitors and/or those that target HR.
ACKNOWLEDGMENTS
We are grateful to Drs. Nathan Berger, George Stark, Peter Harte, and Hua Lou for their comments on the manuscript. This study was supported by Grants R01 CA88939 and P20 CA103736 from the National Institutes of Health and Searle Scholar Award 01-E-109 from the Searle Scholar Program.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-06-0565) on November 5, 2008.
REFERENCES
- Arnaudeau C., Lundin C., Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J. Mol. Biol. 2001a;307:1235–1245. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- Arnaudeau C., Rozier L., Cazaux C., Defais M., Jenssen D., Helleday T. RAD51 supports spontaneous non-homologous recombination in mammalian cells, but not the corresponding process induced by topoisomerase inhibitors. Nucleic Acids Res. 2001b;29:662–667. doi: 10.1093/nar/29.3.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H. E., Helleday T. Poly(ADP-ribose) polymerase inhibitors as potential chemotherapeutic agents. Biochem. Soc. Trans. 2004;32:959–961. doi: 10.1042/BST0320959. [DOI] [PubMed] [Google Scholar]
- Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Byun T. S., Pacek M., Yee M.-C., Walter J. C., Cimprich K. A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheok C. F., Bachrati C. Z., Chan K. L., Ralf C., Wu L., Hickson I. D. Roles of the Bloom's syndrome helicase in the maintenance of genome stability. Biochem. Soc. Trans. 2005;33:1456–1459. doi: 10.1042/BST0331456. [DOI] [PubMed] [Google Scholar]
- Courcelle J., Hanawalt P. C. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol. Gen. Genet. 1999;262:543–551. doi: 10.1007/s004380051116. [DOI] [PubMed] [Google Scholar]
- Courcelle J., Hanawalt P. C. Participation of recombination proteins in rescue of arrested replication forks in UV-irradiated Escherichia coli need not involve recombination. Proc. Natl. Acad. Sci. USA. 2001;98:8196–8202. doi: 10.1073/pnas.121008898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis N. A., Groden J., Ye T. Z., Straughen J., Lennon D. J., Ciocci S., Proytcheva M., German J. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- Farmer H., et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Furuta T., et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 2003;278:20303–20312. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- Garcia P. L., Liu Y., Jiricny J., West S. C., Janscak P. Human RECQ5beta, a protein with DNA helicase and strand-annealing activities in a single polypeptide. EMBO J. 2004;23:2882–2891. doi: 10.1038/sj.emboj.7600301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt P. C. The U.V. sensitivity of bacteria: its relation to the DNA replication cycle. Photochem. Photobiol. 1966;5:1–12. [PubMed] [Google Scholar]
- Hickson I. D. RecQ helicases: caretakers of the genome. Nat. Rev. Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- Holm C., Covey J. M., Kerrigan D., Pommier Y. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res. 1989;49:6365–6368. [PubMed] [Google Scholar]
- Hook G. J., Kwok E., Heddle J. A. Sensitivity of Bloom syndrome fibroblasts to mitomycin C. Mutat. Res. 1984;131:223–230. doi: 10.1016/0167-8817(84)90029-4. [DOI] [PubMed] [Google Scholar]
- Hook S. S., Lin J. J., Dutta A. Mechanisms to control rereplication and implications for cancer. Curr. Opin. Cell Biol. 2007;19:663–671. doi: 10.1016/j.ceb.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiang Y. H., Hertzberg R., Hecht S., Liu L. F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- Hu Y., Lu X., Barnes E., Yan M., Lou H., Luo G. Recql5 and Blm RecQ DNA helicases have nonredundant roles in suppressing crossovers. Mol. Cell. Biol. 2005;25:3431–3442. doi: 10.1128/MCB.25.9.3431-3442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007;21:3073–3084. doi: 10.1101/gad.1609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii Y., Bender M. A. Effects of inhibitors of DNA synthesis on spontaneous and ultraviolet light-induced sister-chromatid exchanges in Chinese hamster cells. Mutat. Res. 1980;79:19–32. doi: 10.1016/0165-1218(80)90144-5. [DOI] [PubMed] [Google Scholar]
- Kanagaraj R., Saydam N., Garcia P. L., Zheng L., Janscak P. Human RECQ5beta helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res. 2006;34:5217–5231. doi: 10.1093/nar/gkl677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao S., Shimamoto A., Goto M., Miller R. W., Smithson W. A., Lindor N. M., Furuichi Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 1999;22:82–84. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- Kohn K. W., Pommier Y. Molecular and biological determinants of the cytotoxic actions of camptothecins. Perspective for the development of new topoisomerase I inhibitors. Ann. NY Acad. Sci. 2000;922:11–26. doi: 10.1111/j.1749-6632.2000.tb07021.x. [DOI] [PubMed] [Google Scholar]
- Koster D. A., Palle K., Bot E. S., Bjornsti M. A., Dekker N. H. Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature. 2007;448:213–217. doi: 10.1038/nature05938. [DOI] [PubMed] [Google Scholar]
- Lambert S., Froget B., Carr A. M. Arrested replication fork processing: interplay between checkpoints and recombination. DNA Repair. 2007;6:1042–1061. doi: 10.1016/j.dnarep.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Lambert S., Lopez B. S. Characterization of mammalian RAD51 double strand break repair using non-lethal dominant-negative forms. EMBO J. 2000;19:3090–3099. doi: 10.1093/emboj/19.12.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel M., Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc. Natl. Acad. Sci. USA. 1998;95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Lundin C., Erixon K., Arnaudeau C., Schultz N., Jenssen D., Meuth M., Helleday T. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol. Cell. Biol. 2002;22:5869–5878. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M., Nitiss J. L. DNA repair functions that control sensitivity to topoisomerase-targeting drugs. Eukaryot. Cell. 2004;3:82–90. doi: 10.1128/EC.3.1.82-90.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M. B., Hodges C. A., Barnes E., Vogel H., Hassold T. J., Luo G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum. Mol. Genet. 2005;14:813–825. doi: 10.1093/hmg/ddi075. [DOI] [PubMed] [Google Scholar]
- Nakayama H. RecQ family helicases: roles as tumor suppressor proteins. Oncogene. 2002;21:9008–9021. doi: 10.1038/sj.onc.1205959. [DOI] [PubMed] [Google Scholar]
- Oakley T. J., Goodwin A., Chakraverty R. K., Hickson I. D. Inactivation of homologous recombination suppresses defects in topoisomerase III-deficient mutants. DNA Repair. 2002;1:463–482. doi: 10.1016/s1568-7864(02)00032-0. [DOI] [PubMed] [Google Scholar]
- Ogburn C. E., Oshima J., Poot M., Chen R., Hunt K. E., Gollahon K. A., Rabinovitch P. S., Martin G. M. An apoptosis-inducing genotoxin differentiates heterozygotic carriers for Werner helicase mutations from wild-type and homozygous mutants. Hum. Genet. 1997;101:121–125. doi: 10.1007/s004390050599. [DOI] [PubMed] [Google Scholar]
- Ohhata T., Araki R., Fukumura R., Kuroiwa A., Matsuda Y., Abe M. Cloning, genomic structure and chromosomal localization of the gene encoding mouse DNA helicase RECQL5beta. Gene. 2001;280:59–66. doi: 10.1016/s0378-1119(01)00740-5. [DOI] [PubMed] [Google Scholar]
- Ozsoy A. Z., Ragonese H. M., Matson S. W. Analysis of helicase activity and substrate specificity of Drosophila RECQ5. Nucleic Acids Res. 2003;31:1554–1564. doi: 10.1093/nar/gkg243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello S., Samper E., Krtolica A., Goldstein J., Melov S., Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzolato J. F., Saltz L. B. The camptothecins. Lancet. 2003;361:2235–2242. doi: 10.1016/S0140-6736(03)13780-4. [DOI] [PubMed] [Google Scholar]
- Pommier Y., et al. Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2006;81:179–229. doi: 10.1016/S0079-6603(06)81005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poot M., Gollahon K. A., Rabinovitch P. S. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum. Genet. 1999;104:10–14. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- Rao V. A., Fan A. M., Meng L., Doe C. F., North P. S., Hickson I. D., Pommier Y. Phosphorylation of BLM, dissociation from topoisomerase IIIalpha, and colocalization with gamma-H2AX after topoisomerase I-induced replication damage. Mol. Cell. Biol. 2005;25:8925–8937. doi: 10.1128/MCB.25.20.8925-8937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A. J., Squires S., Strutt H. L., Johnson R. T. Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res. 1991;19:3295–3300. doi: 10.1093/nar/19.12.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny Y., Delacote F., Vares G., Petitot F., Lambert S., Averbeck D., Lopez B. S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh-Gohari N., Bryant H. E., Schultz N., Parker K. M., Cassel T. N., Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell. Biol. 2005;25:7158–7169. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler J. A., Conti C., Syed A., Aladjem M. I., Pommier Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol. Cell. Biol. 2007;27:5806–5818. doi: 10.1128/MCB.02278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R. G., Cao C. X., Zhang H., Kohn K. W., Wold M. S., Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen C. S., Hansen L. T., Dziegielewski J., Syljuasen R. G., Lundin C., Bartek J., Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- Stark J. M., Pierce A. J., Oh J., Pastink A., Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumberg D., Pilon A. A., Smith M., Hickey R., Malkas L., Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 2000;20:3977–3987. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker J., Ganesh A. N. DNA-break repair, radioresistance of DNA synthesis, and camptothecin sensitivity in the radiation-sensitive irs mutants: comparisons to ataxia-telangiectasia cells. Mutat. Res. 1990;235:49–58. doi: 10.1016/0921-8777(90)90057-c. [DOI] [PubMed] [Google Scholar]
- Yu C. E., et al. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- Zachos G., Rainey M. D., Gillespie D. A. Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol. Cell. Biol. 2005;25:563–574. doi: 10.1128/MCB.25.2.563-574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Gwadry F. G., Reinhold W. C., Miller L. D., Smith L. H., Scherf U., Liu E. T., Kohn K. W., Pommier Y., Weinstein J. N. Transcriptional regulation of mitotic genes by camptothecin-induced DNA damage: microarray analysis of dose- and time-dependent effects. Cancer Res. 2002;62:1688–1695. [PubMed] [Google Scholar]