

Abstract
Intracellullar trafficking of lipids is fundamental to membrane biogenesis. For the synthesis of sphingomyelin, ceramide is transported from the endoplasmic reticulum to the Golgi apparatus by the ceramide transfer protein CERT. CERT is phosphorylated by protein kinase D at S132 and subsequently multiple times in a serine-repeat motif, resulting in its inactivation. However, the kinase involved in the multiple phosphorylation remains unclear. Here, we identify the γ2 isoform of casein kinase I (CKIγ2) as a kinase whose overexpression confers sphingomyelin-directed toxin-resistance to Chinese hamster ovary cells. In a transformant stably expressing CKIγ2, CERT was hyperphosphorylated, and the intracellular trafficking of ceramide was retarded, thereby reducing de novo sphingomyelin synthesis. The reduction in the synthesis of sphingomyelin caused by CKIγ2 was reversed by the expression of CERT mutants that are not hyperphosphorylated. Furthermore, CKIγ2 directly phosphorylated CERT in vitro. Among three γ isoforms, only knockdown of γ2 isoform caused drastic changes in the ratio of hypo- to hyperphosphorylated form of CERT in HeLa cells. These results indicate that CKIγ2 hyperphosphorylates the serine-repeat motif of CERT, thereby inactivating CERT and down-regulating the synthesis of sphingomyelin.
INTRODUCTION
Sphingomyelin (SM) is a ubiquitous membrane lipid in mammalian cells. SM accounts for 5–20% of all phospholipids and is concentrated in the outer leaflet of the plasma membrane (PM). Its saturated hydrocarbon chains pack tightly with cholesterol, which leads to the formation of membrane microdomain “lipid rafts.” Lipid rafts are considered a platform for signal transduction, protein sorting, and membrane transport (Simons and Ikonen, 1997).
The synthesis of SM proceeds from the condensation of l-serine with palmitoyl CoA to the synthesis of ceramide at the cytosolic surface of the endoplasmic reticulum (ER). Thereafter, ceramide is transported from the ER to the trans-Golgi regions, where it is converted to SM by phosphatidylcholine:ceramide choline phosphotransferase (SM synthase) (Huitema et al., 2004; Yamaoka et al., 2004). The ER-to-Golgi transport of ceramide is mediated by the ceramide transfer protein CERT in a nonvesicular manner (Hanada et al., 2003).
CERT consists of three distinct functional domains. The N-terminal pleckstrin homology (PH) domain recognizes phosphatidylinositol 4-monophosphate (PI4P) and recruits CERT to the trans-Golgi regions. The C-terminal START domain is responsible for intermembrane transfer of ceramide. The middle region between the PH and START domains is predicted to form no globular domains but may have various crucial functions. Indeed, the middle region has a FFAT motif (two phenylalanines in an acidic tract), which interacts with VAP, an ER-resident type II membrane protein (Loewen et al., 2003). Efficient trafficking of ceramide from the ER to the Golgi requires both the Golgi-targeting PH domain and ER-interacting FFAT motif (Kawano et al., 2006). Narrow cytoplasmic gaps called membrane contact sites, at which two organelles come into close apposition within ∼10 nm, are speculated to contribute to interorganelle metabolic and functional interaction (Voeltz et al., 2002; Mogelsvang et al., 2004; Holthuis and Levine, 2005). CERT is preferentially distributed to the Golgi region, and the Golgi-associated CERT retains the activity to interact with VAP (Kawano et al., 2006). Based on these results, it has been proposed that CERT-mediated trafficking efficiently occurs at the ER-Golgi membrane contact sites (Kawano et al., 2006; Hanada et al., 2007).
Although the genes that encode enzymes in the biosynthetic pathway of SM have been identified, the genes involved in the regulation of SM synthesis are poorly known. The fact that the treatment of cells with brefeldin A (BFA), which induces fusion between the ER and the Golgi apparatus (Lippincott-Schwartz et al., 1990), increases the synthesis of SM (Fukasawa et al., 1999; Figure 2C) suggests that the CERT-mediated transport of ceramide is a rate-limiting step in the biosynthesis of SM. Thus, the CERT-mediated transport that takes place between distinct membrane compartments is likely to be regulated at multiple points. Knockdown and pharmacological inhibition of phosphatidylinositol 4-kinase IIIβ decreases the amount of CERT recruited to the Golgi and the synthesis of SM, by reducing PI4P levels in the Golgi region (Toth et al., 2006). We previously found that CERT was phosphorylated multiple times at a serine-repeat (SR) motif in the middle region and that the phosphorylation down-regulated the activity of CERT to transport ceramide from the ER to the Golgi site for SM synthesis (Kumagai et al., 2007). Expression or knockdown of protein phosphatase 2Cε, an ER-resident type I membrane protein, whose catalytic domain faces the cytosol, results in dephosphorylation of CERT in the presence of VAP-A expression or attenuates the interaction between CERT and VAP-A, and the synthesis of SM, respectively (Saito et al., 2008). These findings suggest that kinases and phosphatases regulate the function of CERT through phosphorylation and dephosphorylation, and thereby control the synthesis of SM.
Figure 2.

The decreased synthesis of SM in CHO/hCKIγ2 cells is caused by inhibition of the ER-to-Golgi ceramide transport. (A) CHO cell monolayers were incubated with [14C]choline at 37°C. The metabolically labeled lipids were analyzed. PC, phosphatidylcholine. (B) In vitro assay of SM synthase activity was performed with microsomal membranes isolated from CHO/C, CHO/vector, and CHO/hCKIγ2 cells as the enzyme source, and the fluorescent ceramide analogue C6-NBD-Cer as the substrate. Results shown are means ± SD from triplicated experiments. (C) BFA treatment restores SM synthesis in CHO/hCKIγ2 cells to the wild-type level. CHO cell monolayers preincubated with or without 1 μg/ml BFA at 37°C for 30 min were subjected to metabolic labeling with [14C]serine at 37°C for 2 h in the presence or absence of 1 μg/ml BFA. Labeled lipids were analyzed as in Figure 1. The levels of labeled SM are means ± SD from triplicated experiments and represented as a percentage of the level in CHO/C cells in the absence of BFA. (D) Redistribution of C5-DMB-Cer from the ER to the Golgi was retarded in CHO/hCKIγ2 cells. Cell monolayers grown on glass coverslip were pre-labeled with 1 μM C5-DMB-Cer on ice for 30 min and chased at 33°C for 0 (top) or 15 min (bottom). The cells were fixed with 0.125% glutaraldehyde in saline for 5 min on ice and then observed with a fluorescence microscope. For 0-min chase, cells were fixed soon after the prelabeling. The experiments were performed three times, and representative images are shown. (E) Protein transport from the ER to the Golgi complex is not affected in CHO/hCKIγ2 cells. tsO45-VSVG-EGFP was accumulated in the ER by culturing CHO/vector and CHO/hCKIγ2 cells at 40°C for 20 h. The accumulated tsO45-VSVG-EGFP protein was released from the ER by shifting the culturing temperature to 33°C for the indicated time. After the chase, cell lysate was prepared and treated with or without Endo H. Then, the samples were subjected to Western blotting with anti-GFP antibody. R, S, and C indicate the End H-resistant form, End H-sensitive form, and form cleaved by Endo H, respectively.
Here, we report the identification of CKIγ2 as a negative regulator in the synthesis of SM through multiple phosphorylation in the SR motif of CERT. By analyzing a transformant stably expressing CKIγ2, we present in vivo evidence that hyperphosphorylated CERT actually represents the inactivated state, resulting in the decreased synthesis of SM. It has recently been reported that a priming phosphorylation at S132 by protein kinase D (PKD) within the SR motif of CERT is required for decreases in affinity for PI4P and the ceramide-transfer activity (Fugmann et al., 2007). We show that expression of the mutant, which mimics CERT receiving only the priming phosphorylation at S132, also recovered the synthesis of SM in a transformant stably expressing CKIγ2. On the basis of these results, we conclude that the multiple phosphorylation in the SR motif by CKIγ2 triggers the inactivation of CERT, thereby down-regulating the synthesis of SM.
MATERIALS AND METHODS
Reagents
Lysenin was a gift from Dr. Sekizawa (Zenyaku Kogyo, Tokyo). l-[U-14C]Serine (160 mCi/mmol) and [methyl-14C]choline (55 mCi/mmol) were purchased from GE Healthcare (Little Chalfont, Buckinghamshire, United Kingdom) and [γ-32P]ATP (10 Ci/mmol) was from PerkinElmer Life and Analytical Sciences (Boston, MA). 6-[N-(7-nitrobenzo-2-oxa-1,3-diazol-4-yl)amino]caproyl-d-erythro-sphingosine (C6-NBD-Cer) and N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoyl)-d-erythro-sphingosine (C5-DMB-Cer) were from Invitrogen (Carlsbad, CA). BFA, fatty acid-free bovine serum albumin (BSA), and 3-(4,5-dimethylthiazoyl-2-yl)-2,5-diphenyltetrazolium bromide were from Sigma-Aldrich (St. Louis, MO). Thin layer chromatography (TLC) plates (Silica Gel 60) were from Merck (Darmstadt, Germany). λ-Phage protein phosphatase (λPPase) was from New England Biolabs (Ipswich, MA), and endoglycosidase H (Endo H) was from Roche Diagnostics (Basal, Switzerland). The antibodies against hemagglutinin (HA) (3F10), CKIγ2 (N-20), green fluorescent protein (GFP), α-tubulin, and CERT (COL4A3BP) were from Roche Diagnostics, Santa Cruz Biotechnology (Santa Cruz, CA), Invitrogen, Sigma-Aldrich, and Genway (San Diego, CA), respectively.
Cells and Cell Culture
The Chinese hamster ovary (CHO)-K1 cell line (ATCC CCL 61) was obtained from the American Type Culture Cell Collection (Manassas, VA). As parental CHO cells for retroviral infection, CHO-K1 cells were stably transfected with linearized pcDNA3.1 Hyg/mCAT-1 (mCAT1 encodes the mouse retroviral receptor; Albritton et al., 1989), and after the selection of hygromycin-resistant transfectants, a CHO/C cell line was purified by limiting dilution. The CHO cells were routinely maintained in F12/NCS medium (Ham's F-12 medium supplemented with 10% newborn calf serum [NCS], penicillin G [100 U/ml], and streptomycin sulfate [100 μg/ml]) in a 5% CO2 atmosphere at 100% humidity at 33°C. Nutridoma medium (Ham's F-12 medium containing 1% Nutridoma-SP [Roche Diagnostics] and 25 μg/ml gentamicin (Sigma-Aldrich) was used as a serum-free medium. The Flp-In T-REx 293 cell line (FT293) and the expression system containing the vectors for integration into the genome were purchased from Invitrogen. The FT293 cells were routinely maintained in DMEM supplemented with 5% Tet System Approved Fetal Bovine Serum (TF-FBS, BD), penicillin G (100 U/ml), and streptomycin sulfate (100 μg/ml) in a 5% CO2 atmosphere at 37°C.
Isolation of Lysenin-resistant Variants
CHO/C cells were infected with human cDNA-library packaged retroviral particles. The retroviral particles were prepared with a human HeLa retroviral library kit (Clontech, Mountain View, CA) and Plat-E packaging cells (Morita et al., 2000). The infected CHO/C cells (2.5 × 106) were seeded in 15 ml of F12/NCS medium in a 15-cm culture dish and cultured at 33°C overnight. The cells were washed with 10 ml of F-12/NCS medium twice and incubated at 37°C for 1 h in 20 ml of serum-free F-12 medium containing lysenin (25 ng/ml). After incubation, the cells were washed with 5 ml of F12/NCS medium twice and cultured in 20 ml of F12/NCS at 33°C until the surviving cells formed colonies (first screening). The propagated cells were reseeded in 10 ml of F12/NCS medium in a 10-cm dish and underwent a second cycle of lysenin treatment under the same conditions as described above. After three additional cycles of lysenin treatment, the cells that had formed colonies were harvested, and purified by limiting dilution. The cDNA fragments were recovered from the lysenin-resistant variants by genomic polymerase chain reaction (PCR) as described previously (Hanada et al., 2003). The amplified cDNAs were sequenced, and the sequences were subjected to a BLAST search.
Isolation of CHO/hCKIγ2
hCKIγ2's open reading frame (ORF) with its 5′ untranslated region was amplified by PCR (template, pMOPBlue/hCKIγ2; Kitabayashi et al., 1997); forward primer was EcoRI/5′hCKIg2, 5′-CAGAATTCGGCACGAGCAGCAGAATG-3′ and reverse primer was hCKIγ2/XhoI, 5′-GGCTCGAGTCACTTGTGTCGCTGCAGC-3′). The amplified DNA (∼1.5 kbp) was digested with EcoRI and XhoI and cloned into the EcoRI and XhoI sites of a pcDNA3.1 (+) vector (Invitrogen), and sequenced. Then, the cDNA was recloned into the EcoRI and XhoI sites of a pMXs-IP retroviral vector (Kitamura et al., 2003), and the resultant plasmid was named pMXs/5′UTRhCKIγ2-IP. CHO/C cells were infected with retroviral particles prepared from pMXs/5′UTRhCKIγ2-IP, and, after the selection of puromycin-resistant cells, a CHO/hCKIγ2 clone was purified by limiting dilution. As a control cell line, CHO/C cells were infected with retroviral particles prepared from the empty pMXs-IP, and a puromycin-resistant clone was isolated (this clone was named CHO/vector).
Construction of N-Terminally HA-Epitope-tagged Human CKIγ2
To tag hCKIγ2 with a HA epitope N-terminally, the hCKIγ2 ORF amplified by PCR with nHAhCKIγ2-Fw (5′-CCCAAGCTTTTATGGATTTTGACAAGAAAGGA-3′) and hCKIγ2/XhoI, was purified, digested by HindIII and XhoI, and inserted into the HindIII/XhoI sites of pBS-nHAcFL (Kawano et al., 2006), producing pBS/nHAhCKIγ2. The N-terminally HA-tagged hCKIγ2 fragment was transferred from the EcoRI/XhoI and BamHI/XhoI sites of pBS/nHAhCKIγ2 to the former sites of pcDNA3.1(+)neo to construct pcDNA3.1(+)neo/HAhCKIγ2 and the latter sites of pcDNA5 to construct pcDNA5/HAhCKIγ2, respectively.
Construction of CKIγ2 Kinase-Dead Mutant
A kinase-dead mutation (K75R) was inserted into hCKIγ2 by PCR with pBS/nHAhCKIγ2 as a template, and hCKIγ2K75R-Fw (5′-GTGGCTATCAGATTGGAGCCGATC-3′) and -Re (5′-CGGCTCCAATCTGATAGCCACGTA-3′). The absence of unwanted mutations in hCKIγ2 ORF was verified by DNA sequencing. The EcoRI-NcoI fragment in pBS/nHAhCKIγ2 was replaced with the corresponding region of the mutated HAhCKIγ2 sequence to construct pBS/nHAhCKIγ2K75R.
Construction of CERT Mutant
CERT DDD and the DDD S135A mutant were constructed as follows. Residues 130-132 in CERT were replaced with three aspartic acids (DDD) by PCR with pBS/HAhCERT as a template, and hCERT-DDD-Fw (5′-TCCAGCTTGCGTCGAGATGATGATATGGTGTCCCTGGTGTCT-3′) and -Re (5′-CACCAGGGACACCATATCATCATCTCGACGCAAGCTGGATTC-3′) as primers.
The EcoRI-MluI fragment in pBS/HAhCERT was replaced with the corresponding region of the mutated HAhCERT sequence to construct pBS/HAhCERT DDD. The mutation S135A was further inserted into HAhCERT DDD by PCR with pBS/HAhCERT DDD as a template, and DDD S135A-Fw (5′-GATGATATGGTGGCCCTGGTGTCTGGAGCA-3′) and -Re (5′-TCCAGACACCAGGGCCACCATATCATCATC-3′). The HAhCERT DDD or HAhCERT DDD S135A EcoRI-XhoI fragment was transferred to the same site of pET28a(+) or pMXs-IRES-Bsd to purify the recombinant protein from Escherichia coli or to prepare retroviral particles. The absence of unwanted mutations in hCERT sequences described above was verified by DNA sequencing.
Purification of Wild-Type and Kinase-Dead HAhCKIγ2 Protein from Mammalian Cells
FT293 cells expressing HAhCKIγ2 or HAhCKIγ2 K75R (FT293/HAhCKIγ2 or FT293/HAhCKIγ2 K75R cells) were obtained by selecting cells that showed resistance to hygromycin B (100–150 μg/ml) after the transfection of FT293 cells with pcDNA5/HAhCKIγ2 or HAhCKIγ2 K75R, respectively, and pOG44. For purification of the kinase, FT293/HAhCKIγ2 and FT293/HAhCKIγ2 K75R cells were propagated in 15-cm culture dishes to ∼70% confluence and, after the addition of tetracycline at a final concentration of 1 μg/ml, were cultured at 37°C for 4 h. Hereafter, all manipulations were carried out at 4°C or on ice. The cells were harvested with 9 ml of buffer A (40 mM Tris-HCl, pH 7.4, 180 mM NaCl, and 1 mM EDTA) by pipetting, washed with 2 ml of buffer A, and lysed with 2 ml of lysis buffer (50 mM HEPES-NaOH, pH7.5, 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail [Complete; Roche Diagnsotics]) containing 1% Triton X-100. After centrifugation of the lysate at 9100 × g for 10 min, the supernatant fraction was collected. The supernatant fraction was incubated with 150 μl (bed volume) of anti-HA agarose (Sigma-Aldrich) with rotation for 2 h. The kinase-bound HA agaroses was washed five times with 800 μl of lysis buffer containing 0.1% Triton X-100, washed three times with 800 μl of kinase buffer (30 mM HEPES-NaOH, pH7.5, and 7 mM MgCl2), suspended with 100 μl of kinase buffer, and stored at 4°C. The lysate from the cells that did not express HAhCKIγ2 proteins (FT293/pcDNA5) was used to prepare the control fraction, mock-bound anti-HA agarose.
In Vitro Kinase Assay
Recombinant wild-type and mutant CERTs expressed in E. coli cells were purified with a Talon Co2+ affinity column (Clontech) as described previously (Hanada et al., 2003), and the in vitro kinase assay was performed by a modification of previously described methods (Gietzen et al., 1999; Eide et al., 2005). The reactions were performed in 30 μl of kinase buffer containing 200 μM ATP, 1 mM dithiothreitol, 50 μg/ml BSA, 2 μCi of [γ-32P]ATP, 3.5 μg of purified CERT proteins, and 5 μl of the HAhCKIγ2-bound HA agarose suspension prepared as described above. When necessary, HAhCKIγ2 K75R-bound or mock-bound HA agarose was used in place of HAhCKIγ2-bound HA agarose. The reaction mixture was incubated for 30 min at 37°C, and then the reactions were stopped by adding 10 μl of 4× SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer. After SDS-PAGE, the gel was dried, and radioactive bands in the gel were analyzed with a BAS2500 image analyzer (Fujifilm, Tokyo, Japan).
Knockdown of Human CKIγ Isoforms
Small interfering (siRNA) sequences directed against hCKIγ2 (sense strand, 5′-GAAUACGUGGCUAUCAAAUUG-3′; antisense strand, 5′-AUUUGAUAGCCACGUAUUCAU-3′), hCKIγ1 (sense strand, 5′-GGUCGACAAGGCAAUAAGAAA-3′; antisense strand, 5′-UCUUAUUGCCUUGUCGACCAA-3′), and hCKIγ3 (sense strand, 5′-GGGCUACACCAAUAGAAGUGU-3′; antisense strand, 5′-ACUUCUAUUGGUGUAGCCCGU-3′) were designed, selected using the siDirect program at the website of RNAi (http://www.rnai.co.jp/; Hongo, Tokyo) and purchased from the company. A knockdown experiment was carried out as below. One day before transfection of siRNA, HeLa S3 cells were seeded in a six-well plate at a density of 7.5 × 104 cells in 2.5 ml of DMEM supplemented with 10% FBS without antibiotics and cultured at 37°C overnight. After the addition of 500 μl of Opti-MEM I medium (Invitrogen) containing 3 pmol of siRNA complexed with Lipofectamine RNAiMAX (Invitrogen), the transfected cells were further cultured at 37°C for 3 d. Cell lysate was prepared to investigate the effect of hCKIγ's knockdown on the phosphorylation of CERT by Western blotting. The efficiency of the knockdown was determined by real-time PCR.
Real-Time PCR
For real-time PCR, total RNA was isolated with TRIzol Reagent (Invitrogen) from HeLa S3 cells. cDNA was synthesized from 2 μg of total RNA by reverse transcriptase (ReverTra Ace; Toyobo Engineering, Osaka, Japan) and random hexamer (Roche Diagnostics) at 42°C for 50 min and at 95°C for 5 min. Real-time PCR was carried out with a LightCycler, LightCycler-FastStart DNA master SYBR Green I kit (Roche Diagnostics) and sets of primers, according to the manufacturer's protocol. The primers used in the real-time PCR are hCKIγ1RT-Fw (5′-AGCTGCTTTCTCGAATGGAA-3′) and -Re (5′-TGGTTTCGGGGTCAATGTAT-3′), hCKIγ2RT-Fw (5′-CTTCGAGAAGCCCGACTATG-3′) and -Re (5′-AGTTCAACGCCTGGTTTTTG-3′), hCKIγ3RT-Fw (5′-ACTGGGTCTTCATCGTCTGG-3′) and -Re (5′-TACCACAAGGGCCGAAATAG-3′), and hGAPDH_RT-Fw (5′-GAGTCAACGGATTTGGTCGT-3′) and -Re (5′-TTGATTTTGGAGGGATCTCG-3′).
Pulse-Chase Analysis of tsO45-VSVG-EGFP
CHO/vector and CHO/hCKIγ2 cells were transiently transfected with pCDM8.1/tsO45-VSVG-EGFP (Presley et al., 1997) and cultured at 37°C overnight. The cells were reseeded at a density of 2.5 × 105 cells in 5 ml of F12/NCS in 60-mm dishes, cultured at 37°C for 2 d, and further incubated at 40°C for 20 h to accumulate tsO45-VSVG-EGFP in the ER. tsO45-VSVG-EGFP exited the ER with the shift in temperature to 33°C and was chased at 33°C for various periods in 2 ml of F12/NCS supplemented with 200 μg/ml cycloheximide. After being washed with 2 ml of phosphate-buffered saline, cells were harvested in buffer A by scraping. After precipitation by centrifugation, collected cells were suspended in 200 μl of buffer B (10 mM HEPES-NaOH, pH 7.4, buffer containing 250 mM sucrose, 1 mM EDTA, and protease inhibitor cocktail [Complete; Roche Diagnostics]), and lysed by sonication. Ten micrograms of the cell lysate was diluted to 30 μl with buffer B and heated at 95°C for 5 min after the addition of 25 μl of 50 mM sodium acetate buffer, pH 5.5, containing 0.5% SDS, 1% 2-mercaptethanol, and 1 mM (p-amidinophenyl)methanesulfonyl fluoride hydrochloride (Wako Pure Chemicals, Osaka, Japan). The heated lysate (26 μl) was incubated with or without 4 mU of Endo H at 37°C for 16 h. The reaction was stopped by adding 10 μl of 4× SDS sample buffer, and the samples were subjected to SDS-PAGE and Western blotting with an anti-GFP antibody.
Other Methods
Viability of cells exposed to lysenin was determined as described previously (Hanada et al., 1998). Lipid analyses (metabolic labeling, intracellular redistribution of C5-DMB-ceramide, and SM synthase activity) were performed as described previously (Fukasawa et al., 1999; Yasuda et al., 2001). Immunofluorescence microscopy was conducted as described previously (Kawano et al., 2006), except for the following modifications: FT293 cells transfected with phCERT-GFP were grown on glass coverslips coated with poly-lysine for 72 h, and expression of HAhCKIγ2 was induced with 1 μg/ml tetracycline for 4 h before sample preparation. Protein concentrations were determined with a BCA protein assay kit (Pierce Chemical, Rockford, IL), by using bovine serum albumin as the standard.
RESULTS
Screening of Repressors of SM Synthesis
Lysenin is a SM-directed cytolysin (Yamaji et al., 1998). We attempted to isolate the genes whose overexpression conferred lysenin resistance to the cells, because such genes would play roles in down-regulation of the synthesis of SM. We retrovirally transfected CHO-K1 cells expressing the mouse retroviral receptor mCAT-1 (hereafter referred to as the CHO/C cell line) with a HeLa cell cDNA library and selected cell variants resistant to lysenin. From the lysenin-resistant variants with reduced synthesis of SM, cDNAs inserted into the genome were retrieved by genomic PCR. cDNAs encoding the γ2 isoform of human casein kinase I (hCKIγ2) were retrieved from cell clones of two independent screening batches, suggesting that ectopic expression of hCKIγ2 in CHO/C cells was responsible for the lysenin resistance.
Increased Dosage of hCKIγ2 Confers Lysenin Resistance to Cells and Down-Regulates the Synthesis of SM
When hCKIγ2 cDNA was introduced into cells by lipofection using multicopy plasmids, no transfectants were obtained because the transient overexpression of hCKIγ2 was toxic to the cells (including CHO-K1, HeLa S3 and HEK293 cells; data not shown). Nevertheless, the transfection of CHO/C cells with the retroviral vector carrying the open-reading frame of hCKIγ2 with its 5′ untranslated region yielded numerous stable transformants. For further analysis, one of the transformants was purified and named CHO/hCKIγ2. CHO/hCKIγ2 cells exhibited both resistance to lysenin and the down-regulation of SM synthesis (Figure 1, A and C). Western blotting showed that CHO/hCKIγ2 indeed produced hCKIγ2 protein (Figure 1B). These results indicate that the increased dosage of CKIγ2 confers lysenin resistance to the cells and down-regulates the synthesis of SM.
Figure 1.

Overexpression of hCKIγ2 causes resistance to lysenin and decreased synthesis of SM. (A) CHO/hCKIγ2 cells and the parental CHO/C cells were incubated with or without lysenin (25 ng/ml) at 37°C for 1 h. After the treatment, the viability of the cells was determined. Values are means ± SD from triplicated experiments. (B) Lysate prepared from CHO/C and CHO/hCKIγ2 cells was subjected to Western blotting with anti-CKIγ2 antibody. Positions of molecular-weight markers are shown at the left of the panel. (C and D) CHO cell monolayers were incubated with [14C]serine (C) or [3H]sphingosine (D) at 37°C for 2 h. Metabolically labeled lipids of the cells were separated by TLC and analyzed. Left, images of radioactive lipids separated on TLC plates. PS, phosphatidylserine; PE, phosphatidylethanolamine; GlcCer, glucosylceramide; Cer, ceramide; GM3, GM3 ganglioside. Right, means ± SD from triplicated experiments.
Conversion of Ceramide to SM Was Inhibited in CHO/hCKIγ2 Cells
In metabolic labeling with radioactive serine, the rate of de novo synthesis of SM in CHO/hCKIγ2 cells was decreased to ∼15% of that in the parental CHO/C cells and CHO/C cells harboring the empty retroviral vector (CHO/vector). CHO/hCKIγ2 cells exhibited a mild (∼30%) decrease in the labeling of GlcCer and phosphatidylserine (Figure 1C). When the consumption of ceramide is decreased, de novo sphingoid base synthesis seems to undergo a feedback repression, although the mechanism involved is unknown. Hence, we performed a metabolic labeling experiment with [3H]sphingosine to bypass the sphingoid base synthesis (Fukasawa et al., 1999; Hanada et al., 2003; Kawano et al., 2006). The synthesis of SM in CHO/hCKIγ2 cells was decreased to 50% of that in the control cells, whereas ceramide and glycosphingolipids were accumulated (Figure 1D). These results indicate that overexpression of hCKIγ2 decreases the synthesis of SM by inhibiting the conversion of ceramide to SM.
ER-to-Golgi Transport of Ceramide Was Inhibited in CHO/hCKIγ2 Cells
There was no significant difference in the synthesis of phosphatidylcholine, the donor of phosphocholine for SM, between CHO/hCKIγ2 cells and control CHO/C cells (Figure 2A). In addition, the activity of SM synthase was also similar between CHO/hCKIγ2 cells and control cells (Figure 2B). Thus, we examined the possibility that ceramide transport between the ER and the Golgi complex was inhibited in CHO/hCKIγ2 cells. For this, we performed metabolic labeling experiments with [14C]serine in the presence or absence of BFA. The BFA treatment recovered the synthesis of SM in CHO/hCKIγ2 cells to the wild-type level (Figure 2C). We next investigated the movement of C5-DMB-Cer, a fluorescent analogue of ceramide, from the ER to the Golgi complex (Fukasawa et al., 1999; Hanada et al., 2003). In control cells (CHO/C, CHO/vector cells), C5-DMB-Cer was initially distributed to the ER and then moved to the perinuclear Golgi region after the chase. Such ER-to-Golgi movement of C5-DMB-Cer was retarded in CHO/hCKIγ2 cells as well as in LY-A cells, which have a loss-of-function mutation in the CERT gene (Hanada et al., 2003; Figure 2D). These results indicate that the decreased synthesis of SM in CHO/hCKIγ2 cells results from an inhibition of the transport of ceramide from the ER to the Golgi complex.
To examine whether the ER-to-Golgi transport of protein was affected by overproduction of CKIγ2, we performed a pulse-chase analysis of tsO45-VSVG-EGFP protein. The tsO45-VSVG-EGFP accumulated in the ER at a nonpermissive temperature was released from the ER by shifting to the permissive temperature and chased in the presence of the protein synthesis inhibitor cycloheximide. The Endo H-resistance acquisition rate of tsO45-VSVG-GFP was identical between CHO/hCKIγ2 and the parental cells (Figure 2E), indicating that protein transport from the ER to the Golgi complex was not affected by the expression of hCKIγ2.
CERT Is Predominantly Hyperphosphorylated in CHO/hCKIγ2 Cells
CERT is phosphorylated multiple times in the SR motif, which contains a possible sequence recognized by CKI (Kumagai et al., 2007). When the SR motif is hyperphosphorylated, the function of CERT is repressed (Kumagai et al., 2007). Hence, we examined the possibility that expression of hCKIγ2 resulted in hyperphosphorylation of CERT, thereby inhibiting ER-to-Golgi trafficking of ceramide. To this end, after the transfection of CHO/hCKIγ2 cells and parental CHO/C cells with HA-tagged human CERT (HAhCERT), the apparent molecular mass of HAhCERT was examined by Western blotting with an anti-HA-epitope antibody. In CHO/C cells, HAhCERT was detected as ∼70-kDa doublet bands consisting of hyper- and hypophosphorylated forms (Figure 3, lane 1), in agreement with previous results (Kumagai et al., 2007). By contrast, only the hyperphosphorylated form of HAhCERT was detected in CHO/hCKIγ2 cells (Figure 3, lane 3). Treatment of the cell lysate with protein phosphatase changed these bands to those corresponding to the dephosphorylated form (Figure 3, lanes 2 and 4), confirming that HAhCERT in CHO/hCKIγ2 cells is hyperphosphorylated.
Figure 3.

CERT is hyperphosphorylated in CHO/hCKIγ2 cells. CHO/vector and CHO/hCKIγ2 cells were transfected with HA-tagged hCERT. Lysate prepared from the cells was treated with (lanes 2 and 4) or without λPPase (lanes 1 and 3) and subjected to Western blotting with anti-HA antibody (top) and anti-CKIγ2 antibody (bottom). State of CERT is indicated at the left of the figure.
Kinase Activity of hCKIγ2 Is Required for Down-Regulation of SM Synthesis in CHO/hCKIγ2 Cells
When the conserved lysine75 in the nucleotide-binding motif of CKIγ2 is replaced with arginine, the kinase activity is lost (Davidson et al., 2005). To examine the requirement of kinase activity of hCKIγ2 for down-regulation of the synthesis of SM, we established a cell line (CHO/HAhCKIγ2 K75R) that stably expressed the N-terminally HA-tagged hCKIγ2 K75R mutant, a kinase-dead mutant. The rate of de novo SM synthesis was similar between CHO/HAhCKIγ2 K75R cells and CHO/vector cells, and the pattern of phosphorylation of the endogenous CERT was also similar between the two cell types. In CHO/HAhCKIγ2 cells, most of the endogenous CERT was hyperphosphorylated, and the rate of SM synthesis was lower than the control level, although the expression level of HAhCKIγ2 protein was less than that of HAhCKIγ2 K75R (Figure 4). These results indicate that kinase activity of hCKIγ2 is required for the hyperphosphorylation of CERT and the down-regulation of SM synthesis in CHO/HAhCKIγ2 cells.
Figure 4.
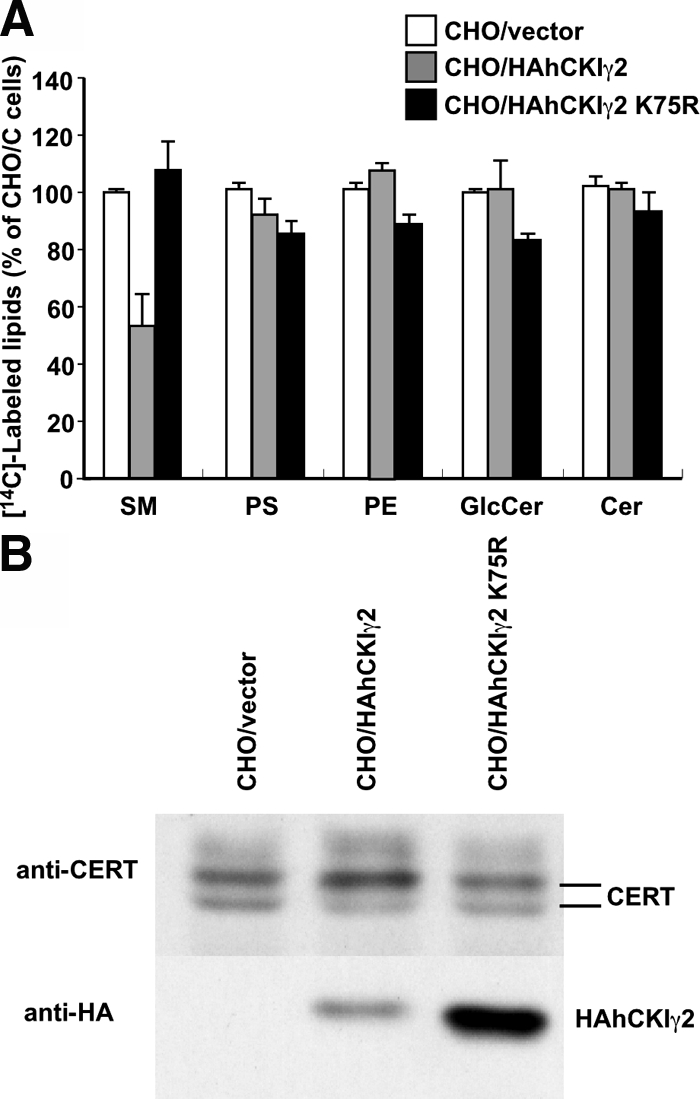
Kinase activity is required for the decreased synthesis of SM and hyperphosphorylation of CERT. (A) CHO cells were incubated with [14C]serine at 37°C for 2 h. Metabolically labeled lipids were analyzed by TLC. Results shown are means ± SD from triplicated experiments. (B) Lysate was prepared from the indicated cells and subjected to Western blotting with anti-CERT and anti-HA antibodies.
CERT Mutant That Is Not Phosphorylated in the SR Motif Restored SM Synthesis in CHO/hCKIγ2 Cells
The typical recognition sequence of CKI is pSer/pThr (or Glu/Asp cluster)-X1-2-Ser/Thr (pSer and pThr are phosphorylated serine and threonine, respectively. The underlined Ser or Thr is to be phosphorylated) (Knippschild et al., 2005). The region that matches the consensus sequence is located in the SR motif of CERT (Figure 5; A; Kumagai et al., 2007). We previously constructed the mutant CERT S132A that does not undergo phosphorylation in the SR motif. CERT S132A was active in the ER-to-Golgi transport of ceramide in a semi-intact cell assay (Kumagai et al., 2007). S132 of CERT is phosphorylated by PKD (Fugmann et al., 2007). When HAhCERT S132A was stably introduced, this mutant protein was phosphorylated in neither wild-type cells nor CHO/hCKIγ2 cells (Figure 5B, lanes 5 and 6), and this hyperphosphorylation-deficient mutant completely restored the synthesis of SM in CHO/hCKIγ2 cells, whereas the introduction of wild-type CERT did not (Figure 5C). These results indicate that the repression of SM synthesis by CKIγ2 is due to the hyperphosphorylation of CERT.
Figure 5.
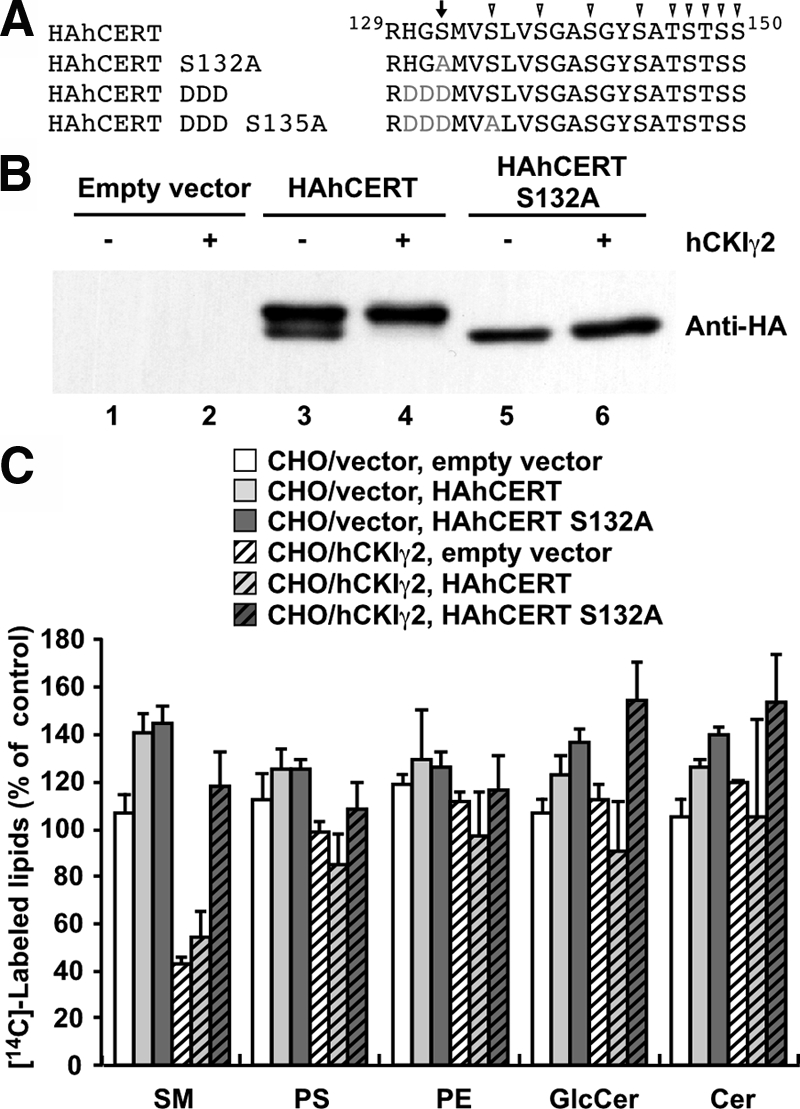
CERT mutant that is not phosphorylated by CKIγ2 restores SM synthesis in CHO/hCKIγ2 cells. (A) Sequence alignment of the SR motif in wild-type CERT and the S132A, DDD, and DDD S135A mutants. The arrow and arrowhead indicate serine/threonine residues phosphorylated by PKD and possibly CKI, respectively. Gray letters indicate the replaced residues in the CERT mutants. (B) Lysate was prepared from the CHO/vector and CHO/hCKIγ2 cells stably transfected with an empty vector (lanes 1 and 2), HAhCERT (lanes 3 and 4), and HAhCERT S132A (lanes 5 and 6) and subjected to Western blotting with anti-HA antibody. (C) The CHO/vector and CHO/hCKIγ2 cells stably transfected with an empty vector, HAhCERT, and HAhCERT S132A were incubated with [14C]serine at 37°C for 2 h. The labeled lipids were extracted and analyzed. Results shown are means ± SD from triplicated experiments. The levels of labeled lipids in CHO/vector cells transfected with the empty vector are set to 100%.
CKIγ2 Phosphorylates Purified CERT Protein in Vitro
We next investigated whether CKIγ2 could directly phosphorylate CERT in a cell-free assay system. To bypass the priming phosphorylation of CERT by PKD, we constructed the CERT S132D, DD, and DDD mutants, in which S132, S132 and G131, and S132 to H130 were replaced with single, double, and triple asparagine(s), respectively (Figure 5A and Supplemental Figure S1A), because CKI also recognizes efficiently a cluster of acidic amino acid or a single acidic amino acids N-terminal of the target serine/threonine (Knippschild et al., 2005). Among them, HAhCERT DDD mutant was most efficiently phosphorylated (Supplemental Figure S1B) and occurred only as a hyperphosphorylated form like a wild-type HAhCERT in CHO/hCKIγ2 cells (Figure 6A, lanes 3 and 7, and Supplemental Figure S1B). Treatment of lysate with a protein phosphatase changed these bands to a dephosphorylated form (Figure 6A, lanes 2, 4, 6, and 8). Thus, the acidic cluster of CERT DDD could be substituted for the phospho-S132 for recognition by CKI, and we used this mutant in the cell-free assay. When recombinant CERT proteins purified from bacteria were incubated with HAhCKIγ2 protein purified from human embryonic kidney (HEK)293 cells in the presence of [γ-32P]ATP, radioactive phosphate was efficiently incorporated into HAhCERT DDD, but not HAhCERT wild-type or S132A proteins (Figure 6B). Purified HAhCKIγ2 K75R, a kinase-dead mutant of CKIγ2, did not phosphorylate HAhCERT DDD in vitro (data not shown). These results indicate that CERT is a substrate of CKIγ2.
Figure 6.

CKIγ2 phosphorylates CERT in vivo and in vitro. (A) The CERT DDD mutant can be phosphosphorylated in CHO/hCKIγ2 cells. Lysate was prepared from the CHO/vector and CHO/hCKIγ2 cells stably transfected with HAhCERT (lanes 1–4) or HAhCERT DDD (lanes 5–8), treated with (even numbered lanes) or without λPPase (odd numbered lanes), and subjected to Western blotting with anti-HA antibody. (B) In vitro kinase assay. Recombinant CERT proteins were incubated with immuno-purified HAhCKIγ2 protein in the presence of [γ-32P]ATP at 37°C for 30 min as described under Materials and methods. After SDS-PAGE, labeled CERT proteins were visualized by image analysis. Positions of molecular standards are shown at the left of the panel. Twenty-five percent of the substrate solution used in the reaction was subjected to Western blotting with anti-HA antibody, and results are shown in the bottom panel.
Effect of Knockdown of CKIγ2 on Phosphorylation of CERT
The CKIγ2 protein has two closely related isoforms, CKIγ1 and γ3, that differ from other members of the CKI family because of a putative palmitoylation site in the C terminus (Davidson et al., 2005). We carried out a knockdown experiment to examine which γ isoform(s) is responsible for the phosphorylation of the endogenous CERT at SR motif. HeLa S3 cells were transfected with each siRNA directed against the γ isoforms of human CKI. The state of the endogenous CERT was analyzed by Western blotting. Unfortunately, the commercially available anti-CKIγ2 antibodies were not able to detect the endogenous CKIγ2. Thus, the knockdown efficiency was determined by real-time PCR. The each siRNA knocked down the target mRNA by ∼80% without any change in mRNA levels of the other isoforms (Figure 7B). The knockdown of γ2 isoform markedly increased the ratio of hypo- to hyperphosphorylated CERT (Figure 7, A and B). The simultaneous knockdown of all three γ isoforms showed an additional increase in the ratio, whereas the knockdown of γ1, γ3, and both increased it slightly (Figure 7, A and B). These results indicate that the γ2 isoform plays a major role in the hyperphosphorylation of SR motif of CERT in HeLa S3 cells.
Figure 7.

Effect of knockdown of CKIγ isoforms on the phosphorylation of CERT. (A) HeLa S3 cells were transfected with or without each siRNA against hCKIγ isoforms and the indicated combination and cultured at 37°C for 3 d. Lysate was prepared from the transfected cells and subjected to Western blotting with anti-CERT antibody and anti-α-tubulin antibody. (B) Levels of mRNA of CKIγ isoforms were determined by real-time PCR in A. Results shown are means ± SD from triplicated experiments. Bars indicate the amount of mRNA as a percentage of that in mock-transfected cells.
Hyperphosphorylated CERT Is Dissociated from the Golgi Complex
Next, we examined the subcellular localization of CKIγ2 and CERT proteins by using a HEK293 cell derivative, in which HA-tagged hCKIγ2 expression is inducible. In the absence of hCKIγ2 overexpression, hCERT-GFP was distributed throughout the cytoplasm with a substantial amount concentrated in the perinuclear region where the Golgi marker GS28 colocalized (Figure 8A), consistent with our previous study (Hanada et al., 2003). When HAhCKIγ2 was expressed, it was localized to the peripheral region of the cell (Figure 8C), whereas hyperphosphorylated hCERT-GFP (Figure 8D, lanes 2 and 4) distributed throughout the cytoplasm without accumulating in the Golgi region (Figure 8B). These results indicate that upon hyperphosphorylation, CERT is dissociated from the Golgi complex.
Figure 8.

Hyperphosphorylation dissociates CERT from the Golgi complex. (A–C) Intracellular distribution of the GFP-fused form of CERT. FT293/pcDNA5 and FT293/HAhCKIγ2 cells were transfected with the expression plasmid encoding hCERT-GFP, and incubated with 1 μg/ml tetracycline for 4 h. Then, the cells were processed for indirect immunostaining for GS28 or HA-epitope. Left, hCERT-GFP. Middle, GS28 (A and B) and HAhCKIγ2 (C). Right, merged images. Bar, 10 μm. (D) Phosphorylation state of hCERT-GFP. Lysate from tetracycline-induced FT293/pcDNA5 and FT293/HAhCKIγ2 cells were treated with or without λPPase. Then, the samples were analyzed by Western blotting with anti-GFP antibody and anti-HA antibody. Positions of molecular standard markers are shown at the left of the panel.
Multiple Phosphorylation within the SR Motif by CKIγ2 Rather than Priming Phosphorylation at S132 Down-Regulates the Function of CERT
Fugmann et al. (2007) have recently shown that the phosphorylation of S132 in CERT by PKD decreases the affinity for PI4P and the ceramide-transfer activity of CERT, whereas we have reported that multiple phosphorylation in the SR motif inactivates both activities. Therefore, we attempted to determine the net effect of the priming phosphorylation on CERT function. To this end, we constructed a CERT DDD S135A mutant (Figure 5A), because the acidic cluster of CERT DDD could be substituted for phospho-S132 for recognition by CKI in vivo and in vitro (Figure 6 and Supplemental Figure S1). In CHO/hCKIγ2 cells, the DDD S135A was detected as a hypophosphorylated form (Figure 9B, lane 7). These results indicate that this mutant can mimic the form of CERT that is phosphorylated only at S132 in the SR motif. In a [14C]serine-labeling experiment, the DDD S135A mutant completely restored the synthesis of SM in CHO/hCKIγ2 cells like the S132A mutant, whereas wild-type CERT and the DDD mutant that occurred in a hyperphosphorylated form did not (Figure 9, A and B, lanes 3 and 11). These results suggest that the CERT phosphorylated only at S132 retains its activity and that the activity is lost after multiple S/Ts within the SR motif are phosphorylated by CKIγ2.
Figure 9.

Multiple phosphorylation at the SR motif by CKIγ2 was necessary to down-regulate the activity of CERT. (A) The CHO/vector cells stably transfected with an empty vector or CHO/hCKIγ2 cells stably transfected with an empty vector, HAhCERT, HAhCERT S132A, HAhCERT DDD S135A, or HAhCERT DDD were incubated with [14C]serine at 37°C for 2 h. The labeled lipids were extracted and analyzed. Results shown are means ± SD from three experiments. The level of labeled SM in CHO/vector cells transfected with the empty vector is set to 100%. (B) Lysate was prepared from the CHO/vector and CHO/hCKIγ2 cells stably transfected with HAhCERT (lanes 1–4), HAhCERT DDD S135A (lanes 5–8), or HAhCERT DDD (lanes 9–12), treated with (even numbered lanes) or without (odd numbered lanes) λPPase, and subjected to Western blotting with anti-HA antibody.
DISCUSSION
In this study, we showed that CKIγ2 hyperphosphorylates the SR motif of CERT, thereby down-regulating the function of CERT. By analyzing the CKIγ2-stable transformant, we found that overexpression of CKIγ2 causes inhibition of the conversion of ceramide to SM without any change in the activity of SM synthase (Figures 1–2B). This inhibition was accompanied by a delay in the ER-to-Golgi movement of C5-DMB-ceramide (Figure 2D). These results led us to investigate the possibility that CERT was inactivated in CHO/hCKIγ2 cells. Consistent with the presence of the consensus sequence for CKI in the SR motif, CERT was indeed hyperphosphorylated in CHO/hCKIγ2 cells (Figure 3). S132 in the SR motif of CERT receives priming phosphorylation by PKD (Fugmann et al., 2007). We showed previously that hyperphosphorylation at the SR motif down-regulates both the PI4P-binding activity and the ceramide transfer activity of CERT (Kumagai et al., 2007). The CERT S132A mutant was not phosphorylated even in CHO/hCKIγ2 cells (Figure 5B) and fully restored the synthesis of SM in these cells (Figure 5C). These results indicate that if S132 is phosphorylated, CKIγ2 hyperphosphorylates the SR motif and that the reduction of SM synthesis in CHO/hCKIγ2 cells results from the hyperphosphorylation of CERT by CKIγ2. Purified CKIγ2 phosphorylated the CERT DDD mutant in vitro (Figure 6). Of three γ isoforms, only the knockdown of CKIγ2 largely increased the ratio of the hypo- to hyperphosphorylated form of CERT in HeLa S3 cells (Figure 7). In previous proteome-scale mapping of human binary protein–protein interactions, the yeast two-hybrid system detected the interaction of CKIγ2 with CERT (Rual et al., 2005). Collectively, we conclude that CERT is a substrate of CKIγ2 and that hyperphosphorylation by CKIγ2 inactivates CERT, thereby down-regulating the synthesis of SM.
We showed here that expression and knockdown of CKIγ2 decreased and increased the ratio of hypo- to hyperphosphorylated CERT, respectively. In addition, expression of CKIγ2 reduced the synthesis of SM, whereas knockdown of endogenous CKIγ2 failed to show the increased synthesis of SM because the knockdown significantly caused cell death. This may reflect its important roles in the regulation of various cellular processes (Davidson et al., 2005; Knippschild et al., 2005; Guo et al., 2008). Instead, we showed that expression of CERT S132A enhances the synthesis of SM in CHO/vector cells (Figure 5, B and C). As shown in the Figure 5, expression of wild-type CERT also enhanced the synthesis of SM, resulting from a nearly equal amount of hypophosphorylated CERT between wild type and S132A. Together with the effect of γ2 knockdown on phosphorylation of CERT (Figure 7), these results suggest the functional consequence of CKIγ2 in the biosynthetic pathway of SM.
Fugmann et al. (2007) have recently shown that the phosphorylation of S132 in CERT by PKD decreases the affinity of CERT for PI4P and reduces ceramide-transfer activity. Because the CERT S132A mutant and wild-type CERT purified from mammalian cells were compared in the report, most of the wild-type CERT seemed to be hyperphosphorylated by CKI. Thus, the net effect of the phosphorylation of S132 by PKD on the affinity for PI4P and ceramide-transfer activity of CERT remained unclear. The apparent molecular mass of the hypophosphorylated form of wild-type CERT seems to be slightly larger than that of the S132A mutant (Figure 5B), suggesting that S132 of wild-type CERT is constitutively phosphorylated in cells. Fugmann et al. (2007) obtained similar results. Our previous analysis showing that most of the phosphopeptides that covered the SR motif possessed a monophosphate group in hypophosphorylated CERT (Kumagai et al., 2007) also supports this suggestion. That a part of wild-type CERT is localized to the Golgi suggests that CERT phosphorylated at only S132 in SR motif retains at least the affinity for PI4P. Consistent with these suggestions, expression of the DDD S135A mutant, but not the DDD mutant, restored the synthesis of SM in CHO/CKIγ2 cells (Figure 9A). On the basis of these results, we conclude that CKIγ2 is the kinase that reduces the affinity for PI4P and the ceramide-transfer activity of CERT through multiple phosphorylations in the SR motif.
CKIγ2 was localized to the PM, consistent with it having C-terminal palmitoylation site (Figure 8C). Human N-ras and H-ras, and the yeast CKI homologue Yck2p are palmitoylated at the Golgi during transport to the PM through exocytic pathways (Apolloni et al., 2000; Babu et al., 2002). Moreover, a de/repalmitoylation cycle regulates the localization and activity of H-, N-ras proteins (Rocks et al., 2005). By analogy, CKIγ2 may phosphorylate CERT on the Golgi membrane during the cycling of CKIγ2 between the Golgi and PM. Palmitoylated proteins are segregated on detergent-resistant membranes (Webb et al., 2000) and palmitoylation may move the protein into cholesterol-rich domains of the membrane (Greaves and Chamberlain, 2007). When SM/cholesterol rafts are disrupted by methyl-β-cyclodextrin or sphingomyelinase treatment, dephosphorylation of the SR motif in CERT is induced (Kumagai et al., 2007). Such treatments may affect the association with rafts and de/repalmitoylation of CKIγ2.
CKIγ2 belongs to a family of monomeric serine/threonine protein kinases that include at least seven isoforms in mammals. Kinase domains are highly conserved within the family. The members further fall into subfamilies that differ significantly in the length and primary sequence of their N- and C-terminal extensions (Price, 2006). Inhibitory autophosphorylation of CKIδ and ε isoforms had been reported previously (Graves and Roach, 1995; Gietzen and Virshup, 1999); however, the regulation of CKIγ isoforms is largely unknown. CKI phosphorylates many different substrates involved in cell differentiation, proliferation, chromosomal segregation, and circadian rhythms (Knippschild et al., 2005), and viral nonstructural proteins (Eichwald et al., 2004; Quintavalle et al., 2006). We showed here that CKIγ2 phosphorylates CERT, a factor involved in sphingolipid metabolism. The regulation of SM/diacylglycerol synthesis in Golgi has recently attracted much attention as a link between lipid homeostasis and membrane transport (Baron and Malhotra, 2002; Hausser et al., 2005; Litvak et al., 2005; Toth et al., 2006; Fugmann et al., 2007). The spatial and temporal regulation of CKIγ2 is a subject for future study.
Supplementary Material
ACKNOWLEDGMENTS
We thank Y. Sekizawa (Zenyaku Kogyo, Tokyo, Japan) for providing lysenin, T. Kitamura (University of Tokyo) for the pMXs series, and J. Lippincott-Schwartz (National Institutes of Health) for the tsO45-VSVG-EGFP–expressing plasmid. This work was supported in part by the Ministry of Education, Culture, Sports, Science and Technology of Japan, by the Japan Health Sciences Foundation, by CREST of JST, and by Toray Science Foundation.
Abbreviations used:
- BFA
brefeldin A
- C5-DMB-ceramide
N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoyl)sphingosine
- CHO
Chinese hamster ovary
- CKI
casein kinase I
- Endo H
endoglycosidase H
- PKD
protein kinase D
- PH
pleckstrin homology
- PI4P
phosphatidylinositol 4-monophosphate
- λPPase
λ-phage protein phosphatase
- SM
sphingomyelin
- SR
serine repeat.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-07-0669) on November 12, 2008.
REFERENCES
- Albritton L. M., Tseng L., Scadden D., Cunningham J. M. A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell. 1989;57:659–666. doi: 10.1016/0092-8674(89)90134-7. [DOI] [PubMed] [Google Scholar]
- Apolloni A., Prior I. A., Lindsay M., Parton R. G., Hancock J. F. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol. Cell. Biol. 2000;20:2475–2487. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu P., Bryan J. D., Panek H. R., Jordan S. L., Forbrich B. M., Kelley S. C., Colvin R. T., Robinson L. C. Plasma membrane localization of the Yck2p yeast casein kinase 1 isoform requires the C-terminal extension and secretory pathway function. J. Cell Sci. 2002;115:4957–4968. doi: 10.1242/jcs.00203. [DOI] [PubMed] [Google Scholar]
- Baron C. L., Malhotra V. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science. 2002;295:325–328. doi: 10.1126/science.1066759. [DOI] [PubMed] [Google Scholar]
- Davidson G., Wu W., Shen J., Bilic J., Fenger U., Stannek P., Glinka A., Niehrs C. Casein kinase 1 γ couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867–872. doi: 10.1038/nature04170. [DOI] [PubMed] [Google Scholar]
- Eichwald C., Jacob G., Muszynski B., Allende J. E., Burrone O. R. Uncoupling substrate and activation functions of rotavirus NSP 5, phosphorylation of Ser-67 by casein kinase 1 is essential for hyperphosphorylation. Proc. Natl. Acad. Sci. USA. 2004;101:16304–16309. doi: 10.1073/pnas.0406691101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide E. J., Kang H., Crapo S., Gallego M., Virshup D. M. Casein kinase I in the mammalian circadian clock. Methods Enzymol. 2005;393:408–418. doi: 10.1016/S0076-6879(05)93019-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugmann T., Hausser A., Schoffler P., Schmid S., Pfizenmaier K., Olayioye M. A. Regulation of secretory transport by protein kinase D-mediated phosphorylation of the ceramide transfer protein. J. Cell Biol. 2007;178:15–22. doi: 10.1083/jcb.200612017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa M., Nishijima M., Hanada K. Genetic evidence for ATP-dependent endoplasmic reticulum-to-Golgi apparatus trafficking of ceramide for sphingomyelin synthesis in Chinese hamster ovary cells. J. Cell Biol. 1999;144:673–685. doi: 10.1083/jcb.144.4.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietzen K. F., Virshup D. M. Identification of inhibitory autophosphorylation sites in casein kinase I ε. J. Biol. Chem. 1999;274:32063–32070. doi: 10.1074/jbc.274.45.32063. [DOI] [PubMed] [Google Scholar]
- Graves P. R., Roach P. J. Role of COOH-terminal phosphorylation in the regulation of casein kinase I delta. J. Biol. Chem. 1995;270:21689–21694. doi: 10.1074/jbc.270.37.21689. [DOI] [PubMed] [Google Scholar]
- Greaves J., Chamberlain L. H. Palmitoylation-dependent protein sorting. J. Cell Biol. 2007;176:249–254. doi: 10.1083/jcb.200610151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Waddell D. S., Wang W., Wang Z., Liberati N. T., Yong S., Liu X., Wang X.-F. Ligand-dependent ubiquitination of Smad3 is regulated by casein kinase 1 gamma 2, an inhibitor of TGF-β signaling. Oncogene. 2008 doi: 10.1038/onc.2008.337. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K., Hara T., Fukasawa M., Yamaji A., Umeda M., Nishijima M. Mammalian cell mutants resistant to a sphingomyelin-directed cytolysin. Genetic and biochemical evidence for complex formation of the LCB1 protein with the LCB2 protein for serine palmitoyltransferase. J. Biol. Chem. 1998;273:33787–33794. doi: 10.1074/jbc.273.50.33787. [DOI] [PubMed] [Google Scholar]
- Hanada K., Kumagai K., Yasuda S., Miura Y., Kawano M., Fukasawa M., Nishijima M. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- Hanada K., Kumagai K., Tomishige N., Kawano M. CERT and intracellular trafficking of ceramide. Biochim. Biophys. Acta. 2007;1771:644–653. doi: 10.1016/j.bbalip.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Hausser A., Storz P., Martens S., Link G., Toker A., Pfizenmaier K. Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIβ at the Golgi complex. Nat. Cell Biol. 2005;7:880–886. doi: 10.1038/ncb1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis J. C., Levine T. P. Lipid traffic: floppy drives and a superhighway. Nat. Rev. Mol. Cell Biol. 2005;6:209–220. doi: 10.1038/nrm1591. [DOI] [PubMed] [Google Scholar]
- Huitema K., van den Dikkenberg J., Brouwers J. F., Holthuis J. C. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004;23:33–44. doi: 10.1038/sj.emboj.7600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M., Kumagai K., Nishijima M., Hanada K. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 2006;281:30279–30288. doi: 10.1074/jbc.M605032200. [DOI] [PubMed] [Google Scholar]
- Kitabayashi A. N., Kusuda J., Hirai M., Hashimoto K. Cloning and chromosomal mapping of human casein kinase I gamma 2 (CSNK1G2) Genomics. 1997;46:133–137. doi: 10.1006/geno.1997.4991. [DOI] [PubMed] [Google Scholar]
- Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 2003;31:1007–1014. [PubMed] [Google Scholar]
- Knippschild U., Gocht A., Wolff S., Huber N., Lohler J., Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17:675–689. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Kumagai K., Kawano M., Shinkai-Ouchi F., Nishijima M., Hanada K. Interorganelle trafficking of ceramide is regulated by phosphorylation-dependent cooperativity between the PH and START domains of CERT. J. Biol. Chem. 2007;282:17758–17766. doi: 10.1074/jbc.M702291200. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Donaldson J. G., Schweizer A., Berger E. G., Hauri H. P., Yuan L. C., Klausner R. D. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- Litvak V., Dahan N., Ramachandran S., Sabanay H., Lev S. Maintenance of the diacylglycerol level in the Golgi apparatus by the Nir2 protein is critical for Golgi secretory function. Nat. Cell Biol. 2005;7:225–234. doi: 10.1038/ncb1221. [DOI] [PubMed] [Google Scholar]
- Loewen C. J., Roy A., Levine T. P. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 2003;22:2025–2035. doi: 10.1093/emboj/cdg201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogelsvang S., Marsh B. J., Ladinsky M. S., Howell K. E. Predicting function from structure: 3D structure studies of the mammalian Golgi complex. Traffic. 2004;5:338–345. doi: 10.1111/j.1398-9219.2004.00186.x. [DOI] [PubMed] [Google Scholar]
- Morita S., Kojima T., Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- Presley J. F., Cole N. B., Schroer T. A., Hirschberg K., Zaal K. J., Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Price M. A. CKI, there's more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 2006;20:399–410. doi: 10.1101/gad.1394306. [DOI] [PubMed] [Google Scholar]
- Quintavalle M., Sambucini S., Di Pietro C., De Francesco R., Neddermann P. The α isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J. Virol. 2006;80:11305–11312. doi: 10.1128/JVI.01465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks O., Peyker A., Kahms M., Verveer P. J., Koerner C., Lumbierres M., Kuhlmann J., Waldmann H., Wittinghofer A., Bastiaens P. I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- Rual J. F., et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Saito S., Matsui H., Kawano M., Kumagai K., Tomishige N., Hanada K., Echigo S., Tamura S., Kobayashi T. Protein phosphatase 2Cepsilon is an endoplasmic reticulum integral membrane protein that dephosphorylates the ceramide transport protein CERT to enhance its association with organelle membranes. J. Biol. Chem. 2008;283:6584–6593. doi: 10.1074/jbc.M707691200. [DOI] [PubMed] [Google Scholar]
- Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Toth B., Balla A., Ma H., Knight Z. A., Shokat K. M., Balla T. Phosphatidylinositol 4-kinase IIIbeta regulates the transport of ceramide between the endoplasmic reticulum and Golgi. J. Biol. Chem. 2006;281:36369–36377. doi: 10.1074/jbc.M604935200. [DOI] [PubMed] [Google Scholar]
- Voeltz G. K., Rolls M. M., Rapoport T. A. Structural organization of the endoplasmic reticulum. EMBO Rep. 2002;3:944–950. doi: 10.1093/embo-reports/kvf202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb Y., Hermida-Matsumoto L., Resh M. D. Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J. Biol. Chem. 2000;275:261–270. doi: 10.1074/jbc.275.1.261. [DOI] [PubMed] [Google Scholar]
- Yamaji A., Sekizawa Y., Emoto K., Sakuraba H., Inoue K., Kobayashi H., Umeda M. Lysenin, a novel sphingomyelin-specific binding protein. J. Biol. Chem. 1998;273:5300–5306. doi: 10.1074/jbc.273.9.5300. [DOI] [PubMed] [Google Scholar]
- Yamaoka S., Miyagi M., Kitano T., Umehara H., Okazaki T. Expression cloning of a human cDNA restoring sphingomyelin synthase-defective lymphoid cells. J. Biol. Chem. 2004;279:18688–18693. doi: 10.1074/jbc.M401205200. [DOI] [PubMed] [Google Scholar]
- Yasuda S., Kitagawa H., Ueno M., Ishitani H., Fukasawa M., Nishijima M., Kobayashi S., Hanada K. A novel inhibitor of ceramide trafficking from the endoplasmic reticulum to the site of sphingomyelin synthesis. J. Biol. Chem. 2001;276:43994–44002. doi: 10.1074/jbc.M104884200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.