

Abstract
To better understand the mechanism controlling nonmuscle myosin II (NM-II) assembly in mammalian cells, mutant NM-IIA constructs were created to allow tests in live cells of two widely studied models for filament assembly control. A GFP-NM-IIA construct lacking the RLC binding domain (ΔIQ2) destabilizes the 10S sequestered monomer state and results in a severe defect in recycling monomers during spreading, and from the posterior to the leading edge during polarized migration. A GFP-NM-IIA construct lacking the nonhelical tailpiece (Δtailpiece) is competent for leading edge assembly, but overassembles, suggesting defects in disassembly from lamellae subsequent to initial recruitment. The Δtailpiece phenotype was recapitulated by a GFP-NM-IIA construct carrying a mutation in a mapped tailpiece phosphorylation site (S1943A), validating the importance of the tailpiece and tailpiece phosphorylation in normal lamellar myosin II assembly control. These results demonstrate that both the 6S/10S conformational change and the tailpiece contribute to the localization and assembly of myosin II in mammalian cells. This work furthermore offers cellular insights that help explain platelet and leukocyte defects associated with R1933-stop alleles of patients afflicted with human MYH9-related disorder.
INTRODUCTION
The aim of our studies is to characterize the mechanism controlling nonmuscle myosin II (NM-II) filament turnover. NM-II isoforms play critical roles in many cellular processes during growth and development, ranging from stabilization of cell polarity, to cell migration, to cell division (Conti and Adelstein, 2008). NM-II filaments are formed by the lateral association of the tails of NM-II “monomers,” which consist of two myosin heavy chains (MHCs), two regulatory light chains (RLC), and two essential light chains (ELCs). The MHC has an amino-terminal globular motor domain that contains an actin-binding site and an ATP-binding site. The carboxyl-terminal portion of the globular head includes two sequential IQ motifs, one that binds the ELC and the other that binds the RLC. The tails of two MHCs interact to form an extended α-helical coiled coil domain. Finally, mammalian isoforms of NM-II also have a carboxyl-terminal nonhelical tailpiece.
One proposed mechanism governing filament assembly involves inhibition of intermolecular lateral tail associations by folding the NM-II monomer tail. Mammalian NM-II isoforms have been shown in vitro to form a 10S hairpin in which the tail folds over and interacts with the RLC of the myosin head in a sequestered state (Trybus et al., 1982;Olney et al., 1996;Salzameda et al., 2006;Burgess et al., 2007). Early studies documented a critical role for this RLC interaction in stabilizing the folded state. For example, Trybus and Lowey (1988) showed that myosin stripped of its RLC was unable to form the sequestered 10S hairpin state, an effect that was reversible upon readdition of RLC. Later work by Ikebe et al. (1994) showed via viscosity measurements that the 10S form of myosin could be abolished by deleting the 16 NH2-terminal residues of the RLC. Furthermore, cross-linking studies have identified the specific residues of the RLC that bind to the MHC in the 10S form (Olney et al., 1996;Salzameda et al., 2006). Taken together these biochemical studies demonstrate that the RLC is critical for folding into the 10S hairpin in vitro. It is therefore widely believed that in live mammalian cells NM-II assembly is controlled via sequestration of the NM-II monomer in the 10S conformation, which unfolds into the assembly-competent 6S form via phosphorylation of the RLC by myosin light-chain kinase (MLCK) or possibly other kinases such as Rho kinase (Craig et al., 1983; Trybus and Lowey, 1984, 1988).
In addition to having a functional role in the 6S–10S transition, the RLC also has a regulatory role in NM-II motor activation. Association of the RLC with MHC inhibits the actin-activated ATPase activity of smooth muscle myosin, and RLC phosphorylation relieves that inhibition (Onishi and Watanabe, 1979; Seidel, 1980; Adelstein et al., 1981). The role of unphosphorylated RLC as an inhibitor of motor activity was also substantiated by studies in Dictyostelium discoideum, where a mutant MHC construct was created that lacked the 30-aa RLC-binding “IQ” motif. This Dictyostelium ΔIQ2-myosin II was found to be fully functional both in vitro and in vivo, with the notable feature that it displays high level constitutive actin-activated ATPase activity and RLC-independent actin filament translocation activity (Uyeda and Spudich, 1993). The construct fully complemented all cellular defects of MHC null cells, including cytokinesis defects and multicellular development. Although these studies validate the concept that similar mutations will yield a functional NM-II in mammalian settings, Dictyostelium NM-II is not believed to undergo a 6S–10S transition, so the amoeba system cannot provide insight into understanding of the roles of 6S–10S transitions for in vivo control of mammalian NM-II assembly.
Another category of possible filament assembly regulatory mechanisms centers on the short nonhelical tailpiece of NM-II isoforms. For example, biochemical and live cell studies have indicated a role for this and adjacent regions as a mediator of NM-IIA assembly control involving binding of the small calcium-binding protein S100A4 (Li and Bresnick, 2006), and via phosphorylation (Straussman et al., 2001; Dulyaninova et al., 2007). The importance of carboxyl-terminal rod and tailpiece-based regulation is further suggested by the collection of human disease syndromes that can result from mutations in this region of the human NMHC-IIA gene (MYH9). Patients with MYH9-related disorders present at an early age with excessively large platelets, low platelet counts, clotting disorders, and leukocyte inclusions (Seri et al., 2003). Approximately 75% of MYH9-related disorders are caused by mutations in the rod of the NM-II tail, and 88% of these mutations occur at only four residues: 1165, 1424, 1841, and 1933 (Franke et al., 2005). Analysis of in vitro paracrystal formation by recombinant tail fragments carrying these rod mutations showed that the mutant tail fragments dominantly interfere with wild-type paracrystal assemblies, causing the formation of amorphous lattices (Franke et al., 2005). How these abnormal assemblies, through their effect on NM-II filament formation in living cells, cause MYH9-related disorders remains unclear. Of particular interest is the mutation at aa 1933, which deletes 28 of the 33-aa nonhelical tailpiece, suggesting an important but undefined role for the nonhelical tailpiece in filament assembly.
To gain further insights into the relative roles of different models of NM-II assembly control and into the importance of the nonhelical tailpiece that is deleted in some MYH9-related disorders, we have constructed a series of NM-II mutants as green fluorescent protein (GFP) fusions. Behavior of these constructs has been analyzed by immunofluorescent microscopy, biochemical fractionation, and fluorescent recovery after photobleaching (FRAP) to evaluate effects on localization and assembly dynamics during lamellar spreading and polarized migration. Our results demonstrate that both the 6S–10S transition and assembly control mediated by the carboxyl-terminal tailpiece contribute, in distinct manners, to the regulation of myosin II assembly in live cells.
MATERIALS AND METHODS
DNA Mutagenesis
The plasmid pEGFP-NMHC-IIA-C3 expresses the full-length corrected human nonmuscle myosin heavy chain under the control of a constitutive cytomegalovirus (CMV) promoter, with enhanced GFP (EGFP) fused at the amino terminus of the NMHC-IIA coding region. This plasmid, a gift from Dr. Anne Bresnick (Albert Einstein College of Medicine, Bronx, NY) (Wei and Adelstein, 2000;Bao et al., 2005;Dulyaninova et al., 2007), was used to express wild-type GFP-NMHC-IIA. Mutant deletion constructs were generated via either PCR-based methods or via deletion using existing internal restriction sites. All DNA segments subjected to PCR were sequenced to confirm absence of PCR-generated errors. pCMV-eGFP-NMHC-IIAΔIQ2 has the same structure, except that the coding segment for aa 804-834 (corresponding to the IQ2 motif that binds RLC) was deleted with the deletion junction marked by an engineered NheI restriction site. pCMV-eGFP-NMHC-IIAΔtailpiece has the same amino-terminal GFP fusion to NMHC-IIA, with a stop codon introduced after codon 1928, eliminating the last 33 amino acids (corresponding to the nonhelical tailpiece). The pCMV-eGFP-NMHC-IIAΔACD has the same structure as NMHC-IIA except that it was digested with SacII and SpeI and rendered blunt via incubation with Klenow fragment, replacing the final 270 amino acids of the tail after codon 1691 (containing the ACD domain) with the sequence LVRPIHRI. The pCMV-eGFP-NMHC-IIAΔIQ2ΔACD has the same structure as the NMHC-IIAΔACD, except it has also had the IQ2 motif removed as described above. The construction of the pCMV-eGFP-NMHC-IIA-S1943A has been described (Dulyaninova et al., 2007).
Cell Culture and Transfections
Human cervical cancer cells (HeLa) were obtained from American Type Culture Collection (Manassas, VA) and maintained in minimal essential media (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 1% l-glutamine, and 1% penicillin/streptomycin. Cells were maintained as monolayer cultures at 37°C, 5% CO2. For live cell imaging, cells were kept in phenol red–free minimal essential media (Invitrogen) supplemented with 10% fetal bovine serum, 1% l-glutamine, and 1% penicillin/streptomycin and 25 mM HEPES. All transfections were carried out by nucleofection (Amaxa Biosystems, Gaithersburg, MD) according to manufacturer's instructions.
Immunostaining
Cells were fixed in PBS supplemented with 2 mM MgCl2, 2 mM EGTA, and 4% formaldehyde. Cells were then permeabilized in PBS containing 0.5% Triton X-100 and actin filaments stained using Alexa Fluor 568 phalloidin (Invitrogen). Nuclei were stained using DAPI (Invitrogen).
Microscopy
All images were acquired using a Plan-Apochromat 63×, 1.4 NA oil objective (Zeiss, Thornwood, NY) on an Axiovert 200M inverted microscope equipped with a confocal LSM 510 META scan head (Zeiss).
Immunoprecipitation and Western Blotting
HeLa cells were transfected with pCMV-eGFP-MHC-IIA or pCMV-eGFP-MHC-IIAΔIQ2, and 48 h after transfection the cells were trypsinized, pelleted, washed, and lysed with an NP-40 lysis buffer (50 mM Tris-HCl, 500 mM NaCl, 5 mM MgCl2, 2 mM ATP, 0.1% NP-40, 10% glycerol, 5 mM EDTA, 5 mM DTT, 1 mM PMSF, 2% PIC I, 2% PIC II (protease inhibitor cocktail I and II, as described previously; Steimle et al., 2001). Insoluble proteins were removed by centrifugation at 51,000 × g. After immunoprecipitation with 20 μl of agarose beads conjugated with monoclonal anti-GFP (MBL International, San Diego, CA; www.mblintil.com), Western blots were performed with a polyclonal rabbit antibody specific to the RLC (Proteintech Group, Chicago, IL; Cat. No. 10324-1-AP). Western blots in the supplemental figure were probed with a polyclonal antibody specific to the total MHC (Biomedical Technologies, Stoughton, MA; BT-561), a polyclonal antibody specific to the nonhelical tailpiece of the NMHC-IIA (Sigma, St. Louis, MO; M8064), or a polyclonal antibody specific to GFP (Invitrogen Molecular Probes, Eugene, OR; A6455).
Salt-dependent Assembly
HeLa cells were transfected with pCMV-eGFP-MHC-IIA or pCMV-eGFP-MHC-IIAΔIQ2, and 48 h after transfection the cells were lysed in situ with cold Triton X-100 lysis buffer (50 mM Tris-HCl, pH 7.4, 5 mM NaCl, 560 mM potassium-acetate, 0.6% Triton X-100, 1 mM EDTA, 1 mM EGTA, 5 mM DTT, 1 mM PMSF, 1% PIC I, and 1% PIC II). To preclear insoluble proteins, 3 mM MgCl2, and 1 mM ATP were added to the lysate, and the lysate was separated by centrifugation at 7800 × g for 15 min at 4°C. The supernatant was then divided in two with each half being diluted four times the original volume in modified lysis buffer (no Triton X-100), one-half being diluted to 400 mM potassium-acetate, and the other half diluted to 140 mM potassium-acetate. Soluble and insoluble proteins were then separated by centrifugation at 51,000 × g for 20 min at 4°C. The pellet was suspended in 2× Laemmli sample buffer. The supernatant was diluted to 70% acetone, the protein in the supernatant was separated by centrifugation at 7800 × g for 15 min, and the pellet was suspended in 2× Laemmli sample buffer. Equivalent portions of pellet and supernatants were subjected to Western blot analysis and probed for GFP.
Triton-insoluble Fractionation
HeLa cells were transfected with GFP-myosin constructs and plated on tissue culture treated plastic Petri dishes. After 48 h cells were lysed in situ with ice-cold Triton X-100 lysis buffer (50 mM Tris-HCl, pH 7.4, 5 mM NaCl, 140 mM potassium-acetate, 0.6% Triton X-100, 1 mM EDTA, 5 mM EGTA, 5 mM DTT, 1 mM PMSF, 1% PIC I, and 1% PIC II). After a 5-min incubation, the soluble and insoluble fractions of the lysate were separated by centrifugation at 7800 × g for 15 min. The supernatant and pellet were resuspended in equal volumes of 2× Laemmli sample buffer. Western blots were probed with rabbit polyclonal antibodies to GFP (Molecular Probes; www.invitrogen.com) and densitometry was performed using Image J version 1.38 (http://rsb.info.nih.gov/ij/).
Radial Intensity Analysis
HeLa cells transfected with a GFP-myosin construct were trypsinized briefly 48 h after transfection and plated at low density on borosilicate chambered coverglass (Nalge Nunc International, Naperville, IL) that had been incubated with 20 μg/ml fibronectin for 1 h. After 1 h at 37°C, cells were fixed and immunostained as described above. Fluorescent images were analyzed using self written analysis scripts in MATLAB (The Mathworks, Natick, MA), which segmented the image to find the cell shape from the actin-stained image. This cell mask was then used to create an index of all pixels equidistant from the cell edge in order to average the intensity of all equidistant pixels from the cell edge for the red (actin) and green (GFP-myosin) images. For plotting, the equidistant average intensities were normalized to the highest average, and the distance was normalized to the longest distance from the cell edge to the cell center, with 0% being the cell edge and 100% being the cell center. The average central cell body intensity was determined by averaging the myosin radial intensity curve between 80 and 92% from the cell edge. The curves are normalized to the highest average intensity, which for NM-IIA and NM-IIAΔtailpiece correspond to the lamella. To get a similar measurement for NM-IIAΔIQ2 we took the average of the normalized myosin radial intensity curve from 80 to 92% from the cell edge and normalized it to the myosin intensity value that corresponded to the actin peak at the lamella.
Polarized Cell Migration
HeLa cells transfected with a GFP-myosin construct were trypsinized 48 h after transfection and plated in a double-blind manner at high density on borosilicate chambered coverglass (Nalge Nunc International) that had been incubated with 20 μg/ml fibronectin for 1 h and had a 2-mm-wide strip of polydimethylsiloxane (PDMS) placed across the middle. After incubation at 37°C for 16 h, the PDMS strip was removed, creating a well-defined wound margin. The cells were allowed to polarize and migrate for 4 h, at which point they were fixed and immunostained as described above. A handmade binary mask made in ImageJ was used to isolate a single cell in each Z-projected confocal image for analysis by self-written scripts in MATLAB. The centroid of cell shape and GFP intensity was determined and a vector was drawn from the center of cell shape to the center of myosin intensity. The length of the vector was then normalized to the perimeter of cell shape.
FRAP
HeLa cells transfected with a GFP-myosin construct were trypsinized briefly 48 h after transfection and plated at low density on a Bioptechs (Butler, PA) Delta T dish that had been incubated with 20 μg/ml fibronectin for 1 h. The cells were maintained at 37°C using a Bioptech Delta T4 Open Culture Dish MicroEnvironmental Control System and a Bioptech objective heater system. FRAP experiments were performed on actively spreading cells between 15 and 60 min after plating. FRAP experiments of full-length myosin mutants were carried out according to Yumura et al. (2005) with time intervals of 4–5 s and a circular bleach spot 2.86 μm in diameter.
For FRAP of ACD constructs, images were collected with an interval of 200 ms and the half-times of recovery were calculated as described (Feder et al., 1996), with α held constant at 0.73 as published previously (Weiss et al., 2004).
RESULTS
To better understand the governing mechanism controlling NM-II filament assembly in mammalian cells, a series of NMHC-IIA mutants were constructed as GFP-fusion proteins (Figure 1). GFP-NMHC-IIAΔIQ2 (Figure 1B) deletes the RLC-binding site and is thus predicted to be incapable of forming a stable 10S hairpin. GFP-NMHC-IIAΔtailpiece (Figure 1C) deletes the 33-aa nonhelical carboxyl-terminal segment implicated in control of assembly. GFP-NMHC-IIAΔS1943A substitutes an alanine for a previously mapped casein kinase II (CK II) phosphorylation site. GFP-NMHC-IIAΔACD (Figure 1D) removes ∼30 kDa of the tail, including the assembly competence domain (ACD) that is essential for filament assembly (Lee et al., 1994;Shoffner and De Lozanne, 1996;Sohn et al., 1997;Nakasawa et al., 2005).
Figure 1.

GFP fusion constructs used to assess myosin IIA assembly models. (A) Wild-type NM-II. (B) The NM-II with the IQ2 region deleted, preventing RLC binding. (C) The NM-II with the nonhelical tailpiece deleted. (D) The NM-II with the assembly competence domain (ACD) deleted. The ACD is required for assembly of the NM-II monomer into bipolar filaments. (E) The NM-II with the ACD deleted and the IQ2 region deleted.
During initial characterization of the mutant constructs after transfection into HeLa cells, flow cytometry–based cell sorting was used to collect populations of cells with typical levels of GFP brightness from transiently transfected populations. These subpopulations were subjected to Western blot analysis revealing that all constructs were expressed at ∼10–20% of the level of the endogenous NMHC-IIA (Supplemental Figure S1).
Deletion of the IQ2 Motif Prevents RLC Binding and Destabilizes the 10S Hairpin
The GFP-NMHC-IIAΔIQ2 construct was designed to eliminate RLC binding to the myosin II holoenzyme. After immunoprecipitation with an antibody to GFP, immunoblots readily detected RLC associated with GFP-NM-IIA but not GFP-NM-IIAΔIQ2 (Figure 2A, bottom panel). This result verifies that deletion of the 30-aa “IQ2” motif from the head of NM-IIA eliminates all detectable RLC binding.
Figure 2.

GFP-NM-IIAΔIQ2ΔACD displays reduced diffusional rates in live cells consistent with destabilization of the 10S folded state. (A) Western blots of immunoprecipitations depicting RLC association with each NM-II. (B) Images depict representative cells transfected with indicated assembly incompetent myosin construct. The live cell diffusional characteristics of each myosin was then measured via FRAP analysis. Plots show average recovery for all experiments (n = 90) plotted in solid black with ±SD shown in dashed gray. Deleting the RLC-binding site increased the half-time of recovery 1.23-fold compared with GFP-NM-IIAΔACD. GFP-NM-IIAΔACD had a half-time of recovery of 0.96 ± 0.45 s. GFP-NM-IIAΔIQ2ΔACD had a significantly slower half-time of recovery of 1.18 ± 0.53 s (p = 0.003). Average half times ± SD. p values were determined by two-tailed t test. Scale bars, 10 μm.
As described in the Introduction, any NM-II lacking an RLC is predicted to be unable to stabilize the folded 10S conformation in vivo. One major aspect of the 6S–10S model is that the 10S species diffuses faster than the 6S species because of its compact shape (Trybus et al., 1982; Trybus and Lowey, 1984; Trybus and Lowey, 1988; Kolega and Taylor, 1993). We therefore tested for diffusional differences between assembly incompetent versions of the GFP-NM-IIA and GFP-NM-IIAΔIQ2 in live cells. These ΔACD constructs still retain the tail regions required to cross-link to the RLC in the 10S hairpin. However, by eliminating the ACD of each construct, we were able to analyze diffusional behavior in live cells in a manner uncomplicated by the presence of an assembled filament pool (Olney et al., 1996;Nakasawa et al., 2005;Salzameda et al., 2006). The GFP-NM-IIAΔACD (Figure 1D) is predicted to still undergo the 6S–10S transition, whereas the GFP-NM-IIAΔIQ2ΔACD (Figure 1E) is predicted to be unable to stabilize the 10S hairpin.
The diffusional properties of GFP-NM-IIAΔACD and GFP-NM-IIAΔIQ2ΔACD were measured via FRAP analysis. Figure 2B shows example cells and the average recovery curves from all experiments (n = 90). GFP-NM-IIAΔIQ2ΔACD had a half-time of recovery of 1.18 ± 0.53 s, which was significantly slower (p = 0.003) than that of GFP-NM-IIAΔACD (0.96 ± 0.45 s). The GFP-NM-IIAΔIQ2ΔACD had a half-time of recovery 1.23-fold greater than GFP-NM-IIAΔACD, which is within expectations for a myosin whose tail has been reduced by 25% and supports the model that elimination of RLC from the myosin II holoenzyme, via the IQ2 deletion, destabilizes the 10S hairpin conformation in live cells.
As a further test of the biochemical properties of GFP-NM-IIA and GFP-NM-IIAΔIQ2 we evaluated assembly behavior in cell lysates that were precleared via ultracentrifugation to remove F-actin and other membrane/cytoskeletal complexes. Previously it has been shown that high-salt concentrations (400 mM) force myosin into the 6S monomeric state by destabilizing myosin filaments and by destabilizing the 10S conformation. At low-salt concentrations (140 mM) wild-type myosin monomers adopt the 10S conformation, preventing the formation of filaments. In our cleared lysates, both wild-type and ΔIQ2 proteins were soluble in 400 mM K-acetate, whereas at 140 mM K-acetate the GFP-NM-IIAΔIQ2 construct displayed dramatically greater assembly than did GFP-NM-IIA (Figure 3). This behavior supports the conclusion that the ΔIQ2 construct cannot form a stable 10S monomer and recapitulates the key observations of Trybus and Lowey (1988) that formed the original evidence that RLC stabilizes the 10S folded state.
Figure 3.

In high-speed cleared cell lysates, GFP-NM-IIAΔIQ2 displayed dramatically greater assembly than did GFP-NM-IIA at 140 mM salt. Essentially all of the GFP-NM-IIA is soluble at 140 mM and at 400 mM salt. However, at 140 mM salt a substantial portion of the GFP-NM-IIAΔIQ2 is assembled and pellets. However, this effect was eliminated at 400 mM salt. This result strongly suggests that the ΔIQ2 mutation causes the myosin to remain filamentous by destabilizing the 10S folded species. Western blot was probed with a GFP antibody.
The Tailpiece and IQ2 Deletions Bias Myosin toward Cytoskeletal Assembly
Although the distribution of wild-type GFP-NM-IIA, GFP-NM-IIAΔtailpiece, and GFP-NM-IIAΔIQ2 along actin stress fibers appeared similar in undisturbed cultured cells (Figure 4), these three proteins had different solubilities in a Triton X-100–insoluble cytoskeletal ghost assay (Figure 5A). Approximately 40% of wild-type GFP-NM-IIA was associated with the Triton-insoluble fraction compared with 52% of GFP-NM-IIAΔtailpiece, and 88% of GFP-NM-IIAΔIQ2 (Figure 5B). These experiments clearly demonstrate that both the Δtailpiece and ΔIQ2 mutations can bias NM-IIA toward greater cytoskeletal association, with the ΔIQ2 mutation causing a more dramatic effect.
Figure 4.

The NM-IIAΔIQ2 and NM-IIAΔtailpiece recombinant proteins can assemble into stress fibers in undisturbed cultured cells. Cells were transiently transfected with GFP-NMHC-IIA constructs and stained with Alexa Fluor 568 phalloidin (actin) and DAPI (DNA stain). Scale bars, 10 μm.
Figure 5.
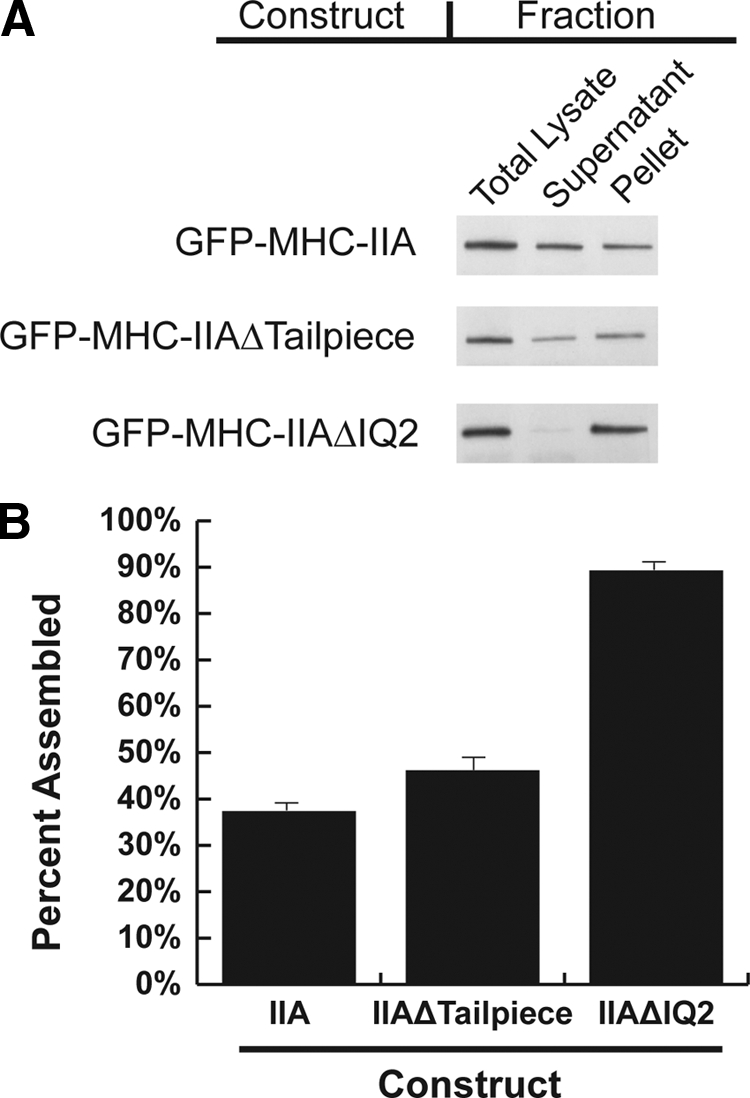
Both NM-IIAΔIQ2 and NM-IIAΔtailpiece display over-assembly in a cytoskeletal ghost assay. (A) Western blots of Triton-insoluble fractions of cells transfected with the indicated construct probed with an anti-GFP antibody. Equivalent fractions were loaded based on cell concentration. (B) Densitometry was performed on blots from seven to eight separate experiments. Error bars, SEs; n = 7 or 8.
The IQ2 Deletion Prevents Normal Localization of NM-IIA during Cell Spreading
Shortly after plating, cells project a lamella uniformly in all directions. The molecular machinery involved in cell spreading is the same machinery involved in leading edge protrusion (Giannone et al., 2004;Dobereiner et al., 2004). Therefore, cell spreading provides a controlled, standardized assay in which to examine NM-IIA localization and assembly dynamics involved in leading-edge protrusion during cell migration. To improve upon the use of linescans to quantify cortical localization and assembly of GFP-tagged myosin constructs, we developed radial-intensity analysis in which all pixels equidistant from the cell edge are averaged and the average is plotted as a function of distance from the cell edge. We performed radial-intensity analysis to analyze the localization, assembly, and effect on the actin cytoskeleton of GFP-NM-IIA, GFP-NM-IIAΔtailpiece, and GFP-NM-IIAΔIQ2 during the initial phase of spreading on fibronectin.
The average intensities of GFP-NM-IIA and GFP-NM-IIAΔtailpiece were at a minimum at the extreme cell edge (lamellipodia) followed by a peak of intensity in the lamella just behind the cell edge, and then the intensity drops off within the central cell body zone (Figure 6, A and B). The average GFP-myosin intensity for both wild-type and Δtailpiece correlates strongly with the actin intensities from each region, as plotted from the Alexa Fluor 568 phalloidin staining (red channel in Figure 6, and as quantified in Table 1). In contrast, GFP-NM-IIAΔIQ2 exhibited localization radically different from the wild type (Figure 6C and Table 1). Although GFP-NM-IIAΔIQ2 cells still displayed strong marginal F-actin localization in these actively spreading cells, the GFP-NM-IIAΔIQ2 protein itself failed to localize efficiently to the spreading margin and instead displayed strong stress fiber localization predominantly in the central cell body zone.
Figure 6.

Altered lamellar localization and assembly of mutant GFP-NM-IIA constructs. Actively spreading cells transfected with the indicated construct were fixed and stained for actin. The pixel intensity values for all equidistant pixels from the cell boundary were averaged for both the myosin image and actin image. Average pixel intensity was then plotted as a function of distance from the cell edge. Distances were normalized to the maximum distance from the cell edge. Therefore, 0% on the x-axis is the cell edge, and 100% is the maximum distance from the cell edge. Images and plots from a typical cell are shown for each construct. For pooled data, see Table 1. Scale bars, 10 μm. Relative to the wild-type GFP-NM-IIA construct (A), the Δtailpiece construct (B), and S1943A construct (D), are recruited to the spreading lamella and display marginal over-assembly relative to the cell center (Table 1). In contrast, GFP-NM-IIAΔIQ2 (C) displayed severe over-assembly in the cell center, and was excluded from the spreading margin.
Table 1.
Both Δtailpiece and ΔIQ2 mutations cause defects in localization and assembly
| Mutant | Correlation coefficient | Average central myosin |
|---|---|---|
| GFP-NMHC-IIA (n = 13) | 0.90 ± 0.02 | 35 ± 4 |
| GFP-NMHC-IIAΔtailpiece (n = 13) | 0.93 ± 0.01 | 18 ± 2* |
| GFP-NMHC-IIA-S1943A (n = 13) | 0.93 ± 0.01 | 19 ± 2* |
| GFP-NMHC-IIAΔIQ2 (n = 11) | −0.06 ± 0.10 | 180 ± 19* |
Distribution of transfected GFP-myosin constructs and actin was determined via radial analysis. The correlation coefficient for the resulting GFP-myosin and actin intensity curves was calculated for each cell, and the averages from all cells ± SE are shown. A correlation coefficient of 1 indicates perfect correlation, 0 indicates no correlation, and −1 indicates anti-correlation. Because the GFP-myosin values were normalized to the maximum average intensity, the average central GFP-myosin intensity is a relative measure of lamellar assembly of myosin; lower average central myosin intensity indicates stronger assembly at the lamella, and higher average central myosin intensity indicates weaker assembly at the lamella. GFP-IIAΔtailpiece, GFP-NMHC-IIA-S1943A had significantly stronger assembly at the lamella (Δtailpiece, p = 0.0021; S1943A, p = 0.004). The GFP-IIAΔIQ2 was significantly different from wild type with p = 0.00000007. Asterisks indicate values significantly different from wild type. Significance determined by a two-tailed t test.
Radial analysis also allowed comparison and quantitation of the relative assembly levels of each construct at the lamella versus central cell body during spreading. Using this comparison, the average central cell body intensity of GFP-NM-IIA from all experiments is 35 ± 4% of its lamellar maximum (Table 1, values ± SE), whereas the central cell body intensity of GFP-NM-IIAΔtailpiece from all experiments is 18 ± 2% of its lamellar maximum. The fact that the normalized central cell body intensity is consistently lower for cells expressing the GFP-NM-IIAΔtailpiece construct indicates that this protein is more highly assembled at the lamellar margin than is the corresponding wild-type construct. In contrast, GFP-NM-IIAΔIQ2 displayed severe over-assembly in the central cell body, with an average intensity 180 ± 19% of its lamellar intensity.
In mammalian systems, tailpiece phosphorylation has been shown to modulate filament turnover in EGF-stimulated cancer cells (Dulyaninova et al., 2005, 2007). NM-IIA has only one mapped phosphorylation site on its nonhelical tailpiece (Murakami et al., 1998). To determine whether complete tailpiece deletion produced equivalent effects on filament behavior as inhibition of tailpiece phosphorylation, we used radial analysis to assess assembly of an unphosphorylatable NM-IIA (GFP-NM-IIA-S1943A) during cell spreading. Notably, the GFP-NM-IIA-S1943A behaved indistinguishably from GFP-NM-IIAΔtailpiece in this assay (Figure 6D and Table 1). This result argues that a main role of the nonhelical tailpiece in assembly regulation may be as a phosphorylation site and additionally that pathology of R1933stop MYH9-related disorder patients may in fact be attributable to loss of phosphorylation of the tailpiece.
The IQ2 Deletion Prevents Recycling of Assembled NM-IIA during Polarized Cell Migration
During polarized cell migration the cytoskeleton undergoes a retrograde flow in which cytoskeletal proteins assemble at the front of the migrating cell and then flow toward the rear of the cell where they are disassembled and recycled back to the leading edge (Bray and White, 1988). During migration of HeLa cells into a wound the centroid of GFP intensity in cells expressing wild-type NM-IIA or NM-IIAΔtailpiece both matched closely to the geometric center of the cell, although on average the wild-type distribution was biased slightly forward relative to the geometric centroid, whereas the Δtailpiece was biased slightly rearward (Figure 7, A and B). In contrast, the centroid of GFP-NM-IIAΔIQ2 intensity displayed a dramatic shift toward the rear of the cell, suggesting that this protein was unable to recycle back to the front of the migrating cell (Figure 7C). This result supports the hypothesis that ability to form the sequestered 10S hairpin is critical for normal trafficking of the myosin monomer within the cell during polarized migration.
Figure 7.

NM-IIAΔIQ2 displays a severe defect in recycling to the leading edge of polarized migrating cells. Cells transfected with the indicated myosin construct (green) were fixed during polarized migration and stained for actin (red). Individual cells from images of the wound margin were outlined by hand and their centroid of GFP intensity relative to the center of cell geometry was determined. The left panels show a typical field of cells with a traced cell (outlined in white) used for analysis. Scale bars, 10 μm. The right panels show compass plots with vectors calculated from all experiments. On the compass plots, 90° is toward the wound, the radial axis is percent of cell perimeter and zero is the center of cell geometry. (A) GFP-NM-IIA exhibits a close localization to the center of cell shape, with a mild forward bias (0.85 ± 0.15% at 27°, n = 11). (B) GFP-NM-IIAΔtailpiece exhibited a mild, but consistent bias behind the geometrical centroid (0.51 ± 0.11% at 318° for IIAΔtailpiece, n = 12), suggesting a possible assembly/disassembly control defect. (C) GFP-NM-IIAΔIQ2 exhibited a pronounced and consistent rearward shift (3.41 ± 0.53% at 284°, n = 11).
Deletion of the IQ2 Motif Accelerates NM-IIA Assembly Turnover Rates as Assessed by FRAP
FRAP analysis at the margin of actively spreading cells (Figure 8 and Table 2, values ± SDs) showed that wild-type GFP-NM-IIA exchanged with the monomer pool with a t1/2 of 125 ± 74 s (Figure 8A and Table 2), a rate statistically indistinguishable from GFP-NM-IIAΔtailpiece (t1/2 = 130 ± 100 s). Deleting the tailpiece did significantly increase the immobile fraction of the myosin to 50 ± 22% (p = 0.04) compared with 41 ± 21% for wild type (Table 2), indicating increased stability of the assembled pool for the Δtailpiece construct. GFP-NM-IIAΔIQ2 displayed an even larger increase in the immobile fraction (65 ± 12%; p = 0.0000007), consistent with the strong over-assembly reported by Triton-insolubility and radial analysis. Surprisingly, GFP-NM-IIAΔIQ2 had a faster turnover rate than wild-type (t1/2 of 90 ± 41 s, p = 0.015, Figure 8C). A model to explain these behaviors is presented in the Discussion.
Figure 8.

FRAP dynamics at the lamellar margin of spreading cells reveals assembly defects for both NM-IIAΔIQ2 and NM-IIAΔtailpiece. FRAP was performed on GFP-myosin assembled at the lamella of actively spreading cells transfected with the indicated construct. Images depict examples of a typical cell transfected with the indicated construct with its associated recovery curve. Scale bars, 10 μm. The Δtailpiece had a similar recovery to wild type, whereas the ΔIQ2 actually had faster halftime (Table 2). Both Δtailpiece and ΔIQ2 had larger immobile fractions compared with wild type, suggesting that they both are biased toward an assembled state.
Table 2.
GFP-NM-IIAΔIQ2 displays faster half-time of recovery in live cells
| Construct | Half-time (s) | Immobile fraction (%) |
|---|---|---|
| GFP-NMHC-IIA (n = 44) | 125 ± 74 | 41 ± 21 |
| GFP-NMHC-IIAΔtailpiece (n = 49) | 130 ± 100 | 50 ± 22* |
| GFP-NMHC-IIAΔIQ2 (n = 33) | 90 ± 41* | 65 ± 12* |
FRAP was performed on GFP-myosin assembled at the lamella of actively spreading cells transfected with the indicated construct. Average half-time of recoveries and immobile fractions are shown ± SD. The immobile fraction is GFP-myosin that did not exchange with the monomer pool during the course of the experiment and is therefore a metric for over assembly. Both Δtailpiece and ΔIQ2 mutants significantly over-assemble compared with wild type, indicated by their increased immobile fractions (Δtailpiece p = 0.04; ΔIQ2 p = 0.0000007). Surprisingly, the ΔIQ mutant had a faster half-time of recovery compared with wild type (p = 0.015). The Δtailpiece myosin recovered with a half time indistinguishable from wild type. Asterisks indicate significantly different values. Significance determined by a two-tailed t test.
DISCUSSION
In this article we provide evidence for a multiple-stage regulatory mechanism governing mammalian nonmuscle myosin II assembly and recycling within the cell (Figure 9). In our model, the 10S hairpin is utilized for sequestering the NM-II as a monomer in order to facilitate long-range recycling such as delivery of disassembled monomers from the rear of migrating cells back to the front. In contrast, the tailpiece clearly influences global assembly levels, but tailpiece-based regulation appears more relevant for driving localized disassembly, such as at leading edge lamellar protrusions. We suggest based on our work (Figure 6), and based on earlier studies (Dulyaninova et al., 2007), that a key role of the tailpiece is to serve as a target for phosphorylation/dephosphorylation at aa S1943. In this scenario phosphorylation of the tailpiece favors dissociation of the myosin molecule from the filament to a 6S monomer. Effects involving binding of S100A4 to the adjacent helical region are also likely important (Dulyaninova et al., 2005). In addition to the mechanisms studied in this work, other mechanisms clearly contribute to spatial control of filament assembly. For example, NM-II motor activity is critical for normal localization and redistribution of NM-II from posterior portions of the lamella to the anterior-most lamellipodial zones (Kolega, 2006).
Figure 9.

Model for in vivo control of myosin IIA assembly in mammalian nonmuscle cells. In this model, sequestration in the 10S form plays important roles in maintaining the normal equilibrium between assembly and disassembled states in live cells. Destabilization of the 10S species by the ΔIQ mutant dramatically shifts the in vivo equilibrium of monomer versus assembled pools toward the assembled state. This in turn severely compromises recycling to leading edge protrusions, which in wild type NM-II would be dependent on RLC dephosphorylation. On the basis of our data and other recent studies (see Discussion), we propose that regulation via the tailpiece specifically plays a role in driving the rapid local removal of myosin IIA subunits from assembled filaments, back to the linear, monomeric, 6S state. We suggest that the fundamental role of the tailpiece in this process is that it serves as a target for phosphorylation at S1943.
We propose that the ΔIQ2 mutation, by eliminating or strongly destabilizing the 10S monomeric or sequestered state, reduces the number of intermediate states or conformations between monomeric form and assembled filament. Extended 6S ΔIQ2 myosin is immediately accessible at all times to anneal to any nearby filament. This effect could explain the accelerated turnover rate observed in FRAP analysis for this mutant (Table 2).
The less severe over-assembly consequences of the NMHC-IIAΔtailpiece mutation, or of the S1943A mutation, compared with the NMHC-IIAΔIQ mutation should not be taken as a lack of importance for tailpiece-based regulation. Our data suggest a cellular mechanism for the pathological effects of R1933stop MYH9-related disorder mutations, and this pathology in turn argues that the over-assembly observed in our assays is physiologically relevant. We suggest that the over-assembly defects observed for the NM-IIAΔtailpiece mutant in our assays reflects assembly control dysfunction that is the basis of the platelet defects in affected patients. Platelet formation from megakaryocytes occurs through a poorly understood budding process that appears to involve blood-flow based shearing forces to help release the nascent platelets and possibly additional shearing at a later stage in the pulmonary capillary beds to produce mature platelets of uniform small size (Junt et al., 2007). We suggest that excessive cortical myosin IIA assembly in patients carrying the tailpiece deletion renders their platelets resistant to the normal shearing process, producing the observed problems in platelet size and abundance. If this hypothesis is correct, treatments designed to reduce overall platelet actomyosin contractility might be effective in ameliorating this MYH9-related disorder symptom.
Our experiments support a model in which phosphorylation of the nonhelical tailpiece of NM-IIA modulates local turnover behavior in a manner similar to phosphorylation of the helical tail of Dictyostelium, although there are interesting contrasts between the role of tail phosphorylation in the amoeba system versus the human system. Earlier studies have clearly established threonine phosphorylation in the carboxyl-terminal portion of the Dictyostelium myosin II tail as the critical and dominant factor that regulates filament assembly in vivo (Egelhoff et al., 1993;Yumura, 2001;Yumura et al., 2005). Unlike mammalian systems, RLC phosphorylation in Dictyostelium has no effect on filament assembly levels (Griffith et al., 1987), and there is no evidence for a RLC-mediated 10S sequestered state. Thus a major regulatory difference between the amoeba system and the mammalian system is the presence of two distinct levels of regulation that can modulate mammalian myosin II filament assembly; an MHC-tail based mechanism, reminiscent of the amoeba system, and a 6S–10S monomer sequestering mechanism, which is not present in the amoeba. We suggest that the development of the additional 6S–10S regulatory module occurred to facilitate the diverse array of biomechanical tasks that are needed in a complex multicellular organism with hundreds of different cell types.
In summary, in this article we provide the first evidence for how different regulatory mechanisms of NM-II assembly interact to regulate NM-IIA behavior within the cell. We propose that the 10S hairpin conformation is used primarily for long range diffusion of myosin from one compartment of the cell to another, whereas the tailpiece is involved in modulating the local filament assembly disassembly reaction, possibly via tail phosphorylation. These studies have implications for understanding human disease MYH9-related disorders and for understanding the evolution of complex cytoskeletal regulatory systems in higher eukaryotes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bob Adelstein and Anne Bresnick, and members of their labs, for gifts of reagents and helpful suggestions at the early stages of this project. This work was supported by National Institutes of Health Grant GM 077224 to T.T.E. We acknowledge National Institute of Diabetes and Digestive and Kidney Diseases training grant support (DK007678) to M.T.B.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-04-0372) on October 29, 2008.
REFERENCES
- Adelstein R. S., Pato M. D., Conti M. A. The role of phosphorylation in regulating contractile proteins. Adv. Cyclic. Nucleotide Res. 1981;14:361–373. [PubMed] [Google Scholar]
- Bao J., Jana S. S., Adelstein R. S. Vertebrate nonmuscle myosin II isoforms rescue small interfering RNA-induced defects in COS-7 cell cytokinesis. J. Biol. Chem. 2005;280:19594–19599. doi: 10.1074/jbc.M501573200. [DOI] [PubMed] [Google Scholar]
- Bray D., White J. G. Cortical flow in animal cells. Science. 1988;239:883–888. doi: 10.1126/science.3277283. [DOI] [PubMed] [Google Scholar]
- Burgess S. A., Yu S., Walker M. L., Hawkins R. J., Chalovich J. M., Knight P. J. Structures of smooth muscle myosin and heavy meromyosin in the folded, shutdown state. J. Mol. Biol. 2007;372:1165–1178. doi: 10.1016/j.jmb.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Conti M. A., Adelstein R. S. Nonmuscle myosin II moves in new directions. J. Cell Sci. 2008;121:11–18. doi: 10.1242/jcs.007112. [DOI] [PubMed] [Google Scholar]
- Craig R., Smith R., Kendrick-Jones J. Light-chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature. 1983;302:436–439. doi: 10.1038/302436a0. [DOI] [PubMed] [Google Scholar]
- Dobereiner H. G., Dubin-Thaler B., Giannone G., Xenias H. S., Sheetz M. P. Dynamic phase transitions in cell spreading. Phys. Rev. Lett. 2004;93:108105-1–108105-4. doi: 10.1103/PhysRevLett.93.108105. [DOI] [PubMed] [Google Scholar]
- Dulyaninova N. G., House R. P., Betapudi V., Bresnick A. R. Myosin-IIA heavy-chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells. Mol. Biol. Cell. 2007;18:3144–3155. doi: 10.1091/mbc.E06-11-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulyaninova N. G., Malashkevich V. N., Almo S. C., Bresnick A. R. Regulation of myosin-IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry. 2005;44:6867–6876. doi: 10.1021/bi0500776. [DOI] [PubMed] [Google Scholar]
- Egelhoff T. T., Lee R. J., Spudich J. A. Dictyostelium myosin heavy chain phosphorylation sites regulate myosin filament assembly and localization in vivo. Cell. 1993;75:363–371. doi: 10.1016/0092-8674(93)80077-r. [DOI] [PubMed] [Google Scholar]
- Feder T. J., Brust-Mascher I., Slattery J. P., Baird B., Webb W. W. Constrained diffusion or immobile fraction on cell surfaces: a new interpretation. Biophys. J. 1996;70:2767–2773. doi: 10.1016/S0006-3495(96)79846-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke J. D., Dong F., Rickoll W. L., Kelley M. J., Kiehart D. P. Rod mutations associated with MYH9-related disorders disrupt nonmuscle myosin-IIA assembly. Blood. 2005;105:161–169. doi: 10.1182/blood-2004-06-2067. [DOI] [PubMed] [Google Scholar]
- Giannone G., Dubin-Thaler B. J., Dobereiner H. G., Kieffer N., Bresnick A. R., Sheetz M. P. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 2004;116:431–443. doi: 10.1016/s0092-8674(04)00058-3. [DOI] [PubMed] [Google Scholar]
- Griffith L. M., Downs S. M., Spudich J. A. Myosin light chain kinase and myosin light chain phosphatase from Dictyostelium: effects of reversible phosphorylation on myosin structure and function. J. Cell Biol. 1987;104:1309–1323. doi: 10.1083/jcb.104.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikebe M., Ikebe R., Kamisoyama H., Reardon S., Schwonek J. P., Sanders C. R., Matsuura M. Function of the NH2-terminal domain of the regulatory light chain on the regulation of smooth muscle myosin. J. Biol. Chem. 1994;269:28173–28180. [PubMed] [Google Scholar]
- Junt T., et al. Dynamic visualization of thrombopoiesis within bone marrow. Science. 2007;317:1767–1770. doi: 10.1126/science.1146304. [DOI] [PubMed] [Google Scholar]
- Kolega J. The role of myosin II motor activity in distributing myosin asymmetrically and coupling protrusive activity to cell translocation. Mol. Biol. Cell. 2006;17:4435–4445. doi: 10.1091/mbc.E06-05-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolega J., Taylor D. L. Gradients in the concentration and assembly of myosin II in living fibroblasts during locomotion and fiber transport. Mol. Biol. Cell. 1993;4:819–836. doi: 10.1091/mbc.4.8.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R. J., Egelhoff T. T., Spudich J. A. Molecular genetic truncation analysis of filament assembly and phosphorylation domains of Dictyostelium myosin heavy chain. J. Cell Sci. 1994;107(Pt 10):2875–2886. doi: 10.1242/jcs.107.10.2875. [DOI] [PubMed] [Google Scholar]
- Li Z. H., Bresnick A. R. The S100A4 metastasis factor regulates cellular motility via a direct interaction with myosin-IIA. Cancer Res. 2006;66:5173–5180. doi: 10.1158/0008-5472.CAN-05-3087. [DOI] [PubMed] [Google Scholar]
- Murakami N., Chauhan V. P., Elzinga M. Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry. 1998;37:1989–2003. doi: 10.1021/bi971959a. [DOI] [PubMed] [Google Scholar]
- Nakasawa T., Takahashi M., Matsuzawa F., Aikawa S., Togashi Y., Saitoh T., Yamagishi A., Yazawa M. Critical regions for assembly of vertebrate nonmuscle myosin II. Biochemistry. 2005;44:174–183. doi: 10.1021/bi048807h. [DOI] [PubMed] [Google Scholar]
- Olney J. J., Sellers J. R., Cremo C. R. Structure and function of the 10 S conformation of smooth muscle myosin. J. Biol. Chem. 1996;271:20375–20384. doi: 10.1074/jbc.271.34.20375. [DOI] [PubMed] [Google Scholar]
- Onishi H., Watanabe S. Chicken gizzard heavy meromyosin that retains the two light-chain components, including a phosphorylatable one. J. Biochem. 1979;85:457–472. doi: 10.1093/oxfordjournals.jbchem.a132352. [DOI] [PubMed] [Google Scholar]
- Salzameda B., Facemyer K. C., Beck B. W., Cremo C. R. The N-terminal lobes of both regulatory light chains interact with the tail domain in the 10 S-inhibited conformation of smooth muscle myosin. J. Biol. Chem. 2006;281:38801–38811. doi: 10.1074/jbc.M606555200. [DOI] [PubMed] [Google Scholar]
- Seidel J. C. Fragmentation of gizzard myosin by alpha-chymotrypsin and papain, the effects on ATPase activity, and the interaction with actin. J. Biol. Chem. 1980;255:4355–4361. [PubMed] [Google Scholar]
- Seri M., et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine. 2003;82:203–215. doi: 10.1097/01.md.0000076006.64510.5c. [DOI] [PubMed] [Google Scholar]
- Shoffner J. D., De Lozanne A. Sequences in the myosin II tail required for self-association. Biochem. Biophys. Res. Commun. 1996;218:860–864. doi: 10.1006/bbrc.1996.0153. [DOI] [PubMed] [Google Scholar]
- Sohn R. L., Vikstrom K. L., Strauss M., Cohen C., Szent-Gyorgyi A. G., Leinwand L. A. A 29 residue region of the sarcomeric myosin rod is necessary for filament formation. J. Mol. Biol. 1997;266:317–330. doi: 10.1006/jmbi.1996.0790. [DOI] [PubMed] [Google Scholar]
- Steimle P. A., Naismith T., Licate L., Egelhoff T. T. WD repeat domains target Dictyostelium myosin heavy chain kinases by binding directly to myosin filaments. J. Biol. Chem. 2001;276:6853–6860. doi: 10.1074/jbc.M008992200. [DOI] [PubMed] [Google Scholar]
- Straussman R., Even L., Ravid S. Myosin II heavy chain isoforms are phosphorylated in an EGF-dependent manner: involvement of protein kinase C. J. Cell Sci. 2001;114:3047–3057. doi: 10.1242/jcs.114.16.3047. [DOI] [PubMed] [Google Scholar]
- Trybus K. M., Huiatt T. W., Lowey S. A bent monomeric conformation of myosin from smooth muscle. Proc. Natl. Acad. Sci. USA. 1982;79:6151–6155. doi: 10.1073/pnas.79.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trybus K. M., Lowey S. Conformational states of smooth muscle myosin. Effects of light chain phosphorylation and ionic strength. J. Biol. Chem. 1984;259:8564–8571. [PubMed] [Google Scholar]
- Trybus K. M., Lowey S. The regulatory light chain is required for folding of smooth muscle myosin. J. Biol. Chem. 1988;263:16485–16492. [PubMed] [Google Scholar]
- Uyeda T. Q., Spudich J. A. A functional recombinant myosin II lacking a regulatory light chain-binding site. Science. 1993;262:1867–1870. doi: 10.1126/science.8266074. [DOI] [PubMed] [Google Scholar]
- Wei Q., Adelstein R. S. Conditional expression of a truncated fragment of nonmuscle myosin II-A alters cell shape but not cytokinesis in HeLa cells. Mol. Biol. Cell. 2000;11:3617–3627. doi: 10.1091/mbc.11.10.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M., Elsner M., Kartberg F., Nilsson T. Anomalous subdiffusion is a measure for cytoplasmic crowding in living cells. Biophys. J. 2004;87:3518–3524. doi: 10.1529/biophysj.104.044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumura S. Myosin II dynamics and cortical flow during contractile ring formation in Dictyostelium cells. J. Cell Biol. 2001;154:137–146. doi: 10.1083/jcb.200011013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumura S., Yoshida M., Betapudi V., Licate L. S., Iwadate Y., Nagasaki A., Uyeda T. Q., Egelhoff T. T. Multiple myosin II heavy chain kinases: roles in filament assembly control and proper cytokinesis in Dictyostelium. Mol. Biol. Cell. 2005;16:4256–4266. doi: 10.1091/mbc.E05-03-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.