

Abstract
Background
Eukaryotic gene expression is a complex process involving multiple cis and trans activating molecules to either facilitate or inhibit transcription. In recent years, many studies have focused on the role of acetylation of histone proteins in modulating transcription, whereas deacetylation of these same proteins is associated with inactivation or repression of gene expression. This study explores gene expression in HepG2 and F9 cell lines treated with Trichostatin A (TSA), a potent histone deacetylase inhibitor.
Results
These experiments show that TSA treatment results in clear repression of genes involved in the cholesterol biosynthetic pathway as well as other associated pathways including fatty acid biosynthesis and glycolysis. TSA down regulates 9 of 15 genes in this pathway in the F9 embryonal carcinoma model and 11 of 15 pathway genes in the HepG2 cell line. A time course study on the effect of TSA on gene expression of various enzymes and transcription factors involved in these pathways suggests that down regulation of Srebf2 may be the triggering factor for down regulation of the cholesterol biosynthesis pathway.
Conclusion
Our results provide new insights in the effects of histone deacetylases on genes involved in primary metabolism. This observation suggests that TSA, and other related histone deacetylase inhibitors, may be useful as potential therapeutic entities for the control of cholesterol levels in humans.
Background
Histone deacetylases (HDACs) are important chromatin remodeling enzymes that are generally involved in transcriptional repression [1]. Mammalian HDACs are classified into three main categories depending on their primary homology to Saccharomyces cerevisiae HDACs (RPD3, HDA1 and SIR2). Histone deacetylase inhibitors (HDACIs) tend to show equal effects on gene activation and repression [2-4]. HDACIs have been shown to induce differentiation, apoptosis or growth arrest in a variety of transformed cell lines [5]. This is generally attributed to the ability of these inhibitors to induce an open chromatin conformation facilitating transcription of regulatory genes like p21 which inhibit tumor cell growth [6]. These qualities make HDACIs promising targets for chemotherapeutic intervention.
Recently many different types of HDAC inhibitors have been discovered (Figure 1). These include short chain fatty acids (sodium butyrate, phenylbutyrate, valproic acid) [7], hydroxamic acids (trichostatin A (TSA), suberoylanilide hydromaxic acid (SAHA), pyroxamide, cyclic hydroxamic acid-containing peptides (CHAPs), cinnamic acid bishydroxamic acid (CBHA) and scriptaid) [8,9], cyclic tetrapeptides (trapoxin, apicidin, depsipeptide) [10-13,13], and benzamides (MS-275)[14,15]. Most HDAC inhibitors (HDACIs) developed to date inhibit both Class I and II HDACs equally with the exceptions being valproic acid (5 fold more selective for HDAC1 vs HDACs 5 and 6) and FK-228 (Class I selective). Class I and II HDACs are inhibited by trichostatin A (TSA) and related compounds whereas Class III HDACs are not. As noted, HDACIs have been shown to promote cell cycle arrest, differentiation, and apoptosis in many transformed cultured cell types. In animal models, HDACIs have been shown to inhibit growth of breast, prostate, lung and stomach cancers, as well as neuroblastomas and leukemias, with little toxicity [16,17]. In a previous study looking at the combination regimen of all trans retinoic acid (RA) with the HDACI, Trichostatin A (TSA), we identified several new targets for HDACIs [18]. We also identified critical differences in gene regulation subsequent to treatment with these two agents and a novel promoter module associated with the regulation of a subset of these differentially regulated genes. These analyses focused on the anticancer therapeutic potential of these compounds alone or in combination. Recent analysis of these data identified certain crucial metabolic pathways that have not previously been shown to respond to HDACI treatment and which may be critical in identifying new therapies for cardiovascular health. In this report we discuss the possible role of HDAC inhibition on cholesterol metabolism.
Figure 1.

Structures of common HDAC inhibitors.
Results
Microarray results from F9 cell treatments
Of the 12,451 mouse genes on the Affymetrix MU74Av2 microarray, 1248 genes (upregulated expression of 489 genes and decreased expression of 759 genes) were found to be significantly differentially expressed following TSA treatment. Of these, only 463 genes were found to be differentially expressed at an arbitrary two-fold or greater level of expression (226 genes up; 237 genes down) (Tables 1 &2, Additional file 1). The raw CEL files for the microarray data are available for download at the Gene Expression Omnibus under series GSE1437. Genes for which up regulated expression was noted were involved in retinoid binding and/or metabolism (e.g., Crabp2, Rbp1, Cyp26), the immune response (H2-Q7, H2-Dma, H2-L, Cmkor1, H-2D4(q), MHC H-2K-f class 1 antigen); extracellular matrix regulation (Col5a1, Col13a1, Gsn, Prhp1, Tuba3, t-PA, Cpe, Tm4sf6, Atp1b2, Dsc2); transcription and maintenance of chromatin structure (Cbx4, Msx2, H1f0, Elf3, Zfpm), signal transduction (Il11ra2, PLD1), apoptosis (Cidea, Zac1), cell growth regulation (IGF-II, Igfbp3, Reck, Meis1, Scgf), and embryonic development (Sema3e, Hoxb1 & 4, Stra8, Hoxa1, Cdx-1). Similarly, genes that were down regulated post-TSA treatment included genes involved in extracellular matrix degradation (MMP10, Adam23), transcriptional regulation (Foxd3, UTF1, SF1/Nr5a1, Msc, Mybbp1a, HMGI-C, lyl1), signal transduction (Tdgf1, Fst, Gna14, Il12rb2, Il5ra, Map3k4, Vegfc), and cell cycle deregulation (Myb, Mybl2, Tal1). Interestingly, we also found down regulated genes involved in pyrimidine biosynthesis (Dhodh) and in the cholesterol metabolism pathway (Mvk, Lss, Hmgcr, Fasn and Sqle) (Figure 2). This latter finding was intriguing and we decided to extend our investigations beyond the pluripotent mouse EC cells. These experiments were repeated using the more relevant human hepatocarcinoma derived HepG2 cells, since these are hepatic in origin and the liver is the primary source for cholesterol and fatty acid metabolism in humans. While these are not primary hepatocytes, this cell line offers the ability to both explore the hereto unknown effects of TSA on cholesterol metabolism and also look at the previously known targets of this drug.
Table 1.
Genes upregulated by TSA treatment in F9 cells (representative genes from total of 226 genes at 2-fold level of expression)
| Treatment type | EtOH | TSA | |||||
| Systematic | FC | Norm | SE | Norm | SE | Gene | GB Acc |
| 93714_f_at | 14.5 | 1.64 | 1.0 | 23.82 | 3.1 | H2-L | AI117211 |
| 100127_at | 18.0 | 1.08 | 0.2 | 19.37 | 3.4 | CRABP2 | M35523 |
| 92770_at | 18.3 | 0.83 | 0.2 | 15.28 | 2.0 | S100A6 | X66449 |
| 95471_at | 23.2 | 0.75 | 0.3 | 17.30 | 2.4 | CDKN1C | U22399 |
| 93981_at | 12.8 | 1.07 | 0.3 | 13.72 | 3.0 | PLAT | J03520 |
| 92275_at | 10.2 | 1.00 | 0.3 | 10.16 | 0.5 | TCFAP2C | X94694 |
| 92502_at | 7.6 | 1.00 | 0.1 | 7.60 | 0.8 | ZAC1 | X95504 |
| 100139_at | 6.6 | 0.92 | 0.3 | 6.11 | 0.7 | PCSK1N | AI841733 |
| 98758_at | 6.0 | 0.82 | 0.2 | 4.92 | 0.4 | ALOX15 | L34570 |
| 160547_s_at | 5.7 | 0.98 | 0.1 | 5.60 | 0.7 | TXNIP | AI839138 |
| 94545_at | 5.5 | 1.02 | 0.2 | 5.60 | 0.1 | RTN1 | AW123115 |
| 99906_at | 4.8 | 0.93 | 0.2 | 4.48 | 0.4 | ESX1 | AF085715 |
| 93875_at | 4.7 | 1.03 | 0.2 | 4.84 | 0.9 | HSP70-3 | M12571 |
| 161482_f_at | 4.7 | 1.21 | 0.2 | 5.65 | 1.1 | PRPH1 | AV068234 |
| 104716_at | 3.8 | 0.99 | 0.2 | 3.82 | 0.1 | RBP1 | X60367 |
| 99642_i_at | 3.6 | 0.94 | 0.2 | 3.40 | 0.2 | CPE | X61232 |
| 94881_at | 3.3 | 0.77 | 0.3 | 2.59 | 0.1 | CDKN1A | AW048937 |
| 99643_f_at | 3.2 | 0.99 | 0.1 | 3.16 | 0.3 | CPE | X61232 |
| 98067_at | 3.2 | 0.77 | 0.2 | 2.43 | 0.1 | CDKN1A | U09507 |
| 92501_s_at | 3.0 | 1.01 | 0.2 | 3.04 | 0.5 | ZAC1 | X95503 |
| 93120_f_at | 2.9 | 1.04 | 0.1 | 3.01 | 0.3 | H2-K; H-2K | V00746 |
| 97487_at | 2.8 | 0.94 | 0.1 | 2.60 | 0.2 | SERPINE2 | X70296 |
| 96704_at | 2.6 | 0.95 | 0.1 | 2.52 | 0.4 | SFN | AF058798 |
| 93888_at | 2.5 | 0.84 | 0.2 | 2.10 | 0.1 | HOXB1; HOX-2.9 | X53063 |
| 95297_at | 2.3 | 1.06 | 0.3 | 2.48 | 0.3 | HOXA1 | M22115 |
| 93278_at | 2.1 | 1.03 | 0.2 | 2.21 | 0.6 | SCP2 | M91458 |
| 104580_at | 2.1 | 0.79 | 0.2 | 1.67 | 0.1 | PLCD | U85711 |
FC: fold change; Norm: Normalized signal, SE: std error of normalized signal; GB Acc: GeneBank Accession number
Table 2.
Genes downregulated by TSA treatment in F9 cells (representative genes from total of 237 genes at 2-fold level of expression)
| Treatment type | EtOH | TSA | |||||
| Systematic | FC | Norm | SE | Norm | SE | Gene | GB Acc |
| 92889_r_at | -25.8 | 1.00 | 0.2 | 0.04 | 0.05 | FOXD3 | AF067421 |
| 100700_s_at | -6.3 | 0.97 | 0.1 | 0.15 | 0.06 | NR5A1 | AB000490 |
| 94712_at | -5.3 | 1.15 | 0.2 | 0.22 | 0.08 | VEGFC | U73620 |
| 101578_f_at | -4.9 | 1.25 | 0.44 | 0.26 | 0.09 | ACTB | M12481 |
| 99963_at | -4.6 | 1.26 | 0.28 | 0.28 | 0.11 | ZFP101 | U07861 |
| 93731_at | -4.5 | 0.96 | 0.13 | 0.21 | 0.07 | FKBP9 | AF090334 |
| 100701_r_at | -3.8 | 0.94 | 0.1 | 0.25 | 0.07 | NR5A1 | AB000490 |
| 99323_at | -3.7 | 1.01 | 0.1 | 0.27 | 0.10 | IL12RB2 | U64199 |
| 98817_at | -3.7 | 1.02 | 0.2 | 0.28 | 0.11 | FST | Z29532 |
| 99058_at | -3.7 | 1.05 | 0.2 | 0.29 | 0.11 | HMGA2 | X99915 |
| 95632_f_at | -3.3 | 0.91 | 0.3 | 0.28 | 0.08 | MVK | AW122653 |
| 93002_r_at | -3.2 | 0.86 | 0.2 | 0.27 | 0.07 | TDGF1 | M87321 |
| 102220_at | -2.8 | 0.90 | 0.1 | 0.33 | 0.08 | UTF1; AI505934 | AB017360 |
| 160737_at | -2.7 | 1.13 | 0.2 | 0.41 | 0.07 | LSS | AW060927 |
| 99425_at | -2.6 | 0.91 | 0.2 | 0.35 | 0.07 | HMGCR | X07888 |
| 93065_at | -2.6 | 1.17 | 0.23 | 0.45 | 0.12 | IL11RA1 | U14412 |
| 103683_at | -2.5 | 0.90 | 0.2 | 0.35 | 0.07 | DHODH | AF029667 |
| 104285_at | -2.3 | 1.02 | 0.2 | 0.44 | 0.08 | HMGCR | M62766 |
| 160832_at | -2.3 | 0.97 | 0.16 | 0.43 | 0.06 | LDLR | Z19521 |
| 98575_at | -2.2 | 1.06 | 0.2 | 0.48 | 0.08 | FASN | X13135 |
| 94322_at | -2.2 | 0.97 | 0.1 | 0.44 | 0.07 | SQLE | D42048 |
| 93234_at | -2.1 | 1.11 | 0.2 | 0.52 | 0.07 | MSC | AF087035 |
FC: fold change; Norm: Normalized signal, SE: std error of normalized signal; GB Acc: GeneBank Accession number
Figure 2.
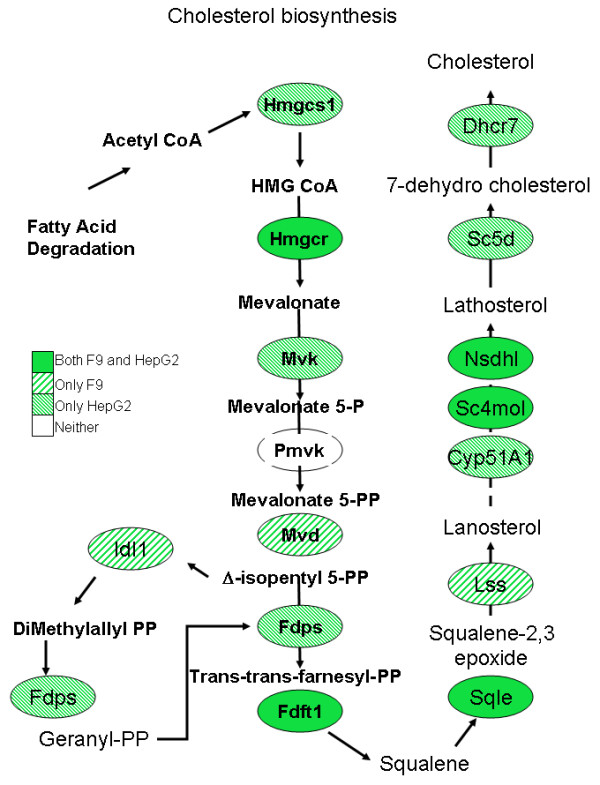
Map of cholesterol biosynthesis. TSA down regulates 9 of 15 genes in this pathway in the F9 embryonal carcinoma model and 11 of 15 pathway genes in the HepG2 cell line.
Microarray results from HepG2 experiments
Of the 54,613 human genes on the Affymetrix HU133 plus 2.0 array, only 6,513 showed significant differential expression following TSA treatment (p value < 0.05). The raw CEL files for the microarray data are available for download at the Gene Expression Omnibus under series GSE4465. TSA treatment of this cell line resulted in 1561 genes being up regulated and 4952 genes being down regulated at this level of significance (Figure 3). This observation was surprising since the current paradigm of HDAC inhibition suggests equal effects on gene activation and repression after treatment with an HDAC inhibitor [3,4]. Furthermore, using a two-fold cutoff, the genelist was reduced to 3229 genes with 254 up and 2975 down regulated (see Tables 3 and 4; Additional file 1) further emphasizing the extreme extent of gene expression down regulation following HDACI treatment. Genes that showed up regulated expression include phospholipid transfer protein (Pltp), tissue inhibitors of metalloproteinase 1 and 2 (Timp1, Timp2) and transforming growth factor beta 1 (Tgfβ1). Perhaps more importantly, the interesting downregulated genes included thymidylate synthetase (Tyms), formyltetrahydrofolate dehydrogenase (Fthfd), dihydroorotate dehydrogenase (Dhodh) and CTP synthase II (CTPSII) (all of which are related to pyrimidine biosynthesis) as well as genes related to lipid transport and fatty acid metabolism including low density lipoprotein receptor (ldlr), enoyl-Coenzyme A hydratase, acyl-Coenzyme A dehydrogenase (Acadm), apolipoproteins A5, C3, L1, high density lipoprotein binding protein (vigilin); and 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 1 (Hmgcs1), farnesyl-diphosphate farnesyltransferase 1 (Fdft1), squalene epoxidase (Sqle), sterol regulatory element binding transcription factor 2 (Srebf2) and 7-dehydrocholesterol reductase (Dhcr7). Notably 11 of these genes are involved in cholesterol metabolism (Figure 2).
Figure 3.
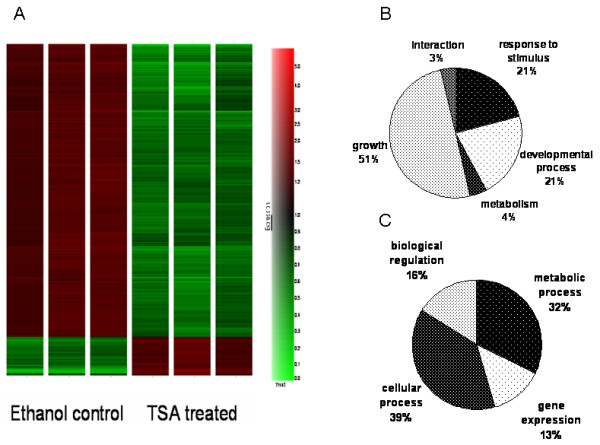
(A)Hierarchical clustering of HepG2 cells treated with ethanol or TSA shows that the majority of genes are down regulated (green) by TSA treatment in contrast with the current paradigm of the role of HDACs in gene repression. Gene Ontology analysis of the terms related too "biological process" (p-value < 0.05) shows a significant difference in the genes being up-regulated (B) or down-regulated (C) by TSA. The down regulation of metabolic processes includes cholesterol, lipid and fatty acid metabolism.
Table 3.
Genes upregulated by TSA treatment in HEPG2 cells (representative genes from total of 254 genes at 2-fold level of expression)
| Treatment type | EtOH | TSA | |||||
| Systematic | FC | Norm | SE | Norm | SE | Gene | GB Acc |
| 214023_x_at | 18.5 | 1.00 | 0.1 | 18.61 | 1.2 | TUBB | AL533838 |
| 201008_s_at | 10.2 | 0.94 | 0.1 | 9.60 | 1.1 | TXNIP | AA812232 |
| 227404_s_at | 9.5 | 1.00 | 0.1 | 9.50 | 0.3 | EGR1 | AI459194 |
| 218280_x_at | 6.5 | 0.92 | 0.1 | 5.97 | 0.4 | HIST2H2AA | NM_003516 |
| 221059_s_at | 4.6 | 0.95 | 0.1 | 4.36 | 0.2 | CHST6 | NM_021615 |
| 208581_x_at | 4.4 | 0.96 | 0.1 | 4.20 | 0.2 | MT1X | NM_005952 |
| 203158_s_at | 3.9 | 1.00 | 0.1 | 3.91 | 0.2 | GLS | AF097493 |
| 206907_at | 3.7 | 1.09 | 0.1 | 4.07 | 0.3 | TNFSF9 | NM_003811 |
| 202075_s_at | 3.6 | 0.98 | 0.1 | 3.48 | 0.2 | PLTP | NM_006227 |
| 201666_at | 2.8 | 1.01 | 0.1 | 2.84 | 0.2 | TIMP1 | NM_003254 |
| 203085_s_at | 2.3 | 1.02 | 0.1 | 2.35 | 0.2 | TGFB1 | BC000125 |
| 203167_at | 2.3 | 1.00 | 0.1 | 2.25 | 0.0 | TIMP2 | NM_003255 |
FC: fold change; Norm: Normalized signal, SE: std error of normalized signal; GB Acc: GeneBank Accession number
Table 4.
Genes downregulated by TSA treatment in HEPG2 cells (representative genes from total of 2975 genes at 2-fold level of expression)
| Treatment type | EtOH | TSA | |||||
| Systematic | FC | Norm | SE | Norm | SE | Gene | GB Acc |
| 205890_s_at | -30.7 | 0.975 | 0.05 | 0.032 | 0.02 | UBD | NM_006398 |
| 220437_at | -12.6 | 0.976 | 0.08 | 0.078 | 0.06 | LOC55908 | NM_018687 |
| 223493_at | -11.5 | 1.056 | 0.07 | 0.092 | 0.06 | FBXO4 | AF129534 |
| 226388_at | -11.4 | 1.015 | 0.06 | 0.089 | 0.03 | TCEA3 | AI675780 |
| 202589_at | -9.6 | 1.07 | 0.09 | 0.11 | 0.04 | TYMS | NM_001071 |
| 203979_at | -8.7 | 1.007 | 0.08 | 0.115 | 0.05 | CYP27A1 | NM_000784 |
| 226216_at | -7.2 | 0.887 | 0.11 | 0.123 | 0.06 | INSR | W84556 |
| 209608_s_at | -6.3 | 1.034 | 0.06 | 0.163 | 0.03 | ACAT2 | BC000408 |
| 203924_at | -6.2 | 0.974 | 0.05 | 0.158 | 0.04 | GSTA2 | NM_000846 |
| 205208_at | -6.9 | 1.03 | 0.08 | 0.15 | 0.06 | FTHFD | NM_012190 |
| 219366_at | -5.6 | 0.954 | 0.14 | 0.169 | 0.06 | AVEN | NM_020371 |
| 205820_s_at | -5.3 | 1.02 | 0.06 | 0.19 | 0.04 | APOC3 | NM_000040 |
| 209546_s_at | -5.2 | 0.99 | 0.06 | 0.19 | 0.07 | APOL1 | AF323540 |
| 224243_at | -4.6 | 1.10 | 0.12 | 0.24 | 0.05 | APOA5; RAP3 | AF202889 |
| 200789_at | -4.0 | 0.96 | 0.11 | 0.24 | 0.03 | ECH1; HPXEL | NM_001398 |
| 221750_at | -3.8 | 0.99 | 0.05 | 0.26 | 0.04 | HMGCS1 | BG035985 |
| 202068_s_at | -3.6 | 1.00 | 0.06 | 0.28 | 0.04 | LDLR; FH; FHC | NM_000527 |
| 225012_at | -3.1 | 0.99 | 0.09 | 0.32 | 0.04 | HDLBP | BE378479 |
| 213577_at | -3.1 | 1.03 | 0.09 | 0.34 | 0.05 | SQLE | AA639705 |
| 202067_s_at | -2.9 | 0.88 | 0.12 | 0.30 | 0.16 | LDLR | AI861942 |
| 202502_at | -2.8 | 0.97 | 0.05 | 0.35 | 0.03 | ACADM; MCAD; | NM_000016 |
| 201248_s_at | -2.6 | 1.00 | 0.08 | 0.39 | 0.04 | SREBF2; SREBP2 | NM_004599 |
| 209218_at | -2.5 | 1.01 | 0.06 | 0.40 | 0.04 | SQLE | AF098865 |
| 222916_s_at | -2.3 | 0.92 | 0.12 | 0.40 | 0.06 | HDLBP | AF116718 |
| 201791_s_at | -2.2 | 0.99 | 0.07 | 0.45 | 0.04 | DHCR7; SLOS | NM_001360 |
FC: fold change; Norm: Normalized signal, SE: std error of normalized signal; GB Acc: GeneBank Accession number
Quantitative PCR results
Sybr green qPCR was used to validate microarray expression data for a subset of the differentially expressed genes. The expression patterns of 10 genes from the F9 microarray data set and 21 from the HepG2 microarray data set (Figure 4) were all confirmed by qPCR. Furthermore we decided to examine the levels of gene expression at early and late time points for 11 of these genes that have a role in cholesterol and lipid metabolism. The relative gene expression was obtained for these genes at 3 h, 6 h, 9 h, 12 h, and 48 h to serve as early and late time frames in comparison to the 24 h treatments (Figure 5). Hmgcr which is the rate limiting enzyme in cholesterol biosynthesis was repressed 2-fold after 12 h of TSA treatment and showed increasing down regulation over 24 h (4 fold) and 48 h (5.3 fold) time points. Hmgcs levels showed increased repression (2.8–3.8 fold) by TSA treatment over 6–24 hours (the amplification reactions failed for the 12 h and 48 h time points). Levels of Mvk (2.1 fold) and Srebf2 (1.8 fold) were down regulated at 3 h with maximal repression at 9 h (13.6 fold and 23.6 fold respectively) after which the levels then came back to normal over the next 39 hours. Srebf2 levels at 6 h, 12 h and 24 h were 2.4 fold, 4.5 fold and 2.4 fold respectively. Genes involved in lipid and fatty acid metabolism such as ApoA5 and Acat2 were found to be maximally down regulated at 12 h (12 fold) and 24 h (16 fold) time points respectively while ApoL1 was down regulated (11 fold) at 12, 24 and 48 h time points. Fabp which is involved in fatty acid metabolism showed increasing down regulation after 12 h (1.9 fold) while Pparγ was found to be increasingly repressed at 9 h (2.8 fold) followed by reversal after 12 h (3.5 fold). The Pparγ levels after 48 h of TSA treatment were still almost 2 fold down regulated as compared to untreated cells. Levels of Cyp27A1 or sterol 27-hydroxylase which participates in the conversion of cholesterol to bile acids was also found to be initially down regulated at 6 h (1.8 fold) and increasingly over the 12 (8.8 fold) and 24 h (10.3 fold) time points. TSA treatment did not show any significant effect on Ldlr expression until 24 h (2.8 fold).
Figure 4.

Real Time qPCR verification of the gene expression levels of various genes involved in (A) lipid transport and fatty acid synthesis, (B) cholesterol metabolism and (C) pyrimidine biosynthesis in HepG2 cells. Fold expression is relative to ethanol control. TSA treatment showed down regulated expression of 14 genes by both qPCR and microarray.
Figure 5.

Time course of gene expression in response to TSA treatment of HepG2 cells for 3, 6, 9, 12, 24 and 48 h. Values are represented as fold change relative to ethanol treated controls at the respective time points.
Discussion
In a previous study we had used microarray analyses to examine the effects of RA and TSA on embryonal carcinoma cell growth and differentiation using the prototypical EC cell line F9 [18]. Results from these studies identified several important genes and pathways differentially regulated by these compounds. In this report we identify new target pathways for TSA treatment based on further analysis of this data. Most importantly, the regulatory pathways that are affected include pyrimidine metabolism and cholesterol biosynthesis. The pyrimidine pathway is of interest because one of the rate limiting enzymes in this pathway, dihydroorotate dehydrogenase (dhodh), has been targeted for inhibition in murine models of rheumatoid arthritis as well as in the human T-lymphoblastoma cell line (A3.01) [21,22]. Dhodh catalyzes the fourth committed step in the de novo biosynthesis of pyrimidines. Activated lymphocytes expand their pyrimidine pool by eightfold during proliferation [23]. In rheumatoid arthritis, inflammation and degradation of synovial tissue are initiated by the influx of lymphocytes (B cells, CD4+, CD8+ and T cells) [24]. Thus, inhibiting activated T-cells by decreasing their supply of pyrimidines via TSA treatment could provide an attractive alternative method for treating rheumatoid arthritis. Interestingly, HDACIs (TSA and phenylbutyrate) were used as treatments in a rat model of rheumatoid arthritis, and resulted in reduced inflammation, and inhibition both of synovial hyperplasia and bone or cartilage destruction [25]. The authors also found that HDACIs inhibited the expression of tumor necrosis factor-α, which functions to stimulate matrix degradation in rheumatoid arthritis [26], therefore suggesting a mechanism by which HDACIs may alleviate some effects of rheumatoid arthritis. Further extending and supporting these results, in this study, we found that TSA itself could significantly inhibit the expression of dhodh even in non-lymphatic cells (60% in F9 and 25% in HepG2), providing an alternative (or synergistic) mechanism by which HDACI might suppress rheumatoid arthritis in both mice and men. Moreover, the mRNA levels of thymidylate synthetase (Tyms), another key enzyme in this pathway, were decreased 8 fold in HepG2 cells (2.4 fold in F9 cells) further potentiating this effect of TSA treatment. A previous study using chondrocytes showed that HDACIs such as TSA and sodium butyrate, blocked the induction of matrix metalloproteinases (MMP-1, MMP-13) as well as aggrecan-degrading enzymes (Adamts4, Adamts5 and Adamts9) [27]. Both of these enzyme families mediate cartilage destruction. In our study with HepG2 cells, we also found that TSA treatment resulted in a modest decrease in the expression of MMPs (MMP-1, MMP-2, MMP-11, MMP-12) (1.2–1.5 fold) and Adamts9 (1.6 fold). This, coupled with increased expression (2.8 fold) of a collagenase inhibitor (tissue inhibitor of MMP: TIMP1, TIMP2), might further promote maintenance of a growth regulating matrix. Interestingly, TSA treatment also resulted in down regulation of LPS induced TNFα levels (2 fold) as well as suppression of the cytokines IL-12 and IL-8 (2.7 and 2 fold respectively). This result is consistent with a report by Leoni et al. [28] on the anti-inflammatory properties of SAHA (another hydroxamic acid based HDACI) in Balb/c mice and human PBMCs induced with LPS.
The pathway most significantly affected by TSA treatment in F9 EC cells is that of cholesterol biosynthesis, most specifically those steps involved in the synthesis of low density lipoprotein. There are two main types of lipoproteins that transport cholesterol in the blood: low density lipoproteins (LDL) and high density lipoproteins (HDL). HDL particles are generally considered to be "good cholesterol", while LDL is considered "bad cholesterol" [29]. Several genes encoding essential enzymes in the LDL synthesis pathway are down regulated by these treatments. Pathway analysis of microarray data using genes showing statistically significant (p < 0.05) differential gene expression indicated that expression levels of 9 enzymes out of the 15 in the cholesterol biosynthesis pathway are decreased following TSA treatment. They include HMG CoA reductase (Hmgcr), mevalonate kinase (Mvk), di-p-mevalonate decarboxylase (Mvd), isopentenyl-PP isomerase (Idi1), squalene synthatase (Fdft1), squalene epoxidase (Sqle), lanosterol synthase (Lss) and lanosterol oxidase (Sc4mol) and NAD(P)-dependent steroid dehydrogenase (Nsdhl) (Figure 2). Of these only 6 of these 9, including Hmgcr, Mvd, Idi1, Sqle, Lss, Sc4mol and Nsdhl, showed a greater than two fold differential expression on TSA treatment. The z-score assigned to each category by MAPPFinder reflects the degree to which the expression of genes in that category was greater than that expected by chance. A positive z-score indicates that a large number of genes in that category are differentially expressed between the compared conditions while a negative Z score indicates that the there are fewer genes meeting the criterion than would be expected by random chance. If the MAPPFinder data truly obeyed the assumptions of the hypergeometric distribution, then a Z score or 1.96 or -1.96 would correlate with a p value of 0.05. The z-score for this pathway was highly significant with values of 4.85 associated with TSA treatment. Real-time qPCR analyses verified decreased expression of 3 of 5 genes (Hmgcr, Mvd, Lss) in this pathway but the expression of neither Mvd nor Lss was significantly down regulated by TSA. Decreasing high cholesterol levels using TSA treatments may work well since repression of few of the detected genes may be sufficient to induce the response, i.e., a reduction in cholesterol intermediates and synthesis.
Following the analysis of these pluripotent EC cells, we decided to investigate these effects in the HepG2 cell line which arose from a carcinoma of the human liver, the primary organ for cholesterol and fatty acid metabolic processes. While we realize that primary hepatocytes would be a better model to evaluate this pathway, we choose the HepG2 cell line as an means to evaluate this phenomenon but allowing for use of the known anti-cancer effects of TSA as a control. Expression data from HepG2 cells also indicated that multiple enzymes in cholesterol biosynthesis and fatty acid synthesis pathways were significantly down regulated (Figure 3). The mRNA transcript levels that were repressed at a greater than 2 fold level of significance included HMG CoA synthase (Hmgcs1), HMG CoA reductase (Hmgcr), sterol receptor binding factor-2 (Srebf2) and lanosterol 14 α-demethylase (Cyp51a1) (involved in cholesterol metabolism), and others including fatty acid synthase (Fasn), fatty acid binding protein (Fabp), farnesyl diphosphate synthase (Fdps), acetyl-coA carboxylase (Acaca), acetyl-coA dehydrogenase (Acadm), acetyl-coA acetyl transferase (Acat2), peroxisome proliferative activated receptor, gamma (Pparγ) and a variety of apolipoproteins that are involved in fatty acid and triglyceride metabolism. Quantitative PCR studies verified that TSA treatment reduced expression of Hmgcr, Hmgcs1, Srebf2, Fabp Fasn, Fdps, Acaca, Acadm, Acat2, ApoA5, C1, E and L1 as well as Cyp27a1, Ldlr, Pparγ and Tyms (Figure 4). The down regulation seems to be a complex phenomenon involving genes that regulate these pathways at different levels. Most evident is the down regulation of Srebf2 which in turn acts as a transcription factor regulating the expression of enzymes like Hmgcr (the target for the statin class of drugs) and Mvd. It is known that Srebf2 overexpression induces all 12 enzymes in the cholesterol biosynthesis pathway and inhibition of Srebf2 by TSA might inhibit the expression of these enzymes [30]. In fact, our microarray data demonstrates that the levels of almost all these enzymes are down (10 genes pass all cutoff filters) following TSA treatment. The repression of Srebf2 occurs at a early time point (around 3 hrs) and continually repressed over 24 hrs (2.4 fold repression at 24 h). This effect is probably responsible for the down regulation of the cholesterol pathway since expression of Hmgcs (repressed at 6 h), Hmgcr (repressed at 12 h) and Ldlr (repressed at 24 h) are all known to be induced by Srebf2 [31]. Srebf-1a and -1c are more involved with regulation of fatty acid synthesis and lipogenesis [30,32]. While we were unable to detect the levels of Srebf1 expression in our microarray experiments, TSA treatment modestly (1.3 fold) decreased expression levels of cytosolic NADP-dependent isocitrate dehydrogenase1 (Idh1) which provides the cytosolic NADPH required for proper functioning of both the cholesterol and fatty acid biosynthetic pathways. The Idh1 promoter is activated by Srebf1a and Srebf2 in human hepatoma cells [33]. Thus there appears to be a concerted down regulation of both pathways through a synergistic effect. Cyp51a1 (lanosterol 14α-demethylase) is another important intermediate in cholesterol metabolism and has in recent years gained importance as a target for the development of hypocholesterolemic agents [34]. Our microarray data showed that levels of Cyp51a1 were also down regulated further adding support to the possible use of this HDACI as a way to decrease plasma cholesterol levels.
Finally, atherosclerosis is the underlying disorder associated with most cardiovascular disease [35]. This disorder is characterized by deposits of fatty substances, cholesterol, cellular waste products, calcium and other substances in the inner lining of an artery (collectively known as plaques) [36]. Cholesterol has been implicated as the major contributor to this condition as atherosclerosis is strongly correlated with an increase in serum cholesterol levels [37,38]. Generally, serum levels should be between 140 and 200 mg per deciliter (mg/dl) whereas high levels surpassing 240 mg/dl indicate one is at high risk for cardiovascular disease [39]. Thus, atherosclerosis is characterized by elevated levels of LDL [40]. The activity of the hepatic LDL receptor (Ldlr) is the primary determinant of plasma LDL cholesterol levels and Ldlr transcription is in turn regulated by Srebf2. When the levels of hepatocellular sterols drop, Srebf2 is activated and this process restores the normal levels by concurrent activation of de novo cholesterol synthesis and increased uptake of plasma cholesterol through Ldlr. LDL receptor is also post transcriptionally regulated by proprotein convertase subtilisn/kexin type 9a (Pcsk9) in an inverse manner [41]. While our microarray and qPCR data shows decreased Ldlr expression following TSA treatment, microarray gene expression levels of Pcsk9 are also down regulated. This suggests existence of a mechanism for potential compensatory increase in Ldlr levels or activity post-transcriptionally. Our time course experiments did not show a significant repression of Ldlr levels until 24 h further highlighting the complex nature of Srebf2 regulation.
Our data with TSA treatment also showed a decrease in the levels of gene expression for a variety of apolipoproteins including apoA1, apoA5, apoB, apoC1, apoE, apoL1. This observation highlights the complex relationship of apolipoprotein levels and lipoprotein metabolism. While elevated levels of apoB and reduced levels of apoA1 are associated with increased cardiac disease, serum levels of apoB100 associated VLDL are regulated in turn by Acat2 which stimulates cholesteryl ester secretion into apoB-containing lipoproteins. Acat inhibitors are being developed as a therapeutic means to lower LDL cholesterol without affecting cholesterol uptake [42,43]. Also apoE deficient mice show high levels of cholesterol and develop spontaneous atherosclerosis while mice with partial or complete deficiency of high-mobility group A2 protein (Hmga2) are able to resist diet-induced obesity [44]. Acat2 inhibition using antisense nucleotides was previously shown to alleviate atherosclerosis in apoB-Ldlr -/- mice [45]. This study also found Acat2 inhibition to be effective in reducing plasma cholesterol, increasing plasma triglycerides, and shifting LDL cholesteryl ester fatty acids to become mainly polyunsaturated. In our study, TSA treatment showed a modest decrease in apoB, apoE and apoA1 in addition to decreased levels of Acat2, Fasn and Hmga2. This indicates that triglyceride metabolism is perturbed by TSA and further studies may be necessary to evaluate the possibility of using TSA and other HDACIs for modulating triglyceride metabolism. Cyp27 has been reported to be regulated by the nuclear receptor subfamily of which PPARγ is a member and levels of both these genes have been found to be high in atherosclerotic lesions. Levels of Cyp27a1 (maximal repression at 24 h) and Pparγ (maximal repression between 9–12 h) were found to be repressed by TSA treatment in both the microarray and qPCR data. This observation adds credence to the potential for development of TSA like HDACIs for atherosclerosis.
Conclusion
Our results show that cholesterol metabolism is significantly down regulated by TSA both directly and indirectly and thus HDACI therapy may be a relatively novel tool to develop for use in controlling cholesterol levels. This study only addresses the effect of TSA treatment on transcript levels of the rate limiting enzymes and transcription factors and further studies evaluating protein expression levels are necessary to derive firm conclusions on regulation of this pathway. Additional studies exploring the different classes of HDACIs with respect to their effects on regulation of the genes in the cholesterol pathway would also help dissect the details of this innovative application for these drugs.
Methods
Cell Culture for Microarray and Quantitative PCR Analysis
F9 mouse embryonal carcinoma cells were cultured as published previously [18] Stock solutions of TSA (3 mM) (Sigma-Aldrich) were freshly prepared in absolute ethanol for each experiment and were diluted in DMEM to a final concentration of 70 nM. Cells were seeded at 2.5 × 106 cells/75 cm2 gelatinized flask and treated with ethanol or TSA for 24 h. All experiments were performed in triplicate using a different preparation of F9 cells for each experiment.
Similarly, HepG2 human hepatoma cells were cultured using DMEM containing 10% FBS and treated with TSA (0.35 μM) or an ethanol control (final concentration 0.2%) for 24 hours before being harvested for RNA isolation. For time course experiments, total RNA was isolated from HepG2 cells treated with an ethanol control or 0.35 μM TSA for 3, 6, 9, 12, 24 or 48 h.
RNA extraction and purification
Both F9 and HepG2 cells were harvested with 4 mL of Tri-reagent (Molecular Research Center, Inc) and RNA isolation was carried out according to the manufacturer's protocol. Total RNA was purified using the RNeasy cleanup kit and protocol (Qiagen), quantified and then analyzed for degradation on a BioAnalyzer (Agilent).
Hybridization of sample to GeneChip Microarrays
RNA was converted to biotinylated cRNA (complimentary RNA) from oligo-dT-primed cDNA using standard Affymetrix protocols. Biotinylated cRNA was used to probe the MU74Av2 (F9 samples) or HU133 Plus 2.0 (HepG2 samples) Affymetrix GeneChip microarrays. A total of six samples (three controls and three TSA treated) for each cell line were analyzed.
Statistical Analysis
The raw data (CEL files) were imported into GeneSpring software (v7.2) for further analysis. A two-step normalization algorithm was implemented to select differential gene expression in response to TSA samples (ethanol treated samples as baseline). In the first normalization step, a global scaling per chip method was used in which the signal of each gene was divided by the mean intensity (50th percentile) of the chip. This normalization step was followed by a per gene normalization which divides each gene by the average intensity of that gene in several control samples. Hierarchical clustering was used to organize the data in discrete expression profiles. Selection of statistically significant genes from each expression profile was done using a p-value cut off of ≤ 0.05 with the cross gene error model (CGEM) combined with Welch t-test. The multiple testing correction (Benjamini and Hochberg false discovery rate) was integrated within each test. Additionally we also analyzed the HepG2 data using both MAS5 (Microarray Suite) as well as RMA (Robust Microarray Analysis) algorithms and genes that passed all criteria from both sets of analyses were used for follow up studies.
Pathway and Functional Cluster Analysis
The differentially expressed genes selected as described above were subjected to functional cluster analysis using MAPPFinder in conjunction with GenMAPP (Gene Microarray Pathway Profiler) 2.0 [19,20]
Quantitative Real-Time PCR
To verify the data obtained from microarrays, 5 μg of total RNA was taken from the same pool of RNA as used for the microarray experiments. The RNA was DNase treated (Ambion) and reverse transcribed to cDNA which served as the template for quantitative PCR (qPCR). Real-time relative qPCR (SYBR Green; Applied Biosystems) was performed in triplicate using a GeneAmp 5700 (F9 samples) or a HT7900 sequence detection system (HepG2 samples) according to the manufacturer's instructions. Primers were specifically designed using Primer express software (Applied Biosystems). 1 μg of cDNA was amplified in 1× SYBR green buffer. PCR conditions were: 10 min at 95°C for AmpliTaq Gold DNA polymerase activation, 45 thermal cycles of 15 sec at 95°C to denature and 1 min at 60°C to anneal and extend. Relative expression levels were analyzed using the method using the GAPDH expression level as a control. Samples treated with TSA were compared to the baseline expression value determined from ethanol treated samples at the respective time points and the fold change is shown. (For primer sequences see Table 5).
Table 5.
Primers used for Sybr green qPCR
| F9 cells | ||
| Gene | Forward | Reverse |
| GAPDH | 5'-GCCAAGAGGGTCATCATCTCC-3' | 5'-TTGGTTCACACCCATCACAAA-3' |
| MVD | 5'-AGCATCGCCCGGCAG-3' | 5'-TGGCCCCTGTAATTTCCCA-3' |
| LSS | 5'-GCGGCTGTGCGATGCT-3' | 5'-AGGTAGCGAACCCGCCA-3' |
| TGFB1 | 5'-TGGAAAGGGCCCAGCAC-3' | 5'-GCAATAGTTGGTATCCAGGGCT-3' |
| IGFII | 5'-AAGAGTTCAGAGAGGCCAAACG-3' | 5'-ATCTCCGAAGAGGCTCCCC-3' |
| MT1 | 5'-TGCTCCACCGGCGG-3' | 5'-TTTGCAGACACAGCCCTGG-3' |
| WNT6 | 5'-GGGCGCTGTCTGAGTCCA-3' | 5'-TGGCCCCTGTAATTTCCCA-3' |
| tPA | 5'-GGCCTGGCACGACACAAT-3' | 5'-CATCACATGGCACCAAGGTC-3' |
| VEGFC | 5'-CAGCTGCGGAAAGGCG-3' | 5'-TTTACACTGTCCCCTGTCCTGG-3' |
| DHODH | 5'-AACACAGGCTACGGGCCAG-3' | 5'-TCCCAGAGGCAGGCCCAT-3' |
| HepG2 cells | ||
| Gene | Forward | Reverse |
| ACADM | 5'-AGCTACCAAGTATGCCCTGGAA-3' | 5'-TAAATGATATTGCTTGGTGCTCTACA-3' |
| ACAT2 | 5'-TGGGCCACCCTCTTGGA-3' | 5'-CCAGTGTGTGTAACAGGGTCACA-3' |
| ACACA | 5'-GCTCCTTGTCACCTGCTTCTG-3' | 5'-TGTAGGCTAGAGATCCCCAAATCA-3' |
| APOA5 | 5'-AGGTGCGCCAGCGACTT-3' | 5'-GCGAGTGAAGGCAGCTATCTG-3' |
| APOC1 | 5'-CAAGGCTCGGGAACTCATCA-3' | 5'-CCCGCATCTTGGCAGAAA-3' |
| APOE | 5'-CGCTGGGTGCAGACACTGT-3' | 5'-AGGCCTTCAACTCCTTCATGGT-3' |
| APOL1 | 5'-TCAGCTGAAAGCGGTGAACA-3' | 5'-CTCTGCTCATTTCCAGGATGCT-3' |
| CYP27A1 | 5'-CCCTGTGGTCCCCACAAA-3' | 5'-GGAAGCCATCAACTTCAATTTCC-3' |
| HMGCR | 5'-CCTGTAACTCAGAGGGTCAAGATGAT-3' | 5'-CCAGCGACTGTGAGCATGAA-3' |
| HMGCS | 5'-TCTTAAATCAAGGCTTGATTCAAGAA-3' | 5'-TGTCCTCTCTGAGCTTCATGTTTT-3' |
| SREBF2 | 5'-CGAATTGAAAGACCTGGTCATG-3' | 5'-TCCTCAGAACGCCAGACTTGT-3' |
| FABP | 5'-CCGCTGGGTCCAAAGTGAT-3' | 5'-CATTGTCTCCAGCTCACATTCC-3' |
| FASN | 5'-GCAAATTCGACCTTTCTCAGAAC-3' | 5'-GGACCCCGTGGAATGTCA-3' |
| LDLR | 5'-AGATAGTGACAATGTCTCACCAAGCT-3' | 5'-CTCACGCTACTGGGCTTCTTCT-3' |
| PPARG | 5'-GCGAAAGCCTTTTGGTGACT-3' | 5'-CAGTGCATTGAACTTCACAGCAA-3' |
| TYMS | 5'-AATCACATCGAGCCACTGAAAA-3' | 5'-AATCCTGAGCTTTGGGAAAGGGT-3' |
For quantitative analysis of the data, CT (threshold-cycle number) values were normalized to those of GAPDH, with use of the ΔΔCT method.
Authors' contributions
SVC conceived the study design for the HepG2 samples and conducted all the analysis for this paper. NSG performed the microarray study on the F9 samples under the guidance of PJM.
Supplementary Material
Full list of statistically significant (p < 0.05) genes differentially expressed (2-fold or greater) in F9 and HepG2 cells on TSA treatment.
Acknowledgments
Acknowledgements
We would like to acknowledge Marcy Kuentzel and David Frank for help with the experiments and to Dr. Martin Tenniswood for his valuable suggestions.
Contributor Information
Sridar V Chittur, Email: schittur@albany.edu.
Niquiche Sangster-Guity, Email: nguity1@jhmi.edu.
Paulette J McCormick, Email: pmc@albany.edu.
References
- Monneret C. Histone deacetylase inhibitors. Eur J Med Chem. 2005;40:1–13. doi: 10.1016/j.ejmech.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Sakajiri S, Kumagai T, Kawamata N, Saitoh T, Said JW, Koeffler HP. Histone deacetylase inhibitors profoundly decrease proliferation of human lymphoid cancer cell lines. Exp Hematol. 2005;33:53–61. doi: 10.1016/j.exphem.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Burton GR, Nagarajan R, Peterson CA, McGehee RE., Jr Microarray analysis of differentiation-specific gene expression during 3T3-L1 adipogenesis. Gene. 2004;329:167–85. doi: 10.1016/j.gene.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Dannenberg LO, Edenberg HJ. Epigenetics of gene expression in human hepatoma cells: expression profiling the response to inhibition of DNA methylation and histone deacetylation. BMC Genomics. 2006;7:181. doi: 10.1186/1471-2164-7-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137–68. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- Mehnert JM, Kelly WK. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J. 2007;13:23–9. doi: 10.1097/PPO.0b013e31803c72ba. [DOI] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo J. 2001;20:6969–78. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WK, O'Connor OA, Marks PA. Histone deacetylase inhibitors: from target to clinical trials. Expert Opin Investig Drugs. 2002;11:1695–713. doi: 10.1517/13543784.11.12.1695. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Furumai R, Nishiyama M, Komatsu Y, Nishino N, Horinouchi S. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother Pharmacol. 2001;48:S20–6. doi: 10.1007/s002800100300. [DOI] [PubMed] [Google Scholar]
- Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem. 1993;268:22429–35. [PubMed] [Google Scholar]
- Yoshida M, Horinouchi S, Beppu T. Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays. 1995;17:423–30. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]
- Kim MS, Son MW, Kim WB, In Park Y, Moon A. Apicidin, an inhibitor of histone deacetylase, prevents H-ras-induced invasive phenotype. Cancer Lett. 2000;157:23–30. doi: 10.1016/s0304-3835(00)00465-1. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S. FR90 a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp Cell Res. 1998;241:126–133. doi: 10.1006/excr.1998.4027. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Ando T, Tsuchiya K, Fukazawa N, Saito A, Mariko Y, Yamashita T, Nakanishi O. Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem. 1999;42:3001–3. doi: 10.1021/jm980565u. [DOI] [PubMed] [Google Scholar]
- Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA. 1999;96:4592–7. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nervi C, Borello U, Fazi F, Buffa V, Pelicci PG, Cossu G. Inhibition of histone deacetylase activity by trichostatin A modulates gene expression during mouse embryogenesis without apparent toxicity. Cancer Res. 2001;61:1247–9. [PubMed] [Google Scholar]
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Sangster-Guity N, Yu LM, McCormick P. Molecular profiling of embryonal carcinoma cells following retinoic acid or histone deacetylase inhibitor treatment. Cancer Biol Ther. 2004;3:1109–20. doi: 10.4161/cbt.3.11.1190. [DOI] [PubMed] [Google Scholar]
- Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- Williamson RA, Yea CM, Robson PA, Curnock AP, Gadher S, Hambleton AB, Woodward K, Bruneau JM, Hambleton P, Moss D, et al. Dihydroorotate dehydrogenase is a high affinity binding protein for A77 1726 and mediator of a range of biological effects of the immunomodulatory compound. J Biol Chem. 1995;270:22467–72. doi: 10.1074/jbc.270.38.22467. [DOI] [PubMed] [Google Scholar]
- Greene S, Watanabe K, Braatz-Trulson J, Lou L. Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem Pharmacol. 1995;50:861–7. doi: 10.1016/0006-2952(95)00255-x. [DOI] [PubMed] [Google Scholar]
- Fairbanks LD, Bofill M, Ruckemann K, Simmonds HA. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. Disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J Biol Chem. 1995;270:29682–9. [PubMed] [Google Scholar]
- Breedveld FC. New insights in the pathogenesis of rheumatoid arthritis. J Rheumatol Suppl. 1998;53:3–7. [PubMed] [Google Scholar]
- Chung YL, Lee MY, Wang AJ, Yao LF. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol Ther. 2003;8:707–17. doi: 10.1016/s1525-0016(03)00235-1. [DOI] [PubMed] [Google Scholar]
- Arend WP, Dayer JM. Inhibition of the production and effects of interleukin-1 and tumor necrosis factor alpha in rheumatoid arthritis. Arthritis Rheum. 1995;38:151–60. doi: 10.1002/art.1780380202. [DOI] [PubMed] [Google Scholar]
- Young DA, Lakey RL, Pennington CJ, Jones D, Kevorkian L, Edwards DR, Cawston TE, Clark IM. Histone deacetylase inhibitors modulate metalloproteinase gene expression in chondrocytes and block cartilage resorption. Arthritis Res Ther. 2005;7:R503–12. doi: 10.1186/ar1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, Dona G, Fossati G, Sozzani S, Azam T, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci USA. 2002;99:2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller NE. Coronary atherosclerosis and plasma lipoproteins: epidemiology and pathophysiologic considerations. J Cardiovasc Pharmacol. 1982;4:S190–5. [PubMed] [Google Scholar]
- Sakakura Y, Shimano H, Sone H, Takahashi A, Inoue N, Toyoshima H, Suzuki S, Yamada N. Sterol regulatory element-binding proteins induce an entire pathway of cholesterol synthesis. Biochem Biophys Res Commun. 2001;286:176–83. doi: 10.1006/bbrc.2001.5375. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Sone H, Shimano H, Sakakura Y, Inoue N, Amemiya-Kudo M, Yahagi N, Osawa M, Suzuki H, Yokoo T, Takahashi A, et al. Acetyl-coenzyme A synthetase is a lipogenic enzyme controlled by SREBP-1 and energy status. Am J Physiol Endocrinol Metab. 2002;282:E222–30. doi: 10.1152/ajpendo.00189.2001. [DOI] [PubMed] [Google Scholar]
- Shechter I, Dai P, Huo L, Guan G. IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells: evidence that IDH1 may regulate lipogenesis in hepatic cells. J Lipid Res. 2003;44:2169–80. doi: 10.1194/jlr.M300285-JLR200. [DOI] [PubMed] [Google Scholar]
- Ekins S, Mankowski DC, Hoover DJ, Lawton MP, Treadway JL, Harwood HJ., Jr Three-dimensional quantitative structure-activity relationship analysis of human CYP51 inhibitors. Drug Metab Dispos. 2007;35:493–500. doi: 10.1124/dmd.106.013888. [DOI] [PubMed] [Google Scholar]
- Kastelein JJ. The future of best practice. Atherosclerosis. 1999;143:S17–21. doi: 10.1016/s0021-9150(99)00103-3. [DOI] [PubMed] [Google Scholar]
- Munro JM, Cotran RS. The pathogenesis of atherosclerosis: atherogenesis and inflammation. Lab Invest. 1988;58:249–61. [PubMed] [Google Scholar]
- Iso H, Jacobs DR, Jr, Wentworth D, Neaton JD, Cohen JD. Serum cholesterol levels and six-year mortality from stroke in 350,977 men screened for the multiple risk factor intervention trial. N Engl J Med. 1989;320:904–10. doi: 10.1056/NEJM198904063201405. [DOI] [PubMed] [Google Scholar]
- Kannel WB, Castelli WP, Gordon T, McNamara PM. Serum cholesterol, lipoproteins, and the risk of coronary heart disease. The Framingham study. Ann Intern Med. 1971;74:1–12. doi: 10.7326/0003-4819-74-1-1. [DOI] [PubMed] [Google Scholar]
- Ranade VV. Significance of cholesterol in health and disease. Int J Clin Pharmacol Ther Toxicol. 1993;31:276–84. [PubMed] [Google Scholar]
- Klag MJ, Ford DE, Mead LA, He J, Whelton PK, Liang KY, Levine DM. Serum cholesterol in young men and subsequent cardiovascular disease. N Engl J Med. 1993;328:313–8. doi: 10.1056/NEJM199302043280504. [DOI] [PubMed] [Google Scholar]
- Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci USA. 2005;102:5374–9. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenn SM, Jr, Parks JS, Immermann FW, Rudel LL. ACAT inhibitors CL 283,546 and CL 283,796 reduce LDL cholesterol without affecting cholesterol absorption in African green monkeys. J Lipid Res. 1995;36:1199–210. [PubMed] [Google Scholar]
- Rudel LL, Lee RG, Parini P. ACAT2 is a target for treatment of coronary heart disease associated with hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2005;25:1112–8. doi: 10.1161/01.ATV.0000166548.65753.1e. [DOI] [PubMed] [Google Scholar]
- Ferguson M, Henry PA, Currie RA. Histone deacetylase inhibition is associated with transcriptional repression of the Hmga2 gene. Nucleic Acids Res. 2003;31:3123–33. doi: 10.1093/nar/gkg403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TA, 3rd, Kelley K, Wilson MD, Sawyer JK, Rudel LL. Dietary fat-induced alterations in atherosclerosis are abolished by ACAT2-deficiency in ApoB100 only, LDLr-/- mice. Arterioscler Thromb Vasc Biol. 2007;27:1396–402. doi: 10.1161/ATVBAHA.107.142802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full list of statistically significant (p < 0.05) genes differentially expressed (2-fold or greater) in F9 and HepG2 cells on TSA treatment.